


Abstract
The aim of this study was to evaluate a unified method for predicting human in vivo intrinsic clearance (CLint, in vivo) and hepatic clearance (CLh) from in vitro data in hepatocytes and microsomes by applying the unbound fraction in blood (fub) and in vitro incubations (fuinc). Human CLint, in vivo was projected using in vitro data together with biological scaling factors and compared with the unbound intrinsic clearance (CLint, ub, in vivo) estimated from clinical data using liver models with and without the various fu terms. For incubations conducted with fetal calf serum (n = 14), the observed CLint, in vivo was modeled well assuming fuinc and fub were equivalent. CLint, ub, in vivo was predicted best using both fub and fuinc for other hepatocyte data (n = 56; r2 = 0.78, p = 3.3 × 10–19, average fold error = 5.2). A similar model for CLint, ub, in vivo was established for microsomal data (n = 37; r2 = 0.77, p = 1.2 × 10–12, average fold error = 6.1). Using the model for CLint, ub, in vivo (including a further empirical scaling factor), the CLh in humans was also calculated according to the well stirred liver model for the most extensive dataset. CLint, in vivo and CLh were both predicted well using in vitro human data from several laboratories for acidic, basic, and neutral drugs. The direct use of this model using only in vitro human data to predict the metabolic component of CLh is attractive, as it does not require extra information from preclinical studies in animals.
Existing methods for the prediction of drug clearance in humans involve the use of in vitro human metabolic stability (intrinsic clearance, CLint) data (Iwatsubo et al., 1997), consideration of preclinical animal data (Boxenbaum, 1982), or a combination of these approaches (Lave et al., 1997a; Naritomi et al., 2001). In vitro drug metabolism kinetic parameters can provide an estimate of in vivo CLint via “scaling” with established biological scaling factors (SFs) e.g., hepatocellularity for isolated hepatocytes, or a SF for microsomes based on incomplete microsomal recovery from human liver tissue using the cytochrome P450 (P450) content in homogenate and microsomes (Houston, 1984). CLint may subsequently be used to provide an estimate of hepatic clearance (CLh) using several liver models (Houston, 1984; Ito and Houston, 2004).
To date, the more extensive analyses of human clearance predictions have concentrated on P450 substrates, and data have therefore been generated in human liver microsomes (Iwatsubo et al., 1997; Obach, 1999; Naritomi et al., 2001). In general, these studies have been less comprehensive in the range of approaches investigated, with only occasional attention given to chemical class (Obach et al., 1997). Interestingly, these reports have also assessed the ability to predict human CLh rather than the more fundamental parameter, CLint, as advocated initially (Houston, 1984; Ito and Houston, 2004). Some controversy also still exists over use of fuinc, with some laboratories having suggested that fuinc and fub may cancel, negating their inclusion in liver models (Obach et al., 1997). Recent reports have challenged this assumption (Obach, 1999; Austin et al., 2002) and perhaps suggest that consideration of CLh rather than CLint, in vivo may desensitize such analyses, particularly to errors associated with higher enzyme activities (Ito and Houston, 2004).
The aim of this study was to investigate direct in vitro-in vivo scaling of human in vitro data generated in hepatocytes and microsomes for predicting human clearance in vivo by applying recently described models for estimating fuinc (Austin et al., 2002, 2005). To provide a more mechanistic insight, in vitro human CLint data were compiled from recent in-house and published studies, and associated in vivo CLint values were derived from published CLh data. Combining datasets permitted extension of models described in previous studies. In addition, the relative drug binding within blood and in vitro incubation matrices is considered further, with respect to their incorporation into liver models.
