


Abstract
Siponimod, a next-generation selective sphingosine-1-phosphate receptor modulator, is currently being investigated for the treatment of secondary progressive multiple sclerosis. We investigated the absorption, distribution, metabolism, and excretion (ADME) of a single 10-mg oral dose of [14C]siponimod in four healthy men. Mass balance, blood and plasma radioactivity, and plasma siponimod concentrations were measured. Metabolite profiles were determined in plasma, urine, and feces. Metabolite structures were elucidated using mass spectrometry and comparison with reference compounds. Unchanged siponimod accounted for 57% of the total plasma radioactivity (area under the concentration–time curve), indicating substantial exposure to metabolites. Siponimod showed medium to slow absorption (median Tmax: 4 hours) and moderate distribution (Vz/F: 291 l). Siponimod was mainly cleared through biotransformation, predominantly by oxidative metabolism. The mean apparent elimination half-life of siponimod in plasma was 56.6 hours. Siponimod was excreted mostly in feces in the form of oxidative metabolites. The excretion of radioactivity was close to complete after 13 days. Based on the metabolite patterns, a phase II metabolite (M3) formed by glucuronidation of hydroxylated siponimod was the main circulating metabolite in plasma. However, in subsequent mouse ADME and clinical pharmacokinetic studies, a long-lived nonpolar metabolite (M17, cholesterol ester of siponimod) was identified as the most prominent systemic metabolite. We further conducted in vitro experiments to investigate the enzymes responsible for the oxidative metabolism of siponimod. The selective inhibitor and recombinant enzyme results identified cytochrome P450 2C9 (CYP2C9) as the predominant contributor to the human liver microsomal biotransformation of siponimod, with minor contributions from CYP3A4 and other cytochrome P450 enzymes.
Introduction
Siponimod (BAF312; Novartis Pharma AG, Basel, Switzerland), a next-generation sphingosine-1-phosphate (S1P) receptor modulator, is currently being evaluated for the treatment of secondary progressive multiple sclerosis (SPMS). S1P signaling pathways play a role in multiple sclerosis pathophysiology, and the therapeutic potential of S1P receptor modulation in multiple sclerosis treatment has been demonstrated using fingolimod (Gilenya), a functional antagonist of S1P1 receptors (Brinkmann et al., 2002; Baumruker et al., 2007; Cohen et al., 2010; Kappos et al., 2010), and siponimod (Selmaj et al., 2013; Kappos et al., 2016, 2018).
Sphingosine-1-phosphate receptors are widely expressed in the body, including in lymphocytes and neural cells such as oligodendrocytes and astrocytes (Brinkmann, 2007). Siponimod has shown nanomolar affinity for S1P1/S1P5 receptors, inducing a profound and long-lasting internalization of S1P1 receptors (Gergely et al., 2012). The internalization of S1P1 receptors renders lymphocytes unresponsive to S1P, depriving them of an obligatory signal to egress from the lymph nodes and recirculate into the central nervous system (CNS), subsequently reducing neuroinflammation (Matloubian et al., 2004). Compared with the first-generation S1P receptor modulators, siponimod does not require phosphorylation in vivo.
Evidence from preclinical models has demonstrated that siponimod readily crosses the blood–brain barrier to enter the CNS. Thus, siponimod may have a direct neurobiologic effect in the CNS, independent from its effects on peripheral lymphocytes, through selective modulation of S1P1 on astrocytes and S1P5 on oligodendrocytes (Tavares et al., 2014). Moreover, in preclinical multiple sclerosis models, siponimod was shown to reverse chronic neurologic paralysis and reduce inflammation and demyelination (Nuesslein-Hildesheim et al., 2009; Choi et al., 2011; Gergely et al., 2012; Brana et al., 2014).
Single-dose and multiple-dose studies of siponimod in healthy volunteers have established 25 mg as the maximum tolerated single dose, with an acceptable safety and tolerability profile at dose levels ranging from 0.3 mg to 20 mg administered daily for 28 days (Novartis data on file). In an adaptive, dose-ranging, phase 2 study, siponimod treatment reduced the combined unique active MRI lesions up to 80% versus placebo in relapsing–remitting multiple sclerosis patients. The MRI dose–response curve indicated near-maximal efficacy at 2 mg (Selmaj et al., 2013). Subsequently, the siponimod 2-mg dose was selected for further development based on the favorable benefit-risk profile. In a recently concluded phase 3 study in patients with SPMS, siponimod (2 mg once daily administration with an initial 6-day dose titration) significantly reduced the risk of confirmed disability progression in an advancing SPMS population.
The detection and identification of all main metabolites in a human absorption, distribution, metabolism, and excretion (ADME) study is essential to plan future studies in case of quantitative and/or qualitative differences in drug metabolism between the animals used in nonclinical safety assessments and humans. As per the M3 (R2) International Conference on Harmonization (ICH) guidance, if a metabolite present in human plasma exceeds a defined threshold (10% of the total drug-related material), an equal or greater steady-state exposure to the metabolite should also be seen in at least one toxicology species to avoid any further metabolite testing (http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf).
Our current study evaluated the ADME of a single 10-mg oral dose of [14C]siponimod in healthy men. The study results also provided data that could be used to estimate the main elimination pathways. In general, enzymes involved in metabolic pathways estimated to contribute to ≥25% of drug elimination should be identified if possible and their in vivo contribution should be quantified (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf). The primary objectives of our study were to: 1) determine the pharmacokinetics of total radioactivity of [14C]siponimod and of any important metabolites in the plasma; 2) identify and quantify siponimod and the metabolites of siponimod in plasma, urine, and feces after a single 10-mg oral dose of [14C]siponimod; 3) determine the rate and routes of excretion and the mass balance of total radioactivity in urine and feces; 4) evaluate the absorption of unchanged drug and total radioactivity from available urinary and fecal excretion data; and 5) elucidate the key biotransformation pathways and clearance mechanisms of siponimod in humans. Furthermore, in vitro experiments were conducted to investigate the major enzymes involved in the oxidative metabolism of siponimod. A brief description of the cholesterol ester conjugate or M17 metabolite, a rare metabolite first identified as a major component in the mouse ADME study with its presence later confirmed in humans in an absolute bioavailability study, is also provided.
Materials and Methods
Study Drug
The radiolabeled drug [14C]siponimod hemifumarate (cocrystal) was synthesized by the Isotope Laboratory of Novartis, Basel, Switzerland. The final drug product was analyzed by the Isotope Laboratory and Pharmaceutical and Analytical Development department of Novartis and was released for human use, according to predefined specifications. The chemical and radiochemical purity of the drug was ≥99.4% with individual impurities accounting for ≤0.2%. Hence, no impurity is expected to confound the metabolite investigations. The nominal specific radioactivity of [14C]siponimod was 0.37 megabecquerel (MBq) per milligram, referring to free base. The chemical structure of the compound and the radiolabel are shown in Fig. 1.

Chemical structure of [14C]siponimod.
Chemicals and Standards
Unlabeled siponimod (reference standard), [D11]siponimod (internal standard), and authentic standards of the metabolites M1 to M8 as well as M17 were synthesized at Novartis Pharma. All other chemicals and solvents were of analytical grade and were obtained from commercial sources.
Study Design and Subjects
This single-center, open-label, single oral dose ADME study enrolled four healthy, cytochrome P450 (P450) 2C9 enzyme (CYP2C9) wild-type (CYP2C9*1/*1), nonsmoking, male Caucasian volunteers who were determined to be in good health according to their past medical history, physical examination, vital signs, ECG, laboratory tests, and urine analysis. Subjects with relevant radiation exposure of greater than 0.2 millisievert within 12 months before the initiation of the study were excluded. The subjects were exposed to a radiation dose of 1.4 millisievert maximally, which was calculated according to the guidelines of the International Commission on Radiologic Protection. The clinical part of the study was performed at PRA International, Zuidlaren, the Netherlands, in accordance with the Good Clinical Practice guidelines and the 1964 Declaration of Helsinki and subsequent revisions. The study protocol and dosimetry calculations were reviewed by the independent ethics committee for the center, and written informed consent was obtained from all subjects before entering the study.
Dosage Administration
After an overnight fast of at least 10 hours, each subject received a single 10-mg oral dose of [14C]siponimod as 10-mg free base (11.12 mg hemifumarate) in a solution containing a radioactive dose of 3.7 MBq. The established solid dosage form of siponimod could not be manufactured with the [14C]-radiolabeled drug substance because of its radiochemical instability. The radiolabeled drug was stable in ethanol solution frozen below −60°C. The stability was ascertained from the period of manufacturing to the dose administration. The concentrate for oral solution in ethanol 94% (wt./wt.) was diluted with water (1:1, v/v) before administration to obtain 4 ml of a 2.5 mg/ml drinking solution. After dose administration, the solution container was rinsed twice with a mixture of 2 ml of ethanol (94%, wt./wt.) and 2 ml of water, which was swallowed by the subjects.
