


Abstract
Fingolimod (FTY720, Gilenya, 2-amino-2-[2-(4-octylphenyl)ethyl]-1,3-propanediol) is a novel drug recently approved in the United States for the oral treatment of relapsing multiple sclerosis. The compound is eliminated predominantly by ω-hydroxylation, followed by further oxidation. The ω-hydroxylation was the major metabolic pathway in human liver microsomes (HLM). The enzyme kinetics in HLM were characterized by a Michaelis-Menten affinity constant (Km) of 183 μM and a maximum velocity (Vmax) of 1847 pmol/(min · mg). Rates of fingolimod metabolism by a panel of HLM from individual donors showed no correlation with marker activities of any of the major drug-metabolizing cytochrome P450 (P450) enzymes or of flavin-containing monooxygenase (FMO). Among 21 recombinant human P450 enzymes and FMO3, only CYP4F2 (and to some extent CYP4F3B) produced metabolite profiles similar to those in HLM. Ketoconazole, known to inhibit not only CYP3A but also CYP4F2, was an inhibitor of fingolimod metabolism in HLM with an inhibition constant (Ki) of 0.74 μM (and by recombinant CYP4F2 with an IC50 of 1.6 μM), whereas there was only a slight inhibition found with azamulin and none with troleandomycin. An antibody against CYP4F2 was able to inhibit the metabolism of fingolimod almost completely in HLM, whereas antibodies specific to CYP2D6, CYP2E1, and CYP3A4 did not show significant inhibition. Combining the results of these four enzyme phenotyping approaches, we demonstrated that CYP4F2 and possibly other enzymes of the CYP4F subfamily (e.g., CYP4F3B) are the major enzymes responsible for the ω-hydroxylation of fingolimod, the main elimination pathway of the drug in vivo.
Introduction
Fingolimod (FTY720, Gilenya, 2-amino-2-[2-(4-octylphenyl) ethyl]-1,3-propanediol) has been recently approved in the United States as a new oral drug for relapsing multiple sclerosis. Treatment with fingolimod results in retention of lymphocytes in the lymph nodes, away from the central nervous system where they are involved in inflammation and tissue damage (Kappos et al., 2006). The compound undergoes reversible biotransformation to the active principle fingolimod phosphate (Billich et al., 2003; Albert et al., 2005; Zollinger et al., 2010). The phosphate acts as a potent agonist at sphingosine 1-phosphate receptors on lymphocytes, inducing receptor internalization and hence depriving the lymphocytes from sensing the stimulus to egress from lymphoid tissue (Brinkmann, 2007). Fingolimod itself shows low or negligible activity at these receptors (Hale et al., 2004; Albert et al., 2005).
Fingolimod is cleared slowly with a terminal half-life of approximately 5 to 6 days in humans (Kovarik et al., 2007, 2009). Even though fingolimod is metabolized by several pathways, including reversible phosphorylation, oxidation, and conjugation with endogenous fatty acids to form ceramide analogs, its elimination occurs predominantly by oxidation, as detailed in an accompanying publication (Zollinger et al., 2010). The oxidative pathway involves ω-hydroxylation at the methyl terminus of the octyl chain, followed by further oxidation to a carboxylic acid and subsequent loss of two carbon units by β-oxidation. The work presented here revealed that the CYP4F subfamily plays a major role in the metabolism of fingolimod.
Of the numerous cytochrome P450 (P450) enzymes identified in the human genome (Chinese Human Liver Proteome Profiling Consortium, 2010), an increasing number are becoming recognized as clinically relevant (Clarke and Jones, 2008). The CYP1, CYP2, and CYP3 enzyme families are well known to be involved in drug metabolism, whereas CYP4 enzymes are known mainly for their role in the metabolism of endogenous fatty acids, prostaglandins, and steroids. The human CYP4F subfamily includes CYP4F2, CYP4F3A, CYP4F3B, CYP4F6, CYP4F8, CYP4F11, CYP4F12, and CYP4F22 (Nelson et al., 2004; Kalsotra and Strobel, 2006; Wang et al., 2009; Chinese Human Liver Proteome Profiling Consortium, 2010). CYP4F2 is expressed in the human liver, intestine, and kidney and catalyzes the ω-hydroxylation of various eicosanoids and related compounds, including arachidonic acid, to form 20-hydroxyeicosatetraenoic acid (20-HETE) (Powell et al., 1998; Lasker et al., 2000; Kalsotra and Strobel, 2006). CYP4F3B is very similar to CYP4F2 in amino acid sequence and displays overlapping catalytic activity. Like CYP4F2, CYP4F3B efficiently oxidizes arachidonic acid (Christmas et al., 2001), and both enzymes contribute to the ω-hydroxylation of very long-chain saturated fatty acids in human liver microsomes (HLM) (Sanders et al., 2006). An alternative transcript of the CYP4F3 gene, CYP4F3A, arises from differential promoter use, leading to the incorporation of an alternative exon that changes substrate preferences to favor the ω-hydroxylation of leukotriene (Christmas et al., 1999). In contrast to CYP4F3B, which is selectively expressed in the liver, kidney, trachea, and gastrointestinal tract (Christmas et al., 2001), CYP4F3A is a nonhepatic enzyme expressed in myeloid cells in peripheral blood and bone marrow (Christmas et al., 2001) and in human polymorphonuclear leukocytes (Kikuta et al., 1993).