Materials and Methods
Data Collection. CLint, in vitro data derived from hepatocyte (Lave et al., 1997a,b; Lau et al., 2002; Shibata et al., 2002; Naritomi et al., 2003) or microsomal incubations (Carlile et al., 1999; Obach, 1999; Naritomi et al., 2001; Andersson et al., 2004) were generated in the authors' laboratory and collated from several published studies (Tables 1, 2, 3). Incubation conditions for data generated in the authors' laboratory have been detailed previously (Austin et al., 2002; McGinnity et al., 2004). Data from microsomal studies reflected a variety of methods including formal Michaelis-Menten kinetic analysis [CLint = Vmax/Km for specific metabolite(s) formation (Carlile et al., 1999; Andersson et al., 2004)] and substrate depletion at low substrate concentrations (Obach, 1999; Naritomi et al., 2001), which was used for all hepatocyte data. Datasets were compiled with several key objectives in mind: to expand existing databases substantially, to provide some assessment of interlaboratory variability, and to complement external datasets in terms of representation from different chemical classes covering a range of physicochemical properties. Particular emphasis was put on hepatocyte data generated in this laboratory for acidic drugs, which were sparsely represented in external datasets. Data produced in the authors' laboratory represent the mean of at least three hepatocyte donors. Replicates and variability in other data can be found in the original reports.
Data for human hepatocyte incubations conducted in the absence of fetal calf serum (corrected for both fub and fuinc)
Data for human hepatocyte incubations conducted in the presence of fetal calf serum corrected for both binding to hepatocytes (fuinc) and fub
Data for human microsome incubations corrected for both fub and fuinc
The hepatocyte data were compiled from several sources: incubations conducted with more simple cell suspensions (Lau et al., 2002; Naritomi et al., 2003; authors' laboratory), and cells cultured in the presence of exogenous protein. Additional protein was either autologous (human) serum (Shibata et al., 2002) or 10% fetal calf serum (FCS) for incubations conducted with cultured cells for up to 72 h (Lave et al., 1997a,b; Schneider et al., 1999). CLint, in vitro was estimated using only an unbound fraction in plasma (fup) correction for incubations that included serum, since binding to plasma proteins was deemed to be greater than any hepatocyte binding (Shibata et al., 2002; Austin et al., 2005). Since interspecies differences in plasma protein binding precluded such a simple correction for incubations conducted in the presence of FCS, a correction was made only for binding to cells, and this dataset was treated separately. For the remaining datasets, the unbound fraction in the incubation (fuinc) was applied either as reported by the authors (Naritomi et al., 2003) or predicted from a consideration of chemical class and either log D7.4 or log P: for microsomes, log(1 – fu/fu) = 0.53 log P/D –1.42 (Austin et al., 2002); and for hepatocytes, log(1 – fu/fu) = 0.40 log P/D –1.38 (Austin et al., 2005).
CLint, ub, in vivo was calculated from values of CLh, the fup, and blood-to-plasma concentration ratio (RB; fraction unbound in blood, fub = fup/RB) reported for each drug according to the well stirred or dispersion liver model (Shibata et al., 2002). Where values of RB were not provided or readily available, this parameter was assumed to be unity for neutral and basic compounds and 0.55 for acids. The assumptions of the various liver models have been detailed previously (Ito and Houston, 2004).
Where the same drug had been studied by several laboratories (Tables 4 and 5), individual values for CLh and fub were used to estimate CLint, ub, in vivo, and CLint, in vitro and fuinc were used to project (scaled) CLint, ub, in vivo. The mean values for key parameters were then used for further modeling and statistical analyses. The sources used to derive key parameters are too extensive to be listed here but can be found in the original references cited.
Human hepatocyte data concordance across different laboratories for dataset studied
Human microsome data concordance across different laboratories for dataset studied
Prediction of the in Vivo Intrinsic Clearance. CLint, in vivo was predicted using SFs for hepatocytes and microsomes to convert the unit of the CLint from μl/min/106 cells or μl/min/mg protein to ml/min/kg using an estimate of human liver hepatocyte content. Hepatocyte content (or hepatocellularity) was routinely 120 × 106 cells/g liver. Some variability was evident in the microsomal protein concentration per gram of liver (45–50 mg protein/g liver) and the human liver weight values (1500–1800 g liver/70 kg) cited, but this was not considered to impact significantly on the conclusions of this study.