Safety Assessments
The safety analysis included the monitoring and recording of all adverse events (AEs), laboratory tests (hematology, blood chemistry, and urinalysis), vital signs, ECG, cardiac monitoring (telemetry), and physical examination.
Sample Collection and Aliquoting
A total of 22 pharmacokinetic blood samples were collected before dose administration* and at 1, 2*, 4*, 6*, 8, 12, 16*, 24, 36*, 48, 72*, 96, 120*, 144, 168*, 192, 216, 240*, 312, 480, and 816 hours after dose administration into ethylenediaminetetraacetic acid (EDTA)-containing vacuum tubes by direct venipuncture or by an indwelling cannula inserted in a forearm vein (blood volume collected: 31 ml at specific time points [*]; 11 ml at all others). Three weighed aliquots of 0.5 ml were removed for radioactivity determination.
The remaining blood was centrifuged at 4°C to obtain plasma. From the total plasma, three weighed aliquots of 0.25 ml each were removed for radioactivity determination, and one aliquot of 0.5 ml was reserved for the analysis of siponimod.
After aliquoting for the different assays, the blood and plasma samples were immediately frozen and stored at ≤ −60°C until analysis. For each subject, predose blank urine was collected on day –1. After the radiolabeled dose, all urine samples were collected in time fractions of 0–6, 6–12, and 12–24 hours and thereafter in 24-hour fractions up to 816 hours. The subjects were asked to collect their urine at home and bring the sample to the study center on days 14, 21, and 35.
Siponimod showed a high adsorption to glass and plastic surfaces in tests with blank urine. Therefore, after each voiding, acetonitrile was added to the initial collection container such that a final concentration of 25% acetonitrile (v/v) was obtained. This provided quantitative recovery of siponimod from all sample containers used in the study. The urine samples were frozen and stored at ≤ −60°C until analysis.
For each subject, a predose blank feces sample was collected on day −1 or −2. After the radiolabeled dose, all feces samples were collected completely during the postdose sample collection period of 816 hours. The subjects were asked to collect their feces at home and bring the sample to the study center on days 14, 21, and 35. The feces samples were quantitatively transferred into a container per subject and per 24-hour interval. A minimum amount of water (1 to 2 weight equivalents) was added. The samples were homogenized using an Ultra Turrax mixer by mixing for at least 2 minutes. After homogenization, one 10-g aliquot was used for determination of the total [14C]-radioactivity, and one 50-g aliquot and one 200-g aliquot were used for metabolite profiling. All samples and the remaining homogenates were stored at −20°C.
The radiometry samples were analyzed at the bioanalytical laboratories of PRA International Early Development Services (Assen, the Netherlands). Metabolism samples were analyzed at Novartis Institutes for BioMedical Research (Basel, Switzerland).
Pharmacokinetic Evaluations
Pharmacokinetic parameters were calculated via noncompartmental methods using WinNonlin software, version 5.2 (Pharsight, Mountain View, CA). The fraction of radioactivity associated with the plasma (Fp) was calculated from blood (Cb) and plasma (Cp) concentrations of radioactivity and from the hematocrit value (H) as follows: Fp(%) = (Cp/Cb) × (1 − H) × 100.
Total Radioactivity Measurement
Radioactivity in blood, plasma, urine, and feces samples was measured using liquid scintillation counting (LSC), with a typical counting time of 10 minutes. Low levels in blood and plasma were counted for 60 minutes. LSC was performed using a LSC model Tri-Carb 3100 TR (Perkin Elmer, Groningen, The Netherlands) equipped with a low-level counting mode using an external standard ratio method for quench correction. Blood samples were analyzed in triplicate (500 µl each, weighed), plasma samples in triplicate (250 μl each, weighed), urine samples in duplicate (1000 µl each), and feces samples in quadruplicate (0.5 g each). Radioactivity in blood samples was measured after solubilization. Radioactivity in plasma and urine samples was measured directly after the addition of LSC cocktail, whereas radioactivity in feces samples was measured after combustion.
With sample counting times of 10 or 60 minutes, the limit of quantification (LOQ) for blood and plasma samples were 25 and 7.5 dpm, corresponding to 2.31 and 1.39 ng-eq/ml (or grams), respectively. For urine and feces samples with counting times of 10 minutes, the LOQs were 10 and 20 dpm, corresponding to 0.46 and 1.85 ng-eq/ml (or grams), respectively.
Determination of Siponimod Concentrations in Plasma
The plasma concentration of siponimod was determined by a validated liquid chromatography with tandem mass spectrometric (LC-MS/MS) method. Aliquots of 50 µl plasma (calibration standards, quality controls, study samples, and blanks), 25 µl of the internal standard working solution (2.00 ng/ml of [D11]siponimod in 50% aqueous methanol [v/v]) or 25 µl of 50% aqueous methanol (v/v), 25 µl of 40% formic acid in water (v/v) and 350 µl of acetonitrile/ethanol/acetic acid (90/10/0.5; v/v/v) were added to the corresponding wells of a 2-ml 96-well assay plate. This was followed by vortex-mixing for approximately 15 minutes on a pulse vortex mixer with a motor speed setting of 60 units. The samples were then centrifuged at 3800 rpm (2700g) for 10 minutes at 10°C. The supernatant (200 µl) was transferred to a clean 1-ml 96-well assay plate, evaporated to dryness under a stream of nitrogen at 45°C, and reconstituted with 200 µl of 70% aqueous acetonitrile. The reconstituted sample extract was vortex-mixed briefly, which was followed by centrifugation at 3500 rpm (2300g) for 5 minutes at 25°C. Thereafter, 10 µl of the reconstituted sample extract was injected onto the LC-MS/MS system for analysis.
The LC-MS/MS system consisted of Shimadzu LC-20AD pumps, a CBM-20A controller, a CTO-20A column oven, a SIL-20ACHT autosampler (with rack changer) and a DGU-20A5 online degasser (Shimadzu, Columbia, MD) with an ACE5 C18 column (50 × 4.6 mm, 5 µm particle size; MAC-MOD Analytical, Chadds Ford, PA) and a AB Sciex API4000 tandem mass spectrometer (AB Sciex, Concord, ON, Canada). Chromatographic elution of the analyte and internal standard was performed using 30% water containing 0.5% formic acid (mobile phase [MP] A) and 70% acetonitrile containing 0.5% formic acid (MP B) at a flow rate of 1.00 ml/min. The eluents were directed to the electrospray ionization source of the MS system between approximately 1.0 and 2.5 minutes. The following MS transitions were monitored: m/z 517.2–416.3 for siponimod and m/z 528.5–427.3 for the internal standard. The lower limit of quantification was 0.250 ng/ml using a 50 µl sample volume.
Determination of Metabolite Profiles in Plasma and Excreta
For metabolite profiling in plasma, the individual samples of subjects taken at 2, 4, 16, 36, 72, and 120 hours after the siponimod dose were analyzed. Each plasma sample was extracted 4 times with methanol and up to 2 times with acetonitrile to yield extraction recoveries of ≥90%. The extracts were combined and evaporated to dryness. The residues were reconstituted sequentially in four different solvents of decreasing polarity.
The first reconstitution was performed with acetonitrile-water (25:75, v/v). After sonication and centrifugation, the supernatant (extract A) was removed, and the pellet was resuspended by sonication in the same solvent mixture of acetonitrile–water (25:75, v/v). After centrifugation, the supernatant (extract B) was removed, and the remaining pellet was reconstituted two more times in the same way using acetonitrile–water (50:50, v/v) (extract C) or acetonitrile (extract D).
Extracts A to D were analyzed together in one high-performance liquid chromatography (HPLC) run after they were injected sequentially into the column in the order D-C-B-A. Using this procedure, both polar and nonpolar components were dissolved and concentrated on the head of the HPLC column before the start of the elution. The recovery of radioactivity from the sample was close to complete (∼90%). However, for the plasma samples taken at 120 hours after dose administration and containing very low amounts of radioactivity, recoveries of 70%–76% were obtained.
Urine samples from each subject were pooled across the collection period of 0–192 hours. An aliquot of each urine pool (15 ml) was evaporated to approximately 350 µl and mixed with 50 µl of acetonitrile. After centrifugation, the supernatant was analyzed using HPLC.
Feces samples from each subject were pooled across the collection period of 0–192 hours. An aliquot of the feces homogenate pool (5 ml) was extracted twice with methanol by sonication and centrifugation. The combined methanol extract of the feces homogenate (100 µl) was mixed with 400 µl of solvent A (ammonium acetate 25 mM; pH 5.4) and injected for HPLC analysis.