CYP4F enzymes, including CYP4F2 and CYP4F3B, were recently shown to be the major enzymes catalyzing the initial O-demethylation of the antiparasitic prodrug pafuramidine by HLM and to catalyze the same reaction in human intestinal microsomes (Wang et al., 2006, 2007). CYP4F11 was found to be able to metabolize a number of drugs, including erythromycin, benzphetamine, ethylmorphine, chlorpromazine, and imipramine (Kalsotra et al., 2004). Hashizume et al. (2002) reported the involvement of intestinal CYP4F12, besides CYP2J2, in the conversion of the prodrug ebastine to the active principle carebastine. Thus, the primal view that CYP4F enzymes are unimportant in drug metabolism is now gradually being revised (Kalsotra and Strobel, 2006).
The objective of the current in vitro study was to identify the enzymes involved in the hydroxylation of fingolimod. This information will provide a basis for assessing the potential effects of comedications and genetic variants on the pharmacokinetics of fingolimod.
Materials and Methods
Study Drug.
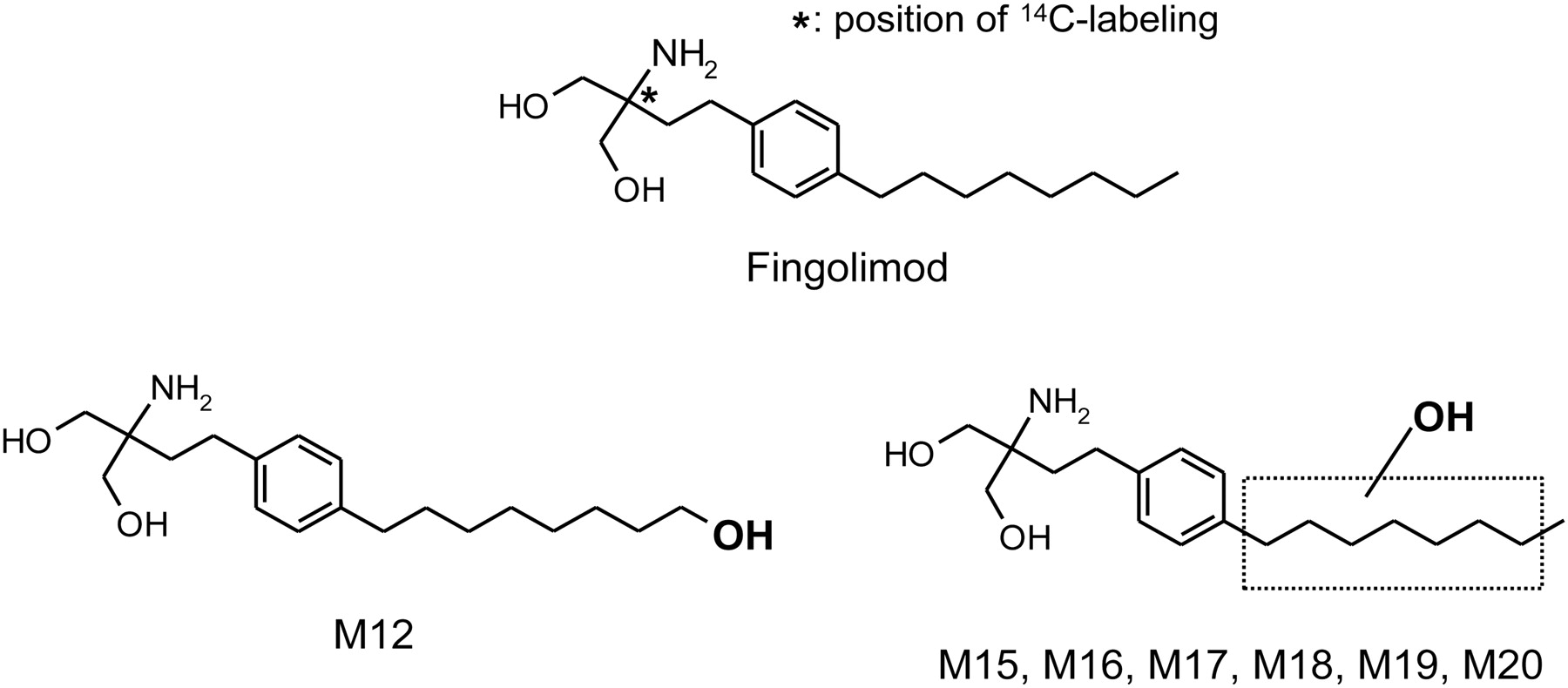
[14C]Fingolimod hydrochloride ([2-14C]2-amino-2-[2-(4-octylphenyl)ethyl]-1,3-propanediol hydrochloride) with a radiochemical purity of 98% was synthesized by the Isotope Laboratory of Novartis (Basel, Switzerland). The structure of the compound and the labeling position are shown in Fig. 1.