Impact of Plasma and in Vitro Binding. In in vitro studies, some drugs bind nonspecifically within the matrix; hence, the kinetic parameters estimated need to be corrected to reflect the unbound drug. Obviously, if fub and fuinc were equivalent, their terms in the equations for liver models would cancel out  where CLint* = (CLint, in vitro × SF) and fuinc is the unbound fraction in hepatocytes or microsomes; Qh = liver blood flow (20 ml/min/kg).
where CLint* = (CLint, in vitro × SF) and fuinc is the unbound fraction in hepatocytes or microsomes; Qh = liver blood flow (20 ml/min/kg).
The role of fuinc and fub was investigated using several approaches. First, the human CLint, in vivo values were predicted assuming fuinc to be unity and compared with CLint, in vivo calculated from “deconvolution” of the well stirred model, acknowledging the potential limitations of this model for highly extracted compounds (Ito and Houston, 2004):  CLint, in vivo data were provided in several reports (Shibata et al., 2002; Naritomi et al., 2003). Relationships obtained using the parallel tube model to compute CLint, in vivo were very similar to those reported (data not shown).
CLint, in vivo data were provided in several reports (Shibata et al., 2002; Naritomi et al., 2003). Relationships obtained using the parallel tube model to compute CLint, in vivo were very similar to those reported (data not shown).
CLint, in vivo values were also compared without consideration of both fuinc and fub. Finally, CLint, in vivo predictions using fuinc were compared with estimates from clinical pharmacokinetics invoking fub (CLint, ub, in vivo).
Prediction of Hepatic (Blood) Clearance. Using the in vivo intrinsic clearance estimates outlined above, CLh, was calculated according to the well stirred liver model as follows: 
Accuracy of Predictions.Quantitative linear regression analysis was not considered appropriate for the predicted CLh data since it is clearly not homoscedastic; i.e., the error in the y data are not even approximately constant across the full range of the data: the spread in the y data decreases with increasing log(predicted clearance). This behavior is a consequence of the format of the well stirred model, which gradually forces compounds with increasingly high intrinsic clearance toward the same value of predicted clearance, that of hepatic blood flow, Qh.
Quantitative regression analyses were performed, however, for the log (predicted) and log (observed) values for CLint, in vivo to obtain the regression equation, correlation coefficient (r2), and a summary of its statistics (standard deviation, S.D., the F statistic, and the p value). The average fold error (afe) of each prediction method was also calculated to provide a measure of bias with equal value to under- and over-predictions: 
Results
Prediction of CLint, ub and CLh from Hepatocyte Incubations.Figure 1 (A–C) shows plots of log(observed CLh) against log(predicted CLh) where hepatic clearance is predicted from the well stirred model, including or excluding the various fu terms. For incubations conducted without FCS, the model with both fuinc and fub corrections shows the best qualitative relationship between log(observed CLh) and log(predicted CLh). The model with an fub correction only (i.e., ignoring fuinc) appears to show the next best qualitative trend, with the uncorrected model showing a very scattered relationship with the acidic compounds becoming separated from the neutral and basic compounds.
Interlaboratory variability in CLint, in vitro was acceptable (≤3-fold) for some compounds studied in hepatocytes under similar conditions (diclofenac, imipramine, naloxone, propranolol, and tolbutamide) and microsomes (diazepam, ibuprofen, and tolbutamide). Variability was more significant for other compounds, possibly due to differences in quality of liver samples, established interdonor differences, and incubation conditions, e.g., potential effects of coincubating hepatocytes with serum. This highlights the challenges in compiling datasets from several sources.