The recovery of radioactivity in the excreta was complete (∼100%). The stability of the siponimod and its metabolites in the biologic samples was demonstrated by repeated extractions and analyses during the study. The stability of siponimod during the sample preparation and HPLC analysis was investigated using blank plasma, urine, and feces spiked with [14C]siponimod. No degradations were observed except for the formation of minor amounts of P73.0.
HPLC Instrumentation for Metabolite Pattern Analysis (Radio-HPLC).
The chromatography was performed on an Agilent 1100 (Agilent Technologies, Waldbronn, Germany) liquid chromatograph equipped with a binary capillary pump, a degasser, and a ultraviolet-visible spectroscopy (UV/VIS) diode array detector with a 13-μl flow cell. The software used was Agilent ChemStation for LC 3D, Rev. B.03.02. The components were separated at 40°C on a NUCLEOSIL 100-5 C18 Nautilus analytical column (4.6 × 250 mm; Macherey-Nagel, Oensingen, Switzerland) protected by a 8.0 × 4.0 mm guard column of the same stationary phase. Volumes up to 500 μl were injected using an HTS PAL autosampler (CTC, Zwingen, Switzerland). Elution was performed with a gradient of ammonium acetate 25 mM in water (pH 5.4; MP A) and acetonitrile (MP B) at a flow rate of 1.00 ml/min (gradient: 0–5 minutes, 5%, MP B; 5–20 minutes, 5%–28%, MP B; 20–65 minutes, 28%–70%, MP B; 65–70 minutes, 70%–100%, MP B; 70–75 minutes, 100%, MP B).
For hydrogen/deuterium exchange experiments the water in MP A was replaced by deuterium oxide. The HPLC effluent was directed into the electrospray liquid chromatography with mass spectrometry (LC-MS) interface with the greater portion directed to UV detection followed by online or offline radioactivity detection.
For online radioactivity detection, the effluent was mixed with Rialuma liquid scintillation mixture (Lumac, Groningen, the Netherlands) at a flow rate of 3.00 ml/min. Radioactivity was detected in an HPLC radioactivity monitor equipped with a flow cell (Berthold Technologies, Bad Wildbad, Germany).
For offline radioactivity detection, the effluent was collected in 0.13- or 0.20-minute fractions in 96-well LumaPlates (Perkin Elmer). The fractions were evaporated to dryness and the radioactivity was counted in a TopCount NXT microplate scintillation counter (Packard Instruments, Meriden, CT).
The amount of siponimod and its metabolites in plasma and excreta was estimated from the radiochromatograms based on the relative peak areas and the concentration/amount of radioactivity in the original biologic samples, reduced by the losses during sample processing and chromatography. The recovery of radioactivity after HPLC was determined for a selected sample of each biologic matrix and was found to be complete (∼100%).
For the separation of the three positional isomers—M4a, M4b, and M4c—an ultraperformance liquid chromatography (UPLC) system (Waters Corporation, Manchester, United Kingdom) was used. Offline radioactivity detection was performed using the Gilson GX-271 Liquid Handler controlled by Trilution LC software version 2.1. The components were separated on an Acquity UPLC HSS T3 1.8 μm analytical column (2.1 × 150 mm; Waters Corporation) protected by a 2.1 × 5.0 mm guard column of the same stationary phase and thermostat at 40°C. Elution was performed with a gradient of 25 mM ammonium acetate (pH 5.4; MP A) and acetonitrile (MP B) at a flow rate of 0.50 ml/min (gradient: 0–1 minutes, 5%, MP B; 1–8 minutes, 5%–30%, MP B; 8–21 minutes, 30%–50%, MP B; 21–22 minutes, 50%–100%, MP B; 22–27 minutes, 100%, MP B). Radioactivity detection was performed by offline solid scintillation counting as described before.
Structural Characterization of Metabolites
The structures of the polar siponimod-related components were characterized by LC-MS, LC-MS/MS, and comparison with the available authentic reference standards M1, M2, M3, M4a, M4b, M4c, M5, M6, M7, and M8 (see the Supplemental Materials).
In brief, the metabolites were analyzed in plasma, urine, and feces by HPLC using sample preparation procedures similar to those described previously for the metabolite profiling and the same HPLC or UPLC methods. For LC-MS and LC-MS/MS analysis after the chromatography, the effluent was split into a ratio of approximately 1:6. The smaller amount was directed into the electrospray LC-MS interface, while the larger part was used for UV detection followed by online or offline radioactivity detection as described earlier. The LC-MS and LC-MS/MS analyses were performed on a quadrupole time of flight Premier or Synapt G2-Si mass spectrometer (Waters Corporation) using electrospray ionization in the positive ion mode.
To determine whether the phase II metabolite M3 is an acyl glucuronide, we investigated its stability in alkaline solution. For this purpose, the metabolite was isolated from urine by preparative HPLC. One aliquot was mixed with 300 μl of phosphate buffer (pH 6.8) and 0.52 mg of β-glucuronidase (Escherichia coli; Sigma G-7396, 125 kU/76 mg). A second aliquot was mixed with 300 μl of borax buffer at pH 10.0 (Fluka 73419, 30 mM Na2B4O7, 42 mM NaOH). Control incubation was performed with a third aliquot, which was mixed with 300 μl phosphate buffer (Fluka 73173, 28 mM KH2PO4, 41 mM Na2HPO4) adjusted at a pH of 6.8 with phosphoric acid. The three samples were incubated at 37°C for 24 hours, followed by HPLC analysis. The identification of the chemical structure was further supported by enzymatic hydrolysis experiments.
Metabolite M17
Experimental data supporting the identification and structural characterization of the nonpolar metabolite M17 in mouse and human studies are provided in the Supplemental Materials.
In Vitro Experiments
Human Liver Microsomes and Recombinant Human P450 Enzymes.
A pool of liver microsomes prepared from 47 individual donors was obtained from BD Biosciences (cat. no. 452161, lot 26; Woburn, MA).
Microsomes prepared from baculovirus-infected insect cells (BTI-TN-5B1-4) expressing human P450 enzymes and insect cell membrane preparations (negative control) were obtained from BD Biosciences.
Incubation of [14C]Siponimod with Human Liver Microsomes in the Presence of Chemical Inhibitors.
Biotransformation of siponimod was investigated at 5 µM in the presence of eight individual chemical inhibitors at two concentrations: 2 and 10 µM for furafylline (CYP1A2 inhibitor), quercetin (CYP2C8 inhibitor), sulfaphenazole (CYP2C9 inhibitor), and tranylcypromine (CYP2C19 inhibitor); 0.1 and 1 µM for quinidine (CYP2D6 inhibitor) and ketoconazole (CYP3A and CYP4F2 inhibitor) (Kovarik et al., 2009; Jin et al., 2011); 5 and 20 µM for triethylenethiophosphoramide (CYP2B6 inhibitor); and 5 and 30 µM for diethyldithiocarbamate (DETC; CYP2E1 inhibitor).
Incubation of [14C]Siponimod with Human Liver Microsomes and Recombinant Enzymes.
The incubation of human liver microsomes (HLMs) and recombinant P450 enzymes (CYP) was performed in 0.1 M phosphate buffer, pH 7.4, at 37°C. Typically, the final incubation volume was 200 µl. After addition of an appropriate volume of siponimod stock solution, the reaction was started by the addition of 20 µl of fresh 10 mM NADPH (1 mM) buffer. The enzyme kinetics were determined by incubating pooled HLMs (0.1 mg/ml) with 15 substrate concentrations ranging from 1 to 300 µM for 90 minutes.
Enzyme mapping experiments were performed by incubating 30 pmol of CYP/ml for 30 minutes at 37°C using 10 and 40 µM substrate concentrations. CYP3A4 enzyme kinetic experiments were performed with 50 pmol/ml (0.345 mg protein/ml) for 30 minutes at 37°C using 13 substrate concentrations ranging from 5 to 300 µM. CYP2C9 enzyme kinetic experiments were performed with 50 pmol/ml (0.12 mg protein/ml) for 60 minutes at 37°C using 13 substrate concentrations ranging from 5 to 300 µM.
Reactions were terminated by the addition of equal volumes (200 or 400 µl) of ice-cooled acetonitrile containing 0.5% formic acid (v/v). After 30 minutes at −80°C (or overnight at −20°C), the samples were centrifuged at 30,000g for 15 minutes to remove protein. Aliquots of the supernatants were diluted with water to a final concentration containing less than 30% of acetonitrile and were analyzed by HPLC with radiodetection.
HPLC with Radiodetection for In Vitro Experiments.