Structures of fingolimod, metabolite M12 (confirmed by comparison with synthetic reference compound), and metabolites M15 to M20 (proposed structures based on mass spectral information).
Reagents and Chemicals.
Paclitaxel (Taxol), quinidine, sulfaphenazole, sodium diethyldithiocarbamate (DETC), and tranylcypromine were purchased from Sigma-Aldrich (St. Louis, MO). Furafylline was obtained from Ultrafine Chemicals (Manchester, UK). Ketoconazole and troleandomycin (TAO) were purchased from Janssen Chemica (Beerse, Belgium), and azamulin was from BD Biosciences (San Jose, CA). NADP disodium salt was from Boehringer (Mannheim, Germany). NADPH, dl-isocitrate, and isocitrate dehydrogenase were from Sigma-Aldrich. The following chemicals were used to prepare the high-performance liquid chromatography (HPLC) eluents: formic acid (analytical grade; Merck, Darmstadt, Germany), acetonitrile (HPLC grade; Merck), methanol (HPLC grade; Merck), and water (HPLC grade; Fluka, Buchs, Switzerland). Other reagents, chemicals, and buffer salts were from Merck or Fluka and were of analytical or HPLC grade. The reference compound for metabolite M12 was obtained from Yoshitomi Pharmaceutical Industries Ltd. (Tokyo, Japan).
Human Liver Microsomes, Recombinant Enzymes, and Inhibitory Antibodies.
Pools of HLM and liver microsomes from 12 individual donors were obtained from BD Biosciences. The microsomes were characterized by the vendor with regard to the levels of enzyme-selective marker activities (CYP1A2, phenacetin O-deethylation; CYP2A6, coumarin 7-hydroxylation; CYP2B6, (S)-mephenytoin N-demethylation; CYP2C8, paclitaxel 6α-hydroxylation; CYP2C9, diclofenac 4′-hydroxylation; CYP2C19, (S)-mephenytoin 4′-hydroxylation; CYP2D6, bufuralol 1′-hydroxylation; CYP2E1, chlorzoxazone 6-hydroxylation; CYP3A4, testosterone 6β-hydroxylation; CYP4A11, lauric acid 12-hydroxylation; and flavin-containing monooxygenase (FMO), methyl p-tolyl sulfide oxidation). Microsomes prepared from baculovirus-infected insect cells (BTI-TN-5B1-4) expressing recombinant human P450 enzymes or FMO3, and control insect cell membrane preparations were supplied by BD Biosciences. Monoclonal antibodies inhibitory to CYP2D6, CYP2E1, and CYP3A4 were obtained from BD Biosciences. A polyclonal antibody against CYP4F2 was purchased from Fitzgerald Industries International, Inc. (Concord, MA). The antiserum was raised in rabbit using purified human CYP4F2 as immunogen and supplied as the lyophilized IgG fraction. The specificity of the polyclonal antibody was determined by Western blotting. The antibody reacted mainly with its corresponding 53-kDa immunogen in HLM. The anti-CYP4F2 was able to inhibit the ω-hydroxylation of oleic acid, arachidonic acid, and leukotriene B4 (all data from the vendor) almost completely. Because of extensive structural homology between CYP4F2 and other CYP4F isoenzymes, reaction of anti-CYP4F2 with other CYP4F isoenzymes could not be excluded. The purity of the antibody was confirmed by SDS-polyacrylamide gel electrophoresis.