Correlation between the observed and predicted human CLh (A–C) and CLint, ub (D–F) for a dataset of 57 drugs from hepatocyte incubations without added FCS. Panels A and D refer to data modeled with fub only; B and E, assuming fub = fuinc; and C and F, incorporating both fub and fuinc terms. Symbols depict different chemical classes (•, acid; ○, base; □, neutral). Dotted lines indicate regression analysis. The equation of best fit for F is given by y = 1.08x + 0.38 (S.D. = 0.38, r2 = 0.78, F = 187.5, p = 3.3 × 10–19, afe = 5.2).
A more appropriate transformation of the data for linear regression analysis is to consider CLint, ub, i.e., log(CLint, in vivo, ub), plotted against log(predicted CLint, ub) as shown in Figs. 1 and 2 (D to F), which also provides a substantial dynamic range with which to study in vitro-in vivo comparisons at a detailed, mechanistic level (Ito and Houston, 2004). These plots show a fairly constant spread in the y data across the full range. Linear regression has been applied to the 3 different scaling models in Fig. 1, D to F, and the resulting statistical parameters are given in Table 6, along with the average fold error of each method. In terms of the statistics of linear regression, the model, which includes both fuinc and fub corrections, provides the best fit to the data as shown by the significantly higher r2 value and lower regression standard deviation (S.D.) compared with the other two models (Table 6). The F statistic and p value of this model are also clearly superior to those of the other two models. The slope of the model including both correction terms (1.08) is close to unity, but the intercept (0.38) reveals a constant bias in the model leading to an approximately 5-fold under-prediction of CLint, ub across the range of compounds. Despite evidence of interlaboratory variability for some data, this model appears largely independent of laboratory and chemical class. In terms of regression statistics, the model utilizing only an fub correction (i.e., omitting fuinc) has the next best performance, with the uncorrected model performing poorly.