Liquid chromatography was performed on an Agilent 1100 liquid chromatograph. Components were separated on a NUCLEOSIL 100 C18 Nautilus analytical column (4.0 × 250 mm; Macherey-Nagel, Germany) protected by an 8.0 × 4.0 mm guard column of the same stationary phase and thermostat at 40°C. Elution was performed with a gradient of 0.5% formic acid and 0.1% trifluoroacetic acid in water (MP A), and 0.5% formic acid and 0.1% trifluoroacetic acid in acetonitrile (MP B) at a flow rate of 1.00 ml/min.
For online radioactivity detection, the effluent was mixed with Rialuma liquid scintillation mixture (Lumac) at a flow rate of 3.00 ml/min. Radioactivity was detected in a HPLC radioactivity monitor equipped with a 0.5 ml flow cell (Berthold Technologies).
Data Analysis.
Enzyme kinetic parameters, Vmax and Km, for the biotransformation by HLMs and major metabolizing enzymes were calculated using SigmaPlot 8.0 (Enzyme Kinetics module version 1.1; SPSS Science, Chicago, IL). The parameters Vmax and Km were determined using the Michaelis-Menten or substrate inhibition model. Intrinsic clearance (CLint) was calculated as Vmax/Km.
Results
Demographic, Safety, and Tolerability Data
All four enrolled men completed the study. The mean age of the study participants was 35.8 years (range: 18–54 years). Their height (mean ± S.D.) was 183.0 ± 6.5 cm (range: 176–189 cm), and weight (mean ± S.D.) was 77.5 ± 8.1 kg (range: 67.2–85.5 kg).
Overall, siponimod was well tolerated without any serious AE or discontinuation due to AEs. The safety profile was consistent with the safety profile of a similar dose level in previous clinical studies with siponimod in healthy subjects (Selmaj et al., 2013; Kappos et al., 2016, 2018). The most commonly reported AEs were headache, somnolence, and asthenia. The majority of the observed AEs were mild to moderate in severity. No clinically significant abnormalities in hematology parameters, biochemistry parameters, or urinalysis were reported for any of the four men. The 24-hour cardiac monitoring revealed intermittent bradycardia in all participants. Intermittent bradycardia and one episode of second-degree atrioventricular block were reported but were clinically well tolerated. In addition, one episode of Wenckebach-type atrioventricular block was reported, starting approximately 2 hours after dosing and lasting intermittently up to 24 hours after dosing. However, these cardiovascular events were considered clinically insignificant by the investigator.
Plasma Concentrations of Total Radioactivity and Siponimod
After oral administration of 10 mg of [14C]siponimod, the peak plasma level (Cmax) of siponimod and total radiolabeled components (radioactivity) in plasma was reached at 4 and 6 hours, respectively. The concentration–time profiles of siponimod were in agreement with the first human single ascending dose study with a time to reach maximum radioactivity (Tmax) of 3–6 hours after dose administration (Novartis data on file), and with the ascending multiple dose study with a Tmax of 2–8 hours (Gergely et al., 2012). The apparent systemic clearance (CL/F) of siponimod was low (mean CL/F = 4.0 l/h). The apparent elimination t1/2 was 56.6 hours. The pharmacokinetic parameters of the [14C]-radioactivity and siponimod in plasma showed moderate interindividual variability. The average pharmacokinetic parameters are presented in Table 1.
Pharmacokinetic parameters of total radioactivity and siponimod in blood and plasma
Values are shown as mean ± S.D. (N = 4) with the exception of tmax values, which are presented as median (range).
Siponimod was moderately distributed within the human body (Vz/F = 291 ± 60 l), with siponimod or its metabolites mainly confined within the plasma compartment. The mean blood/plasma area under the concentration–time curve (AUC) ratio of radioactivity was approximately 0.67, indicating no special affinity of siponimod or its metabolites to blood cells.
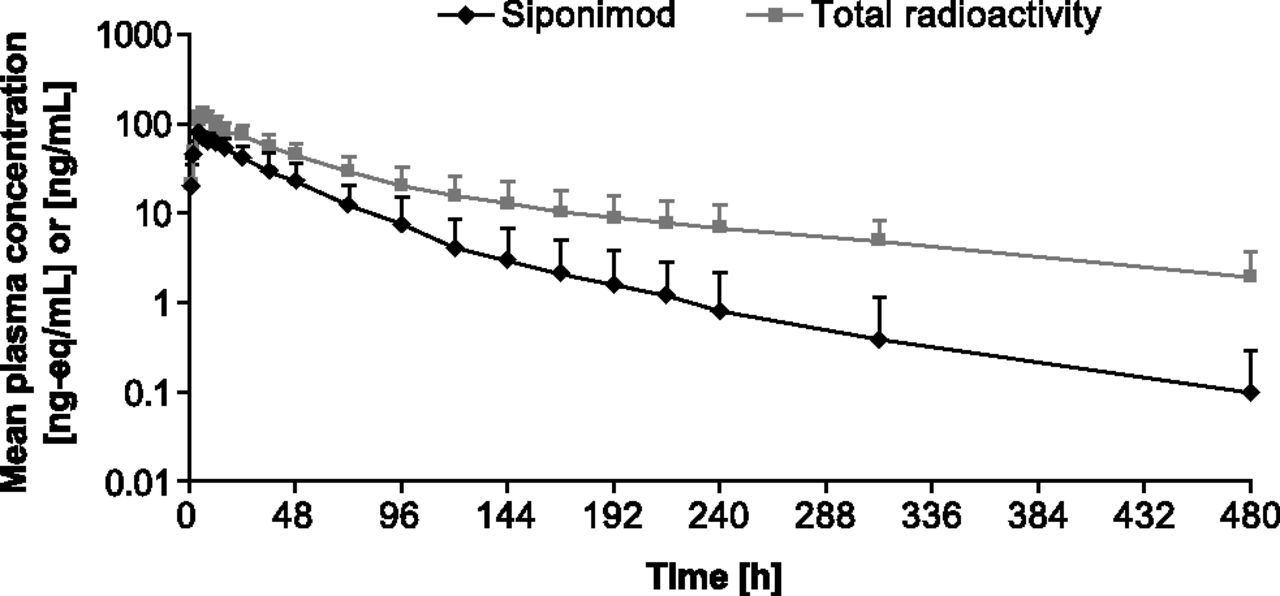
Radioactivity in plasma was detected up to 312–480 hours and was subsequently below the LOQ. Unchanged siponimod was detected in plasma for 216–480 hours after dose administration (Fig. 2). Approximately 38% of the plasma area under the concentration–time curve from 0 to infinity (AUC0–∞) radioactivity was due to siponimod, indicating substantial exposure to metabolites. The estimated terminal half-life (t1/2) of siponimod and total radioactivity in plasma was 56.6 and 171.0 hours, respectively.

Time profiles of total radioactivity and siponimod in plasma of four healthy men treated with a single 10-mg oral dose of [14C]siponimod (mean ± S.D., N = 4; semilogarithmic scale).
Structural Characterization of Metabolites
The structural analysis of the metabolites in plasma and excreta was performed by LC/MS or LC-MS/MS analysis with radioactivity detection for peak correlation with mass spectral data. The metabolite structures were derived from their product ion mass spectra and hydrogen/deuterium exchange experiments, and where possible are supported by comparison of mass spectral data and retention times with chemically and enzymatically synthesized reference standards (see the Supplemental Materials).