Incubations with HLM and Recombinant Enzymes.
Incubations with HLM and recombinant enzymes (P450 and FMO3) were performed in 25 mM sodium citrate buffer (pH 7.5) at 37°C. After addition of different volumes of [14C]fingolimod stock solution, the reactions were started by addition of a mixture consisting of 4 μl of a fresh 10 mM NADPH solution and 20 μl of an NADPH-regenerating system containing isocitrate dehydrogenase (10 U/ml), NADP (10 mM), isocitrate (50 mM), and MgCl2 (50 mM). The final incubation volume was between 0.2 and 0.4 ml. For establishing linear reaction conditions, different concentrations of microsomal protein and different incubation times were tested. For analysis of enzyme kinetics, incubations were performed for 15 min using 0.6 mg of microsomal protein/ml and 13 different substrate concentrations ranging from 1 to 500 μM. Incubations with recombinant P450 enzymes contained 150 pmol of P450/ml. Incubations with recombinant FMO3 contained 0.4 mg protein/ml. CYP4F2 enzyme kinetic experiments were performed with 100 pmol P450/ml for 15 min at 37°C using nine substrate concentrations ranging from 0 to 250 μM. Reactions were terminated by addition of between 0.2 and 0.4 ml of ice-cooled ethanol containing 0.5% formic acid (v/v), followed by centrifugation at 30,000g for 15 min to remove protein. For the kinetic experiments, aliquots of the supernatants were analyzed by high-performance liquid chromatography with radiodetection (HPLC-RA) using HPLC conditions 2 (short gradient; see below). For other investigations the supernatants were analyzed by HPLC-RA using HPLC conditions 1 (longer gradient).
Correlation Analysis.
The rate of fingolimod metabolism was determined in incubations with HLM from 12 individual donors. The incubation time was 24 min, and the protein concentration was 0.7 mg/ml.
Incubations with HLM in the Presence of Chemical Inhibitors.
Biotransformation of 1 and 50 μM [14C]fingolimod by HLM (0.4 mg protein/ml) was investigated in the presence of different chemical inhibitors. Two concentrations of the inhibitors were used. The concentrations were 2 and 10 μM for furafylline (mechanism-based CYP1A2 inhibitor), paclitaxel (CYP2C8), sulfaphenazole (CYP2C9), and tranylcypromine (CYP2C19); 1 and 10 μM for quinidine (CYP2D6) and ketoconazole (CYP3A and CYP4F2); 5 and 10 μM for azamulin (CYP3A); 2 and 20 μM for TAO (CYP3A4 specific mechanism-based inhibitor); and 5 and 30 μM for DETC (mechanism-based CYP2E1 inhibitor). Mechanism-based inhibitors were preincubated with HLM in the presence of NADPH before the start of the reaction. For the determination of the inhibition constant (Ki) of ketoconazole, fingolimod was incubated at eight to nine different concentrations (0–220 μM) with HLM (0.6 mg/ml) for 20 min in the presence of 0, 0.4, 1.3, 4, and 10 μM ketoconazole.
Incubations with HLM in the Presence of P450-Specific Antibodies.
Biotransformation of 50 μM [14C]fingolimod by HLM (0.5 mg protein/ml) was investigated after preincubation with specific antibodies inhibitory to P450 enzymes. The effects of monoclonal antibodies inhibitory to CYP3A4, CYP2E1, and CYP2D6 and of a polyclonal anti-CYP4F2 antibody were investigated. Preincubations of the HLM were performed using different amounts of antibody at 0°C for 15 min. The incubations with fingolimod were started by the addition of NADPH and an NADPH-regenerating system. The incubation time was 20 min.
HPLC with Radiodetection.
HPLC-RA was performed on an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA) using an Advantage Armor C18 precolumn (20 × 2.1 mm, 5-μm particles) and an Advantage Armor C18 column (150 × 3 mm, 5-μm particles) from Analytical Sales and Services (Pompton Plains, NJ). A flow rate of 0.5 ml/min was used. The precolumn and column were held at 35°C. The solvent system consisted of 0.5% (v/v) aqueous formic acid (mobile phase A) and 0.5% (v/v) formic acid in acetonitrile/methanol (1:1, v/v; mobile phase B). Two different gradients of mobile phase were developed. A longer gradient (“conditions 1”: 0 min at 5% B, 10 min at 5% B, 40 min at 80% B, 47 min at 100% B, and 55 min at 100% B) was used for metabolite profiling. A shorter gradient (“conditions 2”: 0 min at 50% B, 6 min at 50% B, 15 min at 85% B, 17 min at 100% B, and 23 min at 100% B) was used for enzyme kinetics, inhibition, and other experiments. For on-line radiodetection, an LB 506 C-1 monitor (Berthold, Wildbad, Germany) and Rialuma liquid scintillator (Lumac, Groningen, The Netherlands) were used.