Regression analysis summary for various models applied to hepatocyte incubations conducted in the presence (+FCS) and absence (-FCS) of fetal calf serum
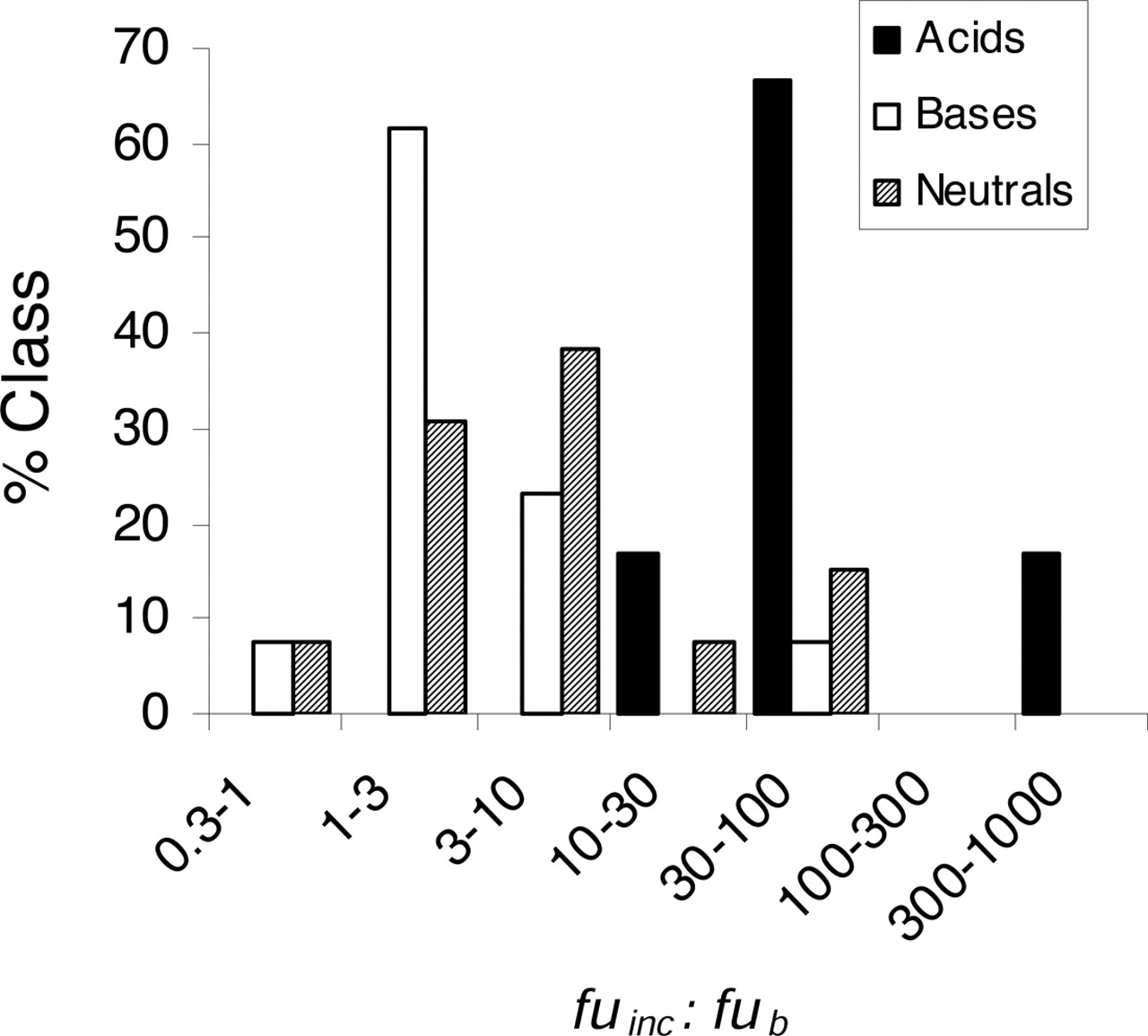
Interestingly, subdividing this dataset into chemical class indicated that the predictions using the model without fub and fuinc corrections were very poor for acidic drugs, in particular: predictions appeared better for basic and neutral compounds. Closer inspection of the ratio between fuinc and fub for each chemical class provided further mechanistic insight (Fig. 3). For the majority of the basic and neutral drugs studied, this ratio was between 1 and 10. However, for some neutral (including the benzodiazepines, diazepam and oxazepam, and the lipophilic calcium channel blocker, felodipine) and lipophilic, basic compounds (for example, mibefradil), the ratio was somewhat larger. By contrast, for acidic drugs, this ratio was much higher on average, reflecting the extensive binding to albumin for many of these compounds in vivo.
Prediction of CLint, ub and of CLb from Hepatocyte Incubations Containing FCS. By contrast, for hepatocyte incubations conducted (up to 72 h) in the presence of FCS, CLb was described well under all conditions, and CLint, ub, in vivo was predicted well from CLint, in vitro (Fig. 3; Table 6). However, models incorporating only fub or both fuinc and fub, although statistically significant, yielded a large bias as evidenced by deviations in the gradient, the intercept, and the associated afe (42- to 86-fold; Table 6). The most robust model (in terms of bias in gradient, intercept, and afe) for CLint, ub, in vivo was derived from ignoring both fuinc and fub.
Prediction of CLint, ub from Microsomal Data. Applying this knowledge to the microsomal data again yielded a robust, highly significant relationship between the scaled CLint, ub, in vivo estimate (using both fub and fuinc) and that derived from clinical pharmacokinetic data (r2 = 0.77, p = 1.22 × 10–12; Fig. 4). The afe (6.1) for this dataset was very similar to the larger human hepatocyte dataset.