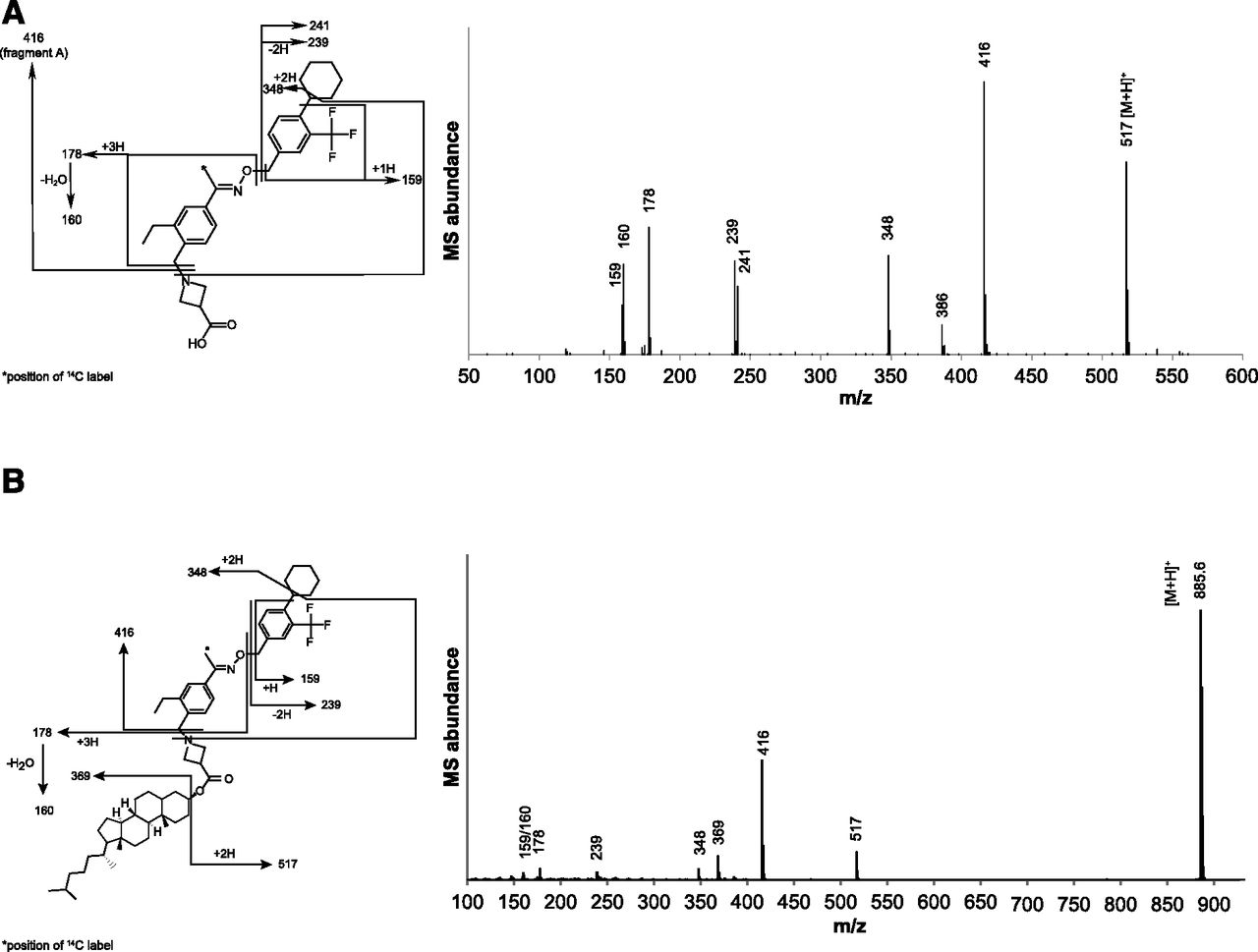
Data from the LC-MS and LC-MS/MS analyses of plasma, urine, and feces extracts are summarized in Table 2. The full scan mass spectrum of siponimod using source-induced dissociation (Fig. 3A) showed the protonated intact molecule [M+H]+ (m/z 517) and a key fragment ion A (m/z 416), resulting from the neutral loss of an acetidine-3-carboxylic acid group. Fragment ions analogous to the key fragment ion A of the parent compound in the mass spectra of the metabolites allowed the assignment of the biotransformation reactions to the substructures of the metabolites (Table 2).
Mass spectral data on siponimod and its metabolites in plasma, urine, and feces
Data are from LC-MS and LC-MS/MS runs of siponimod and metabolites.

(A) Full scan source-induced dissociation mass spectrum of siponimod. Authentic reference standard: quadrupole time of flight (Q-TOF) Premier mass spectrometer, cone voltage of 25 V, collision energy 25 eV and background subtracted. (B) MS/MS collision-induced dissociation mass spectrum of protonated M17. Authentic reference standard; quadrupole time of flight (Q-TOF) Synapt G2-Si mass spectrometer, cone voltage of 40 V, collision energy ramp from 10 to 50 eV.
The chemical structure of the metabolite M3 was further supported by alkaline and enzymatic hydrolysis. The radiochromatogram of the aliquot incubated with β-glucuronidase (E. coli) showed the complete disappearance of M3 and an increase of M5. The radiochromatograms of the metabolite aliquots incubated with borax buffer (pH 10.0) and phosphate buffer (pH 6.8) were stable, confirming M3 as an O-glucuronide of the hydroxylated metabolite M5 but not as an acyl glucuronide.
To further support the chemical structure elucidation, enzymatic synthesis methods were applied to generate metabolite standards. The hydroxylated metabolites M5, M6, and M7 were synthesized from siponimod. Then, in a next enzymatic synthesis step, glucuronic acid or sulfuric acid was introduced and formed the metabolites M3, M4a, M4b, and M4c. The mass spectral and NMR data of the synthesis products (Supplemental Fig. 3) and the comparison with the retention time and mass spectral data of the metabolite data revealed hydroxylation of the cyclohexyl ring and glucuronidation or sulfation of the respective hydroxyl groups.
Furthermore, the compound P73.0, which was only observed in plasma extracts after extensive sample preparation with methanol, was structurally characterized by LC-MS/MS. P73.0 is likely a degradation product of siponimod formed at low levels by methyl-esterification of the carboxyl group during sample processing. The descriptions of the chemical or biochemical synthesis of the reference compounds and characterization by mass spectrometry and NMR spectroscopy are provided in the Supplemental Materials.
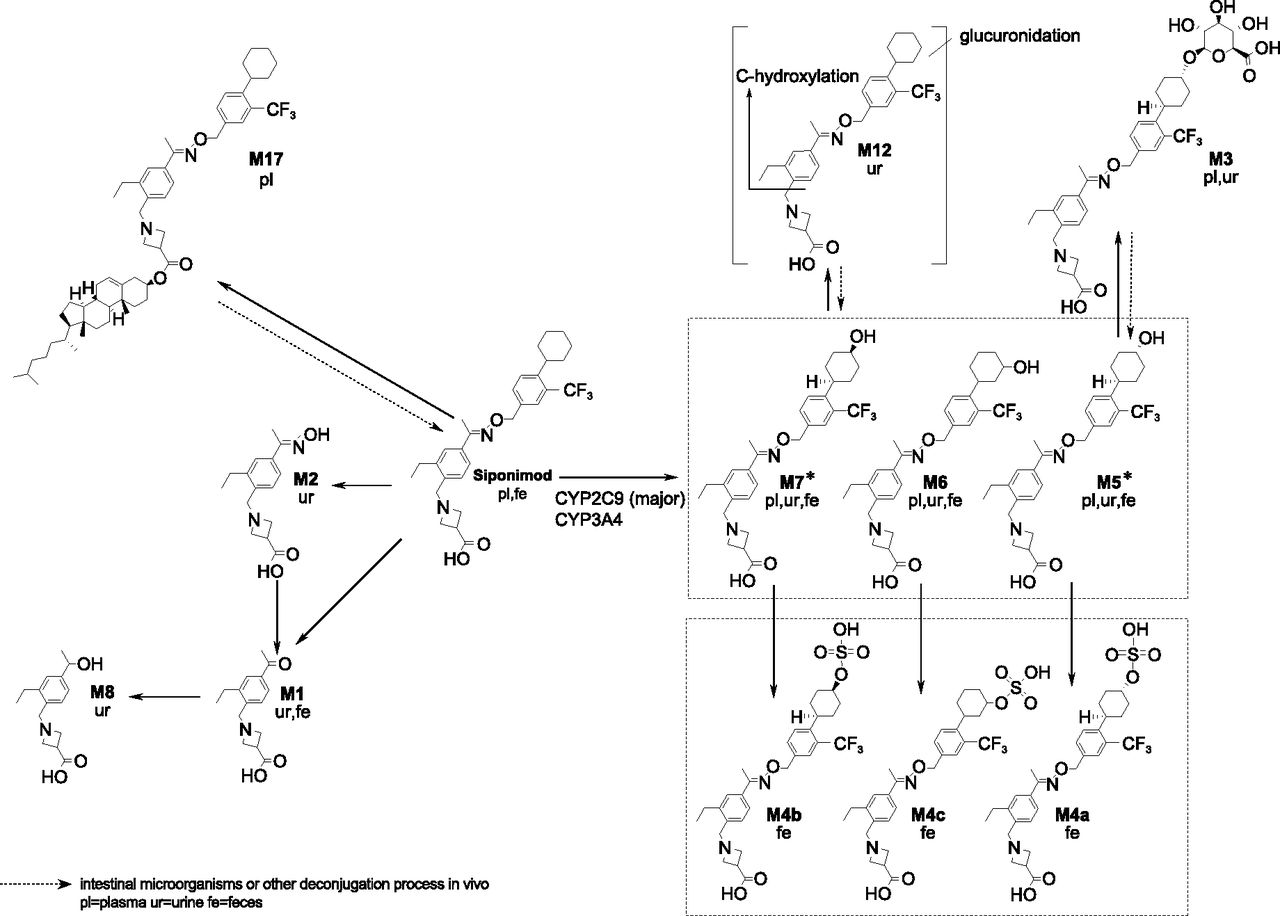
The proposed biotransformation pathways and partial or complete structures of the metabolites are given in Fig. 4.