Structural Characterization of Metabolites by Mass Spectrometry.
For structural characterization, metabolites were generated by incubating 20 μM [14C]fingolimod with HLM and with recombinant human enzymes (CYP2D6, CYP2E1, CYP3A4, CYP4F2, CYP4F3B, and CYP4F12). The enzymatic reactions were stopped by addition of cooled acidified ethanol, the proteins were pelleted by centrifugation, and the supernatants were concentrated by partial evaporation before injection for liquid chromatography-mass spectrometry (LC-MS). The LC-MS instrumentation consisted of an 1100 series capillary pump from Agilent Technologies, a Q-Tof 2 mass spectrometer with an electrospray ion source (Micromass International, Wythenshawe, UK), and an LB 507B detector (Berthold) for parallel radioactivity monitoring. Two different chromatographic conditions were used (“conditions 3” and “conditions 4”). HPLC conditions 3 involved a C18 Advantage Armor column (150 × 2.1 mm, 5-μm particles; Analytical Sales and Services) and an eluent gradient of 0.5% (v/v) aqueous formic acid versus 0.5% (v/v) formic acid in methanol-acetonitrile (1:1, v/v). HPLC conditions 4 involved a Symmetry C18 column (150 × 2.1 mm, 3.5-μm particles; Waters, Milford, MA) and an eluent gradient of 5 mM aqueous ammonium acetate versus acetonitrile. The electrospray interface was operated in the positive ion mode with nitrogen as nebulizer gas (7 bar), desolvation gas (350 l/h), and cone gas (40 l/h). The desolvation temperature was set to 150°C, the source block was heated to 80°C, and the spray capillary was set to 3.0 kV. Fragment ions were generated by up-front collisional activation in the ion source region at elevated cone voltages. The mass resolution was between 7000 and 9000.
Data Analysis.
Velocities of enzymatic reactions are reported as picomoles per minute per milligram of microsomal protein for incubations with HLM and picomoles per minute per nanomole of P450 for incubations with recombinant P450 enzymes. Correlation coefficients (R) were obtained by plotting the metabolic rate of fingolimod versus the marker activity for each P450 enzyme using Microsoft Excel. Inhibitor concentrations producing 50% inhibition (IC50) were determined by linear interpolation. Enzyme kinetic parameters Vmax, Km, and Ki for the biotransformation by HLM and recombinant CYP4F2 were calculated using SigmaPlot 8.0 (Enzyme Kinetics module version 1.1; SPSS Science Inc., Chicago, IL). Vmax and Km were determined by nonlinear regression using the Michaelis-Menten model. The goodness of fit (R2) was 0.995 and 0.807 for HLM and CYP4F2, respectively; 95% confidence intervals of the kinetic parameters were determined by the software. The intrinsic clearance (CLint) was calculated as Vmax/Km.
Results
Metabolite Profiles.
The biotransformation of fingolimod by HLM in the presence of NADPH was slow and produced two detectable metabolites in small amounts. The more abundant of the two was M12; the less abundant was M15 (Fig. 2; Table 1). Additional metabolites (M16–M20) were observed in incubates with some of the recombinant human P450 enzymes (Table 1). No metabolites were detected in incubates with human liver cytosol alone (data not shown).

Radiochromatograms of 30-min incubates of fingolimod with HLM (20 μM substrate and 0.56 mg of microsomal protein/ml) and CYP4F2 (100 μM substrate and 150 pmol of P450/ml).
Chromatographic characteristics and relative abundance of structurally characterized fingolimod metabolites in incubates with HLM and microsomes expressing specific human P450 enzymes
Metabolite Structures.