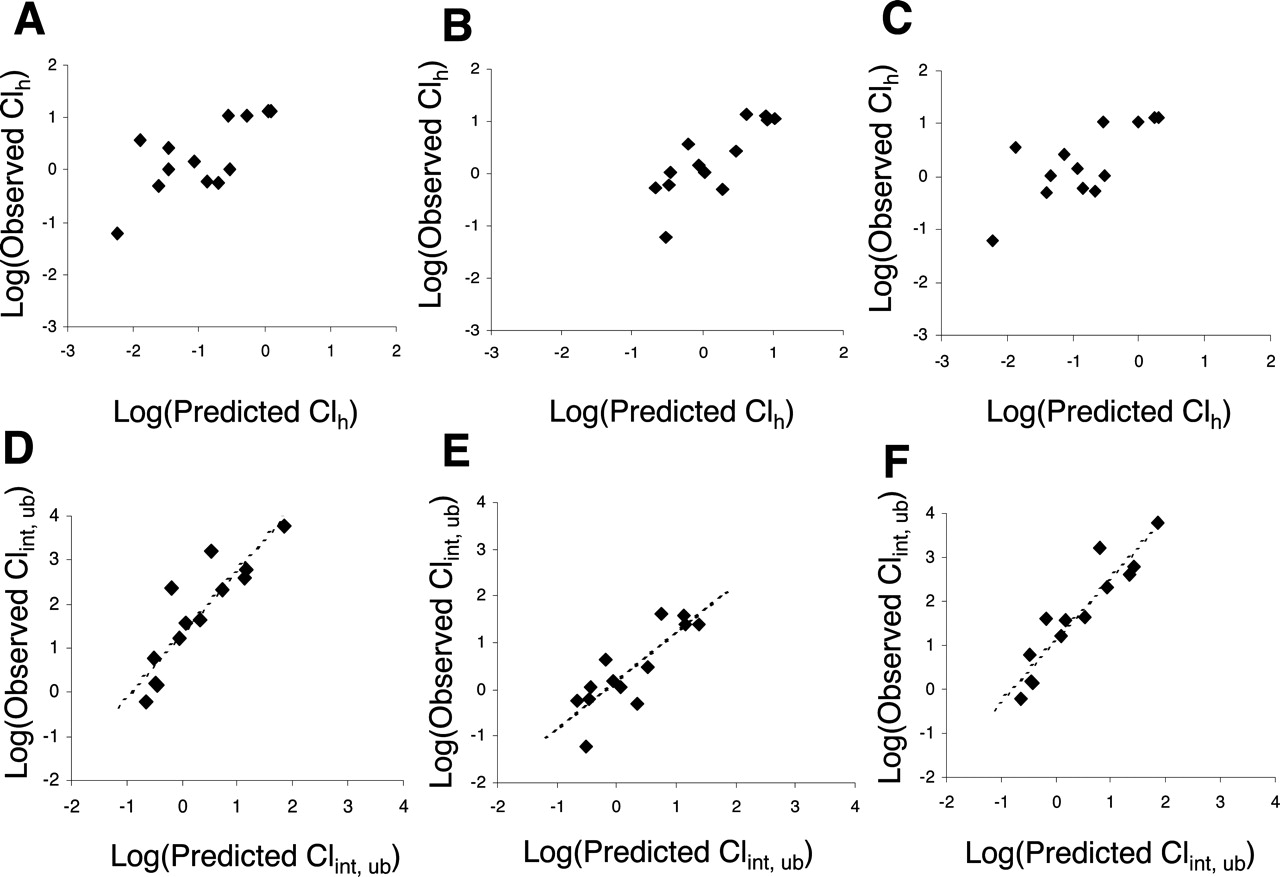
Correlation between the observed and predicted human CLh (A–C) and CLint, ub (D–F) for a dataset of 14 drugs from hepatocyte incubations with added FCS. Panels A and D refer to data modeled with fub only; B and E, assuming fub = fuinc; and C and F, incorporating both fub and fuinc terms. Dotted lines indicate regression analysis.
Further Model Development. For the most extensive hepatocyte dataset, since the fuinc and fub corrected model was highly correlated but with a constant offset, the regression equation was applied to data to evaluate the most precise clearance prediction for test compounds:  and....
and....  Transformation of these predictions of CLint, in vivo to projections of CLb finally give afe values of 1.9 for the fuinc and fub corrected method, 2.0 for the fub corrected method and 15.1 for the uncorrected method. Therefore the model with both correction terms has the potential for making the most precise predictions of CLh for external test compounds.