Proposed biotransformation pathways for siponimod in humans. In vitro experiments demonstrated that CYP2C9 is the main drug-metabolizing enzyme responsible for the catalysis of M5 and M7 formation. *CYP2C9 selectively catalyzes the formation of metabolites M5 and M7.
Excretion and Mass Balance of Radioactivity in Urine and Feces
The [14C] radioactivity was mainly excreted in feces (Fig. 5). The mean total recovery of radioactivity in excreta over 9 days (216 hours after post administration) was 87.7% ± 3.7% of the dose: 84.1% ± 3.5% in feces and 3.6% ± 0.4% in urine. By day 13 (312 hours after dose administration), the recovery of radioactivity was close to complete with a moderate interindividual variability (mean 90.4% ± 2.7% of the dose: 86.7% ± 2.5% in feces and 3.7% ± 0.3% in urine).

Cumulative excretion of radioactivity in urine and feces (mean ± S.D.; N = 4).
Metabolite Profiles in Plasma, Urine, and Feces
Metabolite profiles in plasma were investigated up to 120 hours after dose administration for each participant individually. The quantitative data on siponimod and its metabolites in plasma are given in Table 3. A representative plasma metabolite profile of a subject at 6 hours (Tmax) is depicted in Fig. 6A. In plasma, the parent compound siponimod represented the main proportion of radioactivity (57.1% ± 5.9% of the plasma AUC0–120h). In addition, the metabolite M3 accounted for 18.4% ± 5.0% of the plasma AUC0–120h or 27.6% of the parent drug AUC0–∞. Minor proportions of other metabolite peaks were detected and attributed to the metabolites M5, M6, M7, P29.6, and P30.5, each accounting on average for 1.5%–3.7% of the plasma AUC0–120h. Overall, more than 90% of the detected radioactive components could be covered by the parent drug and structurally characterized metabolites.
Estimated AUC0–120h of siponimod and metabolites in the plasma of healthy men after a single 10-mg oral dose of [14C]siponimod
The AUC0–120 h is mean ± S.D., N = 4 (nmol*h/l), mean values of N = 4 subjects and individual values.

Representative metabolite (excluding M17) patterns in plasma with the total radioactivity Tmax at (A) 6 hours, (B) urine pools 0–192 hours, and (C) feces pools 0–192 hours of healthy men treated with a single 10-mg oral dose of [14C]siponimod.
Metabolite profiles in the urine and feces pools for 0–192 hours were analyzed for each of the four men individually. Quantitative data on siponimod and metabolites in excreta are provided in Table 4. Figure 6, B and C, shows representative radiochromatograms. Only 3.6% ± 0.4% of the administered radioactivity was excreted in urine within 192 hours. Unchanged siponimod in urine was not detected, indicating that siponimod was not renally excreted. The metabolite profiles in urine consisted of the major metabolite M3, amounting to 2.1% ± 0.2% of the dose, and nine minor peaks. The hydroxylated metabolites M5, M6, and M7 were detected in traces (<0.1% each).
Total amount of siponimod and metabolites (% of dose) in the excreta of healthy men after a single 10-mg oral dose of [14C]siponimod
Values are mean ± S.D., N = 4.
The main proportion of the administered radioactivity was excreted in the feces and amounted to 82.3% ± 3.9% of the dose within 192 hours. The metabolite profiles in the feces extracts showed essentially three major peaks and at least six minor components. Unchanged siponimod accounted to 9.2% of the radioactive dose in the feces during 0–192 hours. The major hydroxylated metabolite in feces (M5) accounted for 45.1% ± 9.0% of the dose. Other hydroxylated metabolites M6, M7, and sulfate conjugates of hydroxylated metabolites (M4a, M4b, and M4c) ranged between 1.6% and 6.4% of the administered dose. Overall, 67.3% of the dose was covered by hydroxylated metabolites including sulfate conjugates of hydroxylated metabolites.
Minor metabolites (M1, M2, M8) formed by ether cleavage, hydrolysis, and subsequent reduction (Supplemental Fig. 2) were only detected in urine and represented 2.1% of the dose. No major difference was observed between the men in their urinary and fecal metabolite profiles.
Identification of the Nonpolar Metabolite M17.
Interestingly, M17, a nonpolar metabolite, was identified as the major metabolite in the systemic circulation in a mouse ADME study (conducted after the human ADME study) after single 25 mg/kg oral dose of [14C]siponimod (Supplemental Table 1). The plasma metabolite was identified as a cholesterol ester of siponimod by LC-MS/MS (Table 2; Fig. 3B), and the chemical structure was confirmed by comparison of the retention time and mass spectral data with a reference standard (see the Supplemental Materials). This metabolite represented 23.3% of the total [14C]AUC0–168h or 35.2% of the parent drug exposure.
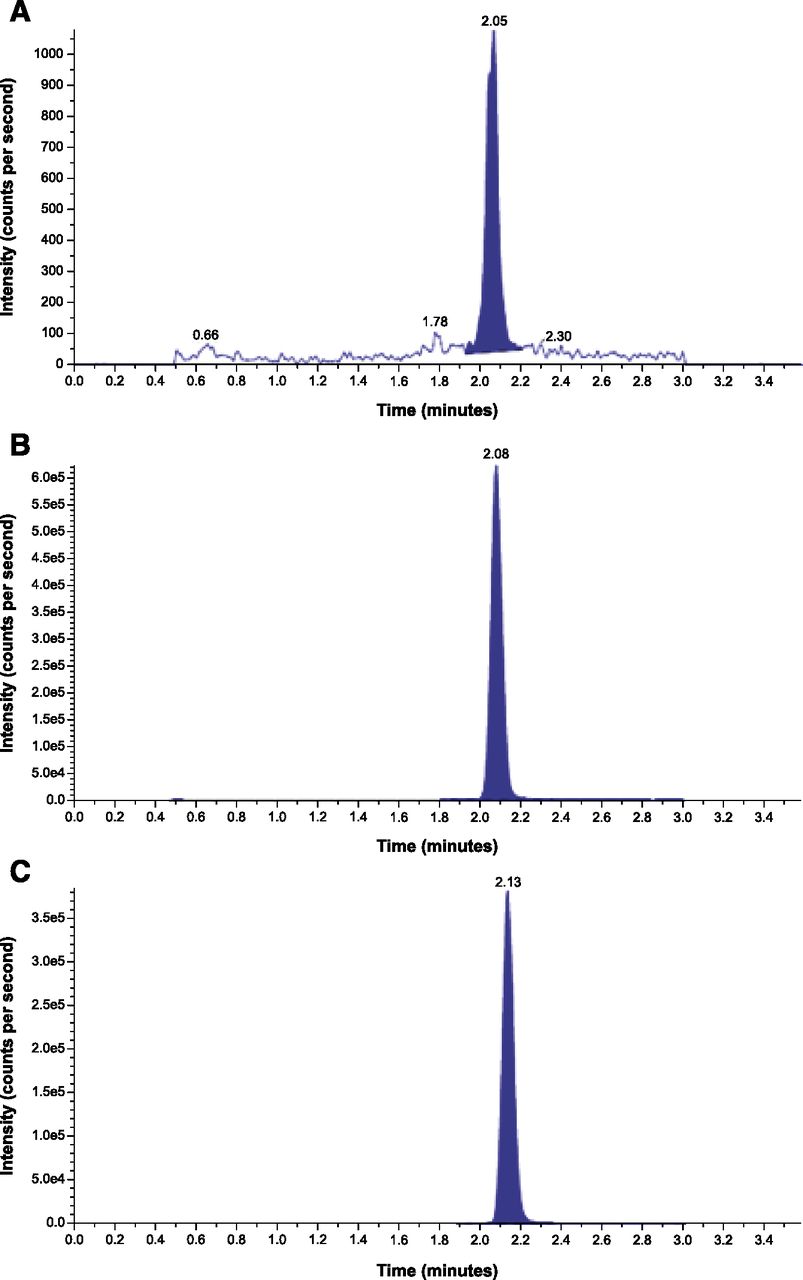
In the human absolute bioavailability study, the presence of the cholesterol ester metabolite M17 with low abundance and a long t1/2 (155 hours) after a single dose was confirmed by a validated bioanalytical assay (Fig. 7). Metabolite M17 represented approximately 81%–97% of the parent exposure (AUC0–∞ 105 – 112 ng*h/ml) and was thus identified as the most prominent systemic metabolite in humans (Supplementary Information). The pharmacokinetic profile of M17 will be reported elsewhere.