Metabolites of fingolimod, formed by HLM and recombinant P450 enzymes, were structurally characterized by LC-MS using up-front collisional activation in the ion source region (elevated cone voltage) and exact mass measurements. Metabolite M12 was identified as the product of a hydroxylation at the terminal methyl group of the octyl chain (Fig. 1). The mass spectra of fingolimod and metabolite M12 are shown in Fig. 3. The spectrum of metabolite M12 as well as its retention times under both HPLC conditions used for LC-MS (conditions 3 and 4) matched those of the respective synthetic reference compound (data not shown). Other metabolites for which structural information could be obtained (M15–M20) (Fig. 1) were also products of a single hydroxylation. Because the mass spectra of these metabolites (not shown) were very similar to the spectrum of M12, it is likely that the metabolites were hydroxylated at the octyl chain as well (Fig. 1). However, the exact sites of hydroxylation could not be determined. Hydroxylation at the aromatic ring seemed unlikely because of prominent collision-induced losses of all three oxygen atoms in the form of water, together with one molecule of ammonia, resulting in the fragment ions at m/z 253 (unlabeled) and m/z 255 (14C-labeled), as observed for M12. Phenols are unlikely to show prominent collision-induced water losses (Ramanathan et al., 2000). Hydroxylation at one of the two carbon atoms between the aromatic ring and the aminopropanediol moiety could not be excluded but seemed unlikely because of the similarity of the mass spectra of M15 to M20 to the spectrum of M12. Some of the metabolites coeluted under the conditions of the radiochromatography (Table 1), but could be distinguished using two different chromatographic systems (conditions 3 and 4) in the LC-MS runs. Under the HPLC conditions 1 (radiochromatography) and 3 (LC-MS analysis), M12 coeluted with M18 and M15 coeluted with M16 and M17. Under the HPLC conditions 4 (LC-MS analysis), M12 coeluted with M17, whereas M15, M16, M18, M19, and M20 eluted as single components. M12 and M17 could be differentiated on the basis of the relative intensity of the fragment ion at m/z 271/273 ([M + H − 2H2O − NH3]+), which was higher in the spectrum of M12 compared with that of M17.

Electrospray mass spectra of 14C-labeled fingolimod (A) and 14C-labeled metabolite M12 (B); degree of 14C-labeling ∼45%. Fragments were generated by up-front collisional activation in the ion source region (60 V cone voltage).
Enzyme Kinetics of Fingolimod Metabolism.
After linear reaction conditions regarding incubation time and protein concentration were established, the dependence of fingolimod metabolism on the substrate concentration was investigated in HLM. Fingolimod, at 13 concentrations ranging from 1 to 500 μM, was incubated with pooled HLM (0.6 mg protein/ml) for 15 min. The plot of velocity (total metabolite formation) versus substrate concentration showed an atypical “bell-shaped” curve (Fig. 4). The decrease in the metabolic rate at high substrate