Transformation of these predictions of CLint, in vivo to projections of CLb finally give afe values of 1.9 for the fuinc and fub corrected method, 2.0 for the fub corrected method and 15.1 for the uncorrected method. Therefore the model with both correction terms has the potential for making the most precise predictions of CLh for external test compounds.
Discussion
Prediction of human pharmacokinetics remains an intense area of research. It is widely accepted that simple allometric methods are generally not suitable for predicting human clearance for compounds, which exhibit low to intermediate extraction via metabolism (Lave et al., 1997a; Nagilla and Ward, 2004). In vitro-in vivo scaling of metabolic clearance has received much attention (Houston, 1984; Houston and Carlile, 1997; Iwatsubo et al., 1997; Naritomi et al., 2001) and also provides input for physiologically based pharmacokinetic models (Theil et al., 2003). Early, extensive investigations of in vitro-in vivo scaling of hepatic metabolic clearance focused largely on the rat as a model species (Houston, 1984; Houston and Carlile, 1997). More recently, several laboratories have extended these analyses of human clearance predictions. These studies have tended to concentrate on a relatively small number of drugs metabolized by P450s, and data have therefore been generated in human liver microsomes (Iwatsubo et al., 1997; Obach et al., 1997). In general, these studies have been less comprehensive in the range of approaches investigated, with only occasional attention given to chemical class (Obach, 1999). Interestingly, these reports have also assessed the ability to predict human CLh rather than the more fundamental parameter, CLint, as advocated in the seminal work by Houston and colleagues (Houston, 1984; Houston and Carlile, 1997; Ito and Houston, 2004).

Distribution of fuinc:fub ratio for drugs studied in hepatocytes classified by chemical class.

Correlation between the observed and predicted human CLh (A) and CLint, ub, in vivo (B) for a dataset of 37 drugs from microsomal incubations. Symbols depict different chemical classes (•, acid; ○, base; □, neutral). The equation of best fit (indicated by the dotted line in B) is given by y = 0.88x + 0.71 (S.D. = 0.48, r2 = 0.77, F = 115.8, p = 1.2 × 10–12, afe = 6.1).
To assess various approaches to predicting human in vivo hepatic metabolic clearance, a database has been collated from in-house and literature sources. Hepatocytes appear to be the most appropriate in vitro system to assess hepatic metabolic stability. Studies in the rat have advocated hepatocytes for more accurate prediction of rapid clearance (Houston, 1984; Houston and Carlile, 1997), and hepatocytes not only contain P450s and the major phase 2 enzymes but also hepatobiliary transporters, which may modulate concentrations of substrate accessible to drug-metabolizing enzymes (Shitara et al., 2003; Liu and Pang, 2005). Advances in cryopreservation technology have recently enabled studies with human hepatocytes to become more routine (Li et al., 1999; McGinnity et al., 2004). The present analysis considered primarily in vitro and in vivo CLint estimates, the latter obtained from deconvoluting CLh to generate a wide range of values to allow detailed, mechanistic comparisons. Predictions of the kinetic parameter CLh were also evaluated.
The role of plasma protein binding in clearance prediction has been the subject of some controversy. Whereas the basic tenet of pharmacokinetics states that the unbound drug concentration in the plasma dictates tissue distribution, some reports using microsomes have suggested that in vitro CLint may provide a better estimate of in vivo clearance of total rather than unbound drug (Obach et al., 1997; Lin et al., 1999). Presumably, the assumption was that fub and fuinc effectively nullified in the liver model calculation, negating the measurement of either process. However, measurements of in vitro binding have shown that drug binding within these two matrices is not equivalent and hence fuinc should not be ignored, in principle (Obach, 1999; Austin et al., 2002, 2005).