Representative LC-MS/MS chromatograms of M17 in human plasma: (A) lower LOQ sample, (B) upper LOQ sample, and (C) unknown study sample. LC-MS/MS validated quantitative assay.
In Vitro Metabolism Experiments
Metabolite Profile in Human Liver Microsomes.
To further elucidate siponimod metabolism, in vitro experiments were conducted to investigate the enzymes involved in the oxidative metabolism of siponimod. The incubation of siponimod with HLMs in the presence of NADPH produced three detectable metabolites in small amounts, which were confirmed as M5, M6, and M7. No metabolites were detected in the control incubates without HLMs.
Enzyme Kinetics of Siponimod Metabolism in Human Liver Microsomes.
After establishing linear reaction conditions regarding incubation time and protein concentration, we investigated the enzyme kinetics of siponimod metabolism. Siponimod, at 15 concentrations ranging from 1 to 300 µM, was incubated with pooled HLMs (0.1 mg protein/ml) for 90 minutes. The rates of biotransformation were analyzed by nonlinear curve-fitting using the Enzyme Kinetics module of SigmaPlot, considering different kinetic models (Michaelis-Menten, Hill, isoenzyme, random substrate activation, and substrate inhibition). The substrate inhibition model provided the best fit for the data when plotted as the Michaelis-Menten plot and the Eadie-Hofstee plot. Nonlinear regression analysis of the rate of metabolism versus siponimod concentration revealed a Km of 50.3 ± 9.7 µM and a Vmax of 191 ± 25 pmol/min per milligram. The derived CLint of the hepatic metabolism of siponimod was 3.8 µl/mg per minute.
Inhibition of Siponimod Metabolism by CYP-Specific Chemical Inhibitors.
Sulfaphenazole (2 and 10 µM), a known in vitro CYP2C9 inhibitor, exhibited strong inhibition of siponimod metabolism in human liver enzymes (65%–77%). Ketoconazole (1 µM) exhibited 25% of inhibition, whereas quercetin and tranylcypromine inhibited siponimod metabolism by 11%–31% and 11%–29%, respectively. No significant inhibition of siponimod metabolism was observed with furafylline, triethylenethiophosphoramide, quinidine, or DETC (Fig. 8).

Depletion rate of [14C]siponimod (10 and 40 µM) by recombinant human P450 enzymes.
Biotransformation of Siponimod by Recombinant Human CYP Isoenzymes.
Microsomes prepared from baculovirus-infected insect cells expressing a single human P450 isoenzyme were used to assess the involvement of specific enzymes in the biotransformation of siponimod. Incubations with a panel of 21 recombinant human P450 enzymes (CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C9*1, CYP2C18, CYP2C19, CYP2D6*1, CYP2E1, CYP2J2, CYP3A4, CYP3A5, CYP3A7, CYP4A11, CYP4F2, CYP4F3A, CYP4F3B, CYP4F12, and CYP19) were conducted under similar conditions for each isoenzyme using 10 and 40 µM siponimod and 30 pmol CYP/ml. CYP2C9*1 showed significant metabolic activity and contributed predominantly to the total CLint in HLMs (Fig. 9).

Inhibition of siponimod metabolism in HLMs by chemical inhibitors. Results are the mean of two incubations.
The metabolite profile obtained with CYP2C9 was qualitatively similar to that observed in HLMs (Supplemental Fig. 1). CYP2C9 mainly produced the metabolites M5 and M7, whereas M6 was produced to a small extent with this enzyme and formed predominantly by other enzymes such as CYP3A4, CYP2C8, and CYP3A5. M5 and M7 can therefore be considered as CYP2C9-selective or specific metabolites of siponimod.
Low metabolic activity was also observed with CYP3A4, whereas only traces of metabolites were detected with other CYP isoenzymes: CYP2B6, CYP2C8, CYP2C19, CYP2J2, CYP3A5, and CYP1A1. The enzyme kinetics parameters of the isoenzymes CYP2C9 and CYP3A4 for siponimod metabolism were determined by incubating different concentrations of substrate with the enzyme. Kinetic constants for both CYP3A4 (Km: 85.1 ± 8.6 µM; Vmax: 706 ± 31 pmol/min per nmol) and CYP2C9 (Km: 34.5 ± 5 µM; Vmax: 2596 ± 233 pmol/min per nmol) were determined. The derived CLint (Vmax/Km) of the total metabolite formation of siponimod was 8.3 and 75.2 µl/nmol per minute for CYP3A4 and CYP2C9, respectively (Table 5).
Enzyme kinetic constants and estimated contribution of various CYPs to the metabolic clearance of siponimod in human liver microsomes
Enzyme kinetic parameters Vmax and Km of CYP3A4 and CYP2C9 were obtained from kinetic experiments. The values of other isoenzymes were estimated by solving the Michaelis-Menten equation for the two concentrations of substrate and their rate numbers.
For minor siponimod metabolizing CYP isoforms, the kinetic constants Km and Vmax were estimated by solving two linear equations with two variables, which allowed the estimation of enzymatic efficiency, or Clint, for various enzymes.
With regard to their importance in hepatic metabolic clearance, we calculated a CLint relative to their abundance in HLMs (Yeo et al., 2004). CYP2C9 contributed predominantly (79.2%) to the total CLint in HLMs (Table 5), and CYP3A4 contributed to 18.5% of the CLint. The contributions of other known drug-metabolizing enzymes (CYP2B6, 2C8, 3A5, and 2C19) were low.
Discussion
A single oral dose of 10 mg [14C] siponimod in humans showed medium to slow absorption (median Tmax: 4 hours) and moderate distribution (Vz/F: 291 ± 60 l, mean ± S.D.) and was mainly cleared through biotransformation. On average, approximately 9.2% of recovered radioactivity in the feces pool consisted of unchanged drug with only trace amounts of unchanged siponimod in urine, indicating extensive metabolism. Assuming that the drug and metabolites were stable against intestinal bacterial enzymes, the extent of oral absorption was estimated to be higher than 70% of the administered dose—that is, approximately 3.6% renally excreted radioactivity and approximately 73% of radioactivity in feces representing metabolites.
The absorbed part of the siponimod oral dose was cleared mainly by C-hydroxylations on the cyclohexyl moiety (M5, M6, and M7). The hydroxylated metabolites underwent further phase II reactions involving sulfation (M4a, M4b, and M4c) and glucuronidation (M3 and M12). Cleavage or hydrolysis at the oxime ether bond (M1 and M2) and further reduction (M8) was only a minor pathway. In vitro, the metabolites M5, M6, and M7 were also detected in incubations with HLMs.
In plasma, the parent drug represented the most abundant radioactive component (57.1%). In line with the in vitro metabolism studies, metabolite M3 formed by glucuronidation of the hydroxylated metabolite M5 was the main circulating metabolite (18.4%). The hydroxylated metabolites M5, M6, and M7 accounted for less than 3% of the radioactivity.
An additional nonpolar metabolite, M17, which was detected as a major component in the systemic circulation in the mouse ADME study after a single oral administration of 25 mg/kg of [14C]siponimod hemifumarate, represented 23.3% of the total [14C]AUC0–168h. It should be noted that M17 was unknown at the time the human ADME study was conducted, so the presence of M17 was not investigated. A very low amount of drug-related radioactivity in plasma, the reduced recovery of the radioactivity after sample preparation at late sampling time points, and the low concentration of drug-related components may have been the limiting factor that prevented the detection of M17 in the human ADME study. Moreover, the single-dose nature of the human ADME study may not have been suitable for detection of metabolites such as M17 with low abundance and a long half-life (t1/2 of M17: 150–155 hours).
Nevertheless, formation of M17 in humans was considered possible due to the longer half-life of radioactivity than for the parent compound that was observed in the human ADME study. Consequently, a validated quantitative analytical method (described in the Supplemental Methods) was developed and appended to the human bioavailability study to measure the quantities of M17 present in human plasma to satisfy the ICH M3 (R2) and EMA (drug-drug interactions) requirements (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf; http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf). M17 was identified as the most prominent systemic metabolite based on AUC0–∞. Despite the high systemic exposure to M17, cholesterol ester formation was not considered a primary elimination pathway of siponimod, which was confirmed by the complete analytical recovery of total radioactivity for urine and feces in the human ADME study (Table 4) and the previously reported hydrolysis of cholesterol esters in the liver before elimination. In mammals, the relevance of such types of metabolic transformation (direct esterification and hydrolysis) in the maintenance of cholesterol homeostasis in the body has been studied previously (Rudel and Shelness, 2000). Although rare, this metabolic transformation of a xenobiotic acid into a cholesterol ester has been previously reported in the literature (Fears et al., 1982; Miyamoto et al., 1986).