concentrations suggested P450 inhibition or inactivation. The reason for the atypical kinetics was not investigated. An Eadie-Hofstee plot over the 35 to 220 μM concentration range showed a straight line (Fig. 4, left inset), suggesting a single Km. Nonlinear regression analysis of the rate of metabolism versus fingolimod concentration in the range of 35 to 220 μM revealed a Km of 183 ± 19 μM (mean ± S.E.; 95% confidence interval: 134–232 μM) and a Vmax of 1847 ± 110 pmol/(min · mg) [mean ± S.E.; 95% confidence interval: 1565–2129 pmol/(min · mg)]. The intrinsic clearance in HLM was low [10 μl/(min · mg)].
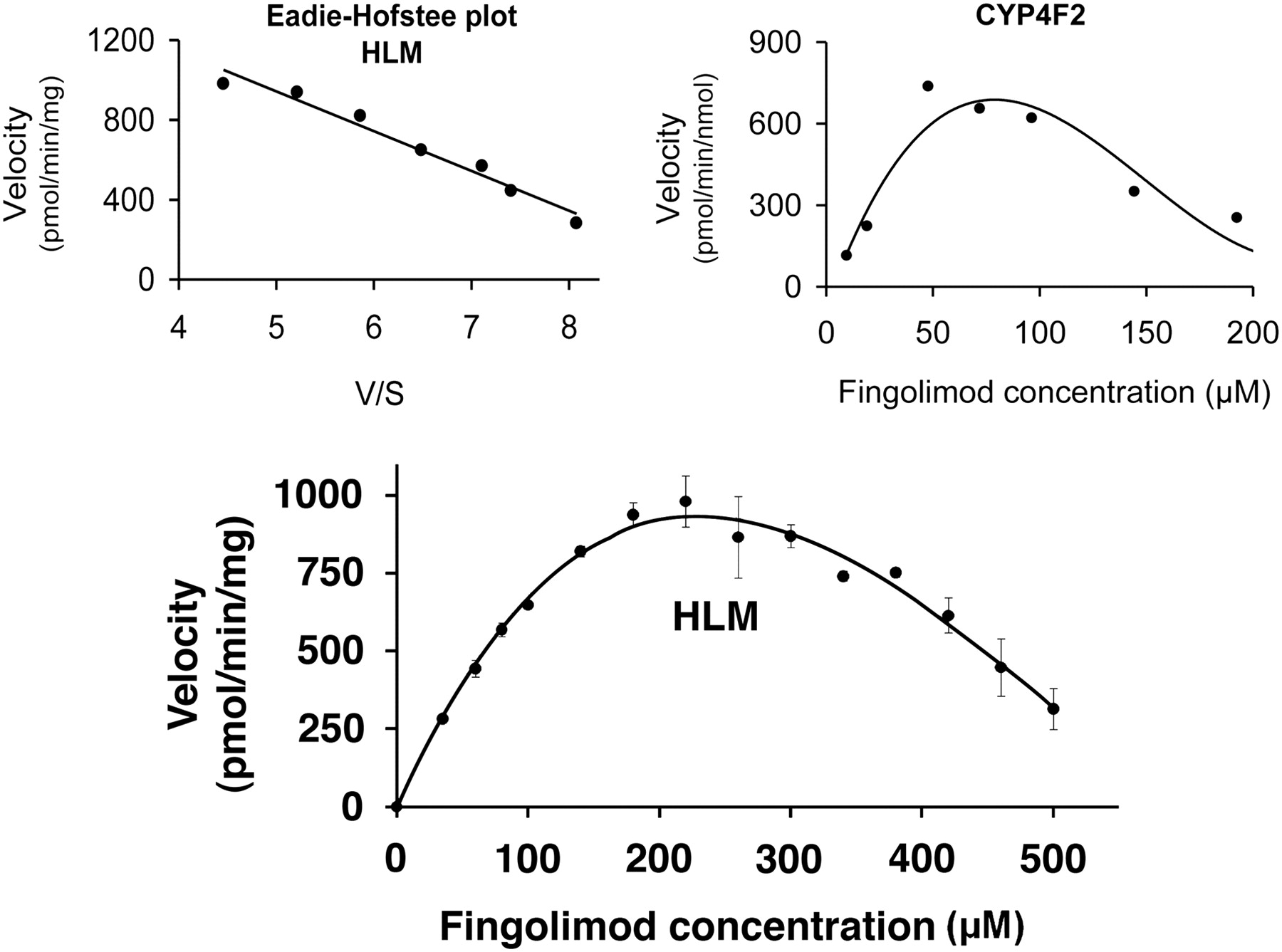
Enzyme kinetic analysis of fingolimod metabolism by HLM. The reaction mixture contained 0.6 mg/mg HLM and 35 to 500 μM [14C]fingolimod. Reactions were performed for 15 min. Symbols denote mean ± S.D. of triplicate incubations. An Eadie-Hofstee plot (left inset) was obtained for substrate concentrations between 35 and 220 μM. The right inset shows the enzyme kinetic plot for recombinant CYP4F2 (100 pmol/ml) obtained with a 15-min incubation time and 10 to 200 μM [14C]fingolimod.
Correlation Analysis of Fingolimod Metabolism Using a Panel of HLM from Individual Donors.
HLM from 12 individual donors were tested for their ability to metabolize fingolimod. The metabolic rates varied only a little between the donors (2- to 3-fold; data not shown). No correlation between these rates and any of the enzyme-specific probe substrate activities was found. All correlation values (R) were less than 0.5 (Table 2). These results did not provide an indication for an involvement of the enzymes investigated (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, CYP4A, and FMO) in the metabolism of fingolimod in HLM.
Correlation analysis of fingolimod metabolism rates with marker enzyme activities in a panel of HLM from individual donors
Biotransformation of Fingolimod by Recombinant Human P450 Enzymes.
Microsomes prepared from baculovirus-infected insect cells expressing single human P450 isoenzymes were used to assess the involvement of these enzymes in the biotransformation of fingolimod. Incubations with a panel of 21 recombinant human P450 enzymes (CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP2D6, CYP2E1, CYP2J2, CYP3A4, CYP3A5, CYP3A7, CYP4A11, CYP4F2, CYP4F3A, CYP4F3B, CYP4F12, and CYP19) were conducted under similar conditions for each isoenzyme using 5 and 100 μM fingolimod and 150 pmol P450/ml. Incubations with FMO3 were performed with 0.4 mg protein/ml. Five recombinant enzymes (CYP2D6, CYP2E1, CYP3A4, CYP4F2, and CYP4F12) showed notable metabolic activity. Low activity was found also with CYP4F3B, whereas only traces of metabolites or none at all were detected with the rest of the enzymes investigated. The metabolite profiles obtained with selected P450 enzymes and HLM are summarized in Table 1. In addition to the two metabolites observed in HLM incubates (M12 and M15), further hydroxylated metabolites (M16–M20) (Fig. 1) were produced by the recombinant CYP2D6, CYP3A4, and CYP4F12.
The metabolite patterns obtained with CYP4F2 and CYP4F3B (Table 1) were most similar to those observed in HLM among all isoenzymes investigated, with M12 being the major metabolite. In addition, small amounts of M15 were detected with CYP4F2, as with HLM. The rate of metabolism by CYP4F3B was relatively low, representing approximately one-third of that by CYP4F2. CYP2D6 metabolized fingolimod to at least four hydroxylated metabolites (M12, M15, M16, and M18), with M15 as the most abundant metabolite. CYP2E1 produced M15 as the only metabolite of fingolimod. In the incubations with recombinant CYP3A4, six hydroxylated metabolites were characterized by LC-MS (M15–M20) of which M20 was the major one. M15 was a very minor component, accounting for only 3% of the total metabolites formed by CYP3A4. CYP4F12 metabolized fingolimod to M15 and M17 in a 2:3 ratio. Enzyme kinetic parameters for recombinant CYP4F2 were determined from incubations with fingolimod at different substrate concentrations (Fig. 4, right inset). As with HLM, an atypical “bell-shaped” curve was found with a decrease in the metabolic rate at high substrate concentrations. A Km of 45 μM and a Vmax of 1049 pmol/(min · nmol P450) were determined by nonlinear regression analysis of the metabolism rate versus fingolimod concentration using the concentration range of 10 to 100 μM. The intrinsic clearance was 23 μl/(min · nmol P450).
Inhibition of Fingolimod Metabolism by P450-Specific Chemical Inhibitors.
P450-selective chemical inhibitors were used as the next approach for enzyme phenotyping (Fig. 5). The metabolism of fingolimod in HLM was most strongly inhibited by ketoconazole (1 and 10 μM), a known CYP3A4 inhibitor. The inhibition was almost complete using 10 μM ketoconazole with 1 μM substrate concentration. However, there was no inhibition by TAO (2 and 20 μM), a mechanism-based inhibitor for CYP3A4. Azamulin (5 and 10 μM), a potent and irreversible CYP3A inhibitor (Stresser et al., 2004), showed only slight inhibition (13–27%) of fingolimod metabolism in HLM. No significant inhibition of fingolimod metabolism was observed with furafylline, paclitaxel, sulfaphenazole, tranylcypromine, quinidine, and DETC (Fig. 5).

Effect of P450 isoenzyme-selective chemical inhibitors on fingolimod metabolism in HLM. [14C]Fingolimod was incubated with HLM (0.4 mg/ml) in the presence of different inhibitors, and the rates of total metabolite formation (M12 plus M15) were determined. Reactions were performed for 20 min. The data are shown as mean and range of duplicate determinations. The concentration of fingolimod was 1 μM (□) and 50 μM (■) for all assays with the exception of azamulin inhibition for which the fingolimod concentration was 30 μM (□) and 60 μM (■). Control reactions (100% activity) were run with inhibitor vehicle (methanol).
Because no relevant role of CYP3A4 was found in the experiments with recombinant P450 enzymes (Table 1) and the strongest inhibition was observed with ketoconazole, which is known to inhibit CYP4F2 and CYP4F12 (Stresser et al., 2004), besides CYP3A4, the effects of ketoconazole on fingolimod metabolism by recombinant CYP4F2 and CYP4F12 were investigated. The biotransformation of 10 μM fingolimod by 100 pmol/ml CYP4F2 and 50 pmol/ml CYP4F12 was readily inhibited by ketoconazole with IC50 values of 1.6 and 0.6 μM, respectively.
The inhibition of the metabolism of fingolimod in HLM by ketoconazole was further investigated at different substrate concentrations in the presence (0.4–10 μM) or absence of ketoconazole under linear conditions with regard to incubation time and protein concentration (Fig. 6). The rates of biotransformation were analyzed by nonlinear curve-fitting using the Enzyme Kinetics module of SigmaPlot, considering different inhibition types: full or partial competitive, noncompetitive, uncompetitive, and mixed inhibition. The models were ranked by the goodness of fit (Akaike's information criterion). The partial competitive inhibition model provided the best fit of the data with a Ki of 0.74 ± 0.09 μM.