Most models were reasonable for neutral and basic drugs, particularly for CLh, as reported previously (Davis and Riley, 2004; McGinnity et al., 2004; Riley and Kenna, 2004). However, examination of the hepatocyte data indicated the need to consider both fuinc and fub for incubations not conducted in the presence of exogenous protein to include all chemistries. Previous studies have shown that, as for microsomes, fuinc for hepatocytes may be substantial for lipophilic neutral and basic compounds and can be predicted from consideration of charge at physiological pH and lipophilicity (Austin et al., 2005). Some neutral compounds (e.g., the benzodiazepines diazepam and oxazepam) and lipophilic bases (e.g., mibefradil; Fig. 2) also showed a similar effect, as suggested previously (Riley and Kenna, 2004). By contrast, hepatocyte binding for most acidic drugs is low compared with their binding to distinct sites on albumin. Inclusion of both fuinc and fub terms resulted in a unified model, the robustness of which was indicated by its ability to translate across both laboratories and chemical classes.
Interestingly, several reports on relatively small numbers of compounds have indicated that data generated from hepatocyte incubations containing exogenous protein (either FCS or serum) may yield more direct estimates of CLint, in vivo and CLh (Lave et al., 1997a,b; Shibata et al., 2003). However, the nonphysiological composition of such assays and effects other than those attributable to protein binding (Blanchard et al., 2005) remain the subject of much controversy and may contribute to some of the variability depicted in Table 4. Furthermore, such methodology poses challenges in terms of determining low CLint values accurately and may necessitate long incubation periods (up to 72 h) with cultured cells, which may incur a (differential) loss of P450 activity.
Previous reports have debated the pros and cons of inclusion of fuinc for data from human liver microsomes; hence, this topic was not analyzed in depth here. Inclusion of both fuinc and fub yielded a highly significant correlation between predicted and observed CLint, ub with a bias or offset similar to that shown in Fig. 1F, as suggested previously for both CLh and CLint (Iwatsubo et al., 1997; Obach, 1999; Naritomi et al., 2001).
A model incorporating in vivo and in vitro data from preclinical and clinical studies has the potential advantage of providing a drug-specific factor that would theoretically correct for any systematic difference between in vitro and in vivo parameters (Naritomi et al., 2001, 2003). However, further work using larger databases is required to validate this approach and evaluate whether various “factors” (which may be passive or active in origin) translate routinely across a range of species.
In summary, human in vitro CLint provided accurate predictions of CLint, ub and CLh with more robust models resulting from incorporation of fuinc for both hepatoyctes and microsomes. Using the standard biological SFs alone to scale in vitro CLint to provide in vivo CLint estimates resulted in a systematic under-prediction for data from both hepatocytes and microsomes, an observation consistent with other studies using human tissues. Interestingly, previous analyses of rat predictions from both in vitro systems did not show such a systematic bias (Ito and Houston, 2004). Although a scientific rationale for this observation is lacking currently for these specific datasets, likely contributors include active transport processes in vivo not reflected adequately in vitro (Liu and Pang, 2005; Shitara et al., 2005); incorrect assumptions within the liver models; interindividual variability atypical kinetics, e.g., for CYP3A4 substrates (Houston and Galetin, 2004); quality of tissue used for hepatocyte preparations; or some extrahepatic metabolism in vivo. Future studies will aim to provide a systematic analysis and mechanistic interpretation, which could then be applied to modify existing scaling strategies further.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.004259.
-
ABBREVIATIONS: CLint, intrinsic clearance; SF, scaling factor; P450, cytochrome P450; CLh, hepatic clearance; CLint, in vitro, in vitro intrinsic clearance; CLint, in vivo, in vivo intrinsic clearance; fup, plasma unbound fraction; RB, blood-to-plasma concentration ratio; fuinc, unbound fraction in incubations in vitro; fub, unbound fraction in blood; FCS, fetal calf serum; Qh, hepatic blood flow; afe, average fold error.
- Received February 14, 2005.
- Accepted May 26, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}