The first human ascending single-dose trial demonstrated the mono exponential decay of siponimod, with a geometric mean apparent terminal t1/2 of between 27.0 and 56.7 hours (Novartis data on file). The apparent elimination t1/2 of metabolites M3, M5, M6, and M7 obtained from semiquantitative data ranged between 29.3 and 35.2 hours. Therefore, an unexpectedly high accumulation of siponimod or these polar metabolites was not anticipated after daily oral administration. However, for the nonpolar metabolite M17, a significant accumulation after multiple dosing could be expected.
The predominant elimination route of siponimod and its metabolites was fecal excretion in the form of metabolite M5 (hydroxylation) and M4a–c (hydroxylation and sulfation). In urine, excretion of radioactivity was low and mainly in the form of hydroxylated glucuronide M3. No unchanged siponimod was recovered in urine. Excretion of radioactivity was close to complete after 13 days, with more than 80% of the dose (>90% of total radioactivity in 0–192 hours’ urine or feces pools) being covered by siponimod and structurally characterized metabolites.
The in vitro experiments in HLMs and recombinant human CYP enzymes investigated the enzymes involved in the oxidative metabolism of siponimod. The biotransformation of siponimod in HLMs was slow, with an apparent CLint of 3.8 µl/mg per minute. The apparent Km and Vmax were 50.3 µM and 191 pmol/min per milligram, respectively, and aligned with the substrate inhibition model. Based on kinetic results with recombinant CYP isoforms and the relative abundance values of the currently known CYP isoforms, it was estimated that CYP2C9 was the predominant contributor (79.2%) to the oxidative metabolism of siponimod in HLMs. Furthermore, CYP2C9 produced metabolite profiles similar to those produced by HLMs, with M5 and M7 being the major metabolites. As both metabolites were produced mainly by CYP2C9 and only to a small extent by other enzymes, M5 and M7 could therefore be considered as CYP2C9-selective or specific metabolites of siponimod. The enzyme phenotyping experiments using both the chemical inhibition and recombinant P450 enzymes methods confirmed the results of additional phenotyping approaches with correlation analysis and CYP2C9-genotype sensitivity analysis (Jin et al., 2018).
These experiments demonstrated the predominant contribution of CYP2C9 in the in vitro metabolism, accurately predicting the in vivo pharmacogenetic effect on siponimod clearance. CYP2C9 is a major CYP enzyme that is involved in the metabolic clearance of approximately 15% of all drugs that undergo phase I biotransformation (Rettie and Jones, 2005). The in vitro metabolism correlates well to the in vivo situation. The two major and selective metabolites produced by CYP2C9, M5 and M7, and their secondary metabolites M3, M4a, M4b, and M4c were also found in vivo as the predominant metabolites in human excreta and in the systemic circulation.
The metabolic activity of CYP3A4 was low and contributed 18.5% toward the biotransformation of siponimod. Enzymes of the CYP3A subfamily are the most abundant CYP enzymes in HLMs, although the levels vary enormously (>10-fold) among individuals, whereas CYP3A5 expression is relatively low in HLMs (10%–30% of human livers) (Shimada et al., 1994; Yeo et al., 2004). CYP3A4/5 enzymes play a key role in the biotransformation of numerous drugs, metabolizing approximately 50% of the drugs that are known to be metabolized by CYP enzymes (Li et al., 1995; Rodriguez-Antona and Ingelman-Sundberg, 2006; Ingelman-Sundberg et al., 2007). In line with these results, the metabolism of siponimod in HLMs was strongly inhibited by the CYP2C9-selective chemical inhibitor sulfaphenazole, and partly by ketoconazole, a potent inhibitor of CYP3A4.
CYP2C9 is subject to significant genetic polymorphism (CYP2C9*1, CYP2C9*2, and CYP2C9*3), varying largely between different ethnic populations. Because the biotransformation of siponimod to its hydroxylated metabolites was mainly catalyzed by CYP2C9, a significant effect of genetic polymorphism of this isoenzyme on human metabolism could be anticipated (Jin et al., 2018). To reduce the variability due to genetic polymorphism, only four men with the wide-type CYP2C9*1/*1 genotype were selected for the current human ADME study.
In conclusion, the absorption of orally administered siponimod was moderate to slow but almost complete, with Cmax achieved at 4 hours after dose administration. The extent of oral absorption was estimated to be higher than 70% of the administered dose. Siponimod had moderate distribution, with an apparent distribution volume, Vz/F, at steady state of 291 ± 60 l. Siponimod and its metabolites were confined primarily within the plasma compartment with no special affinity to erythrocytes.
The parent drug was extensively metabolized and systemic exposure to metabolites was substantial. The metabolites M3 and M17 were above the action limit recommended in the ICH M3 (R2) guidance (http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf) and warranted further consideration. Indeed, adequate exposure of the major human plasma metabolites was obtained in toxicological evaluations (Novartis data on file).
The biotransformation of siponimod occurred by the following pathways: phase I metabolic reactions involving C-hydroxylations on the cyclohexyl moiety (M5, M6, and M7, predominantly catalyzed by CYP2C9). Cleavage or hydrolysis at the oxime ether bond (M1 and M2) and further reduction yielding metabolite M8 was only a minor pathway. Phase II reactions of hydroxylated metabolites involved sulfation (three positional isomers M4a, M4b, and M4c) and glucuronidation (M3 and M12). The most abundant radioactive component in plasma was unchanged siponimod. The metabolite M3 formed by glucuronidation of the hydroxylated metabolite M5 was identified as a main metabolite. Although the metabolite M17 was identified as the most prominent systemic metabolite in humans, the cholesterol ester formation was not a major elimination pathway of siponimod. The excretion of siponimod and its metabolites was slow but essentially complete after 13 days. The elimination of siponimod was predominantly through feces.
Acknowledgments
We would like to thank the participants from whom data were taken for analysis. We would also like to thank Albrecht Glänzel and Thomas Mönius (Isotope Laboratory, Novartis Pharma AG, Basel, Switzerland) and Frederic Zecri and Virginie Tucconi (Global Discovery Chemistry, Novartis Pharma AG, Basel, Switzerland) for the preparation of the radiolabeled drug or the synthesis of reference compounds, and Claudia Sayer and Janine Wank (Biotransformation Laboratory, Novartis Pharma AG, Basel, Switzerland) as well as Fabian Eggimann and Liliane Porchet Zemp (Bioreactions Group, Novartis Pharma AG, Basel, Switzerland) for their excellent technical support. We would also like to thank Rahul Bajirao Birari and Richa Chhabra (Novartis Healthcare Pvt. Ltd, Hyderabad, India) for providing medical writing assistance on this manuscript. All authors edited the manuscript for intellectual content, provided guidance during manuscript development, and approved the final version submitted for publication.
Authorship Contributions
Participated in research design: Glaenzel, Jin, Nufer, Schroer, Adam-Stitah, Peter van Marle, Legangneux, James, Camenisch.
Conducted experiments: Glaenzel, Jin, Nufer, Li, Schroer, Peter van Marle, Legangneux, Borell, James.
Contributed new reagents or analytic tools: Glaenzel, Jin, Schroer.
Performed data analysis: Glaenzel, Jin, Nufer, Schroer, Borell, James, Meissner, Gardin.
Wrote or contributed to the writing of the manuscript: Glaenzel, Jin, Nufer, Li, Schroer, Adam-Stitah, Legangneux, Meissner, Camenisch, Gardin.
Footnotes
- Received November 20, 2017.
- Accepted April 20, 2018.
This study was funded by Novartis Pharma AG, Basel Switzerland.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ADME
- absorption, distribution, metabolism, and excretion
- AE
- adverse event
- AUC
- area under the concentration–time curve
- CL
- systemic clearance
- CLint
- intrinsic clearance
- CNS
- central nervous system
- CYP
- cytochrome P450 enzyme
- DETC
- diethyldithiocarbamate
- F
- bioavailability
- HLM
- human liver microsomes
- HPLC
- high-performance liquid chromatography
- ICH
- International Conference on Harmonization
- LC-MS
- liquid chromatography with mass spectrometry
- LC-MS/MS
- liquid chromatography with tandem mass spectroscopy
- LOQ
- limit of quantitation
- LSC
- liquid scintillation counting
- MP
- mobile phase
- P450
- cytochrome P450
- S1P
- sphingosine-1-phosphate
- SPMS
- secondary progressive multiple sclerosis
- Tmax
- time to reach maximum radioactivity
- UPLC
- ultraperformace liquid chromatography
- Vz
- apparent volume of distribution during the terminal phase
- Copyright © 2018 The Author(s).
This is an open access article distributed under the CC BY Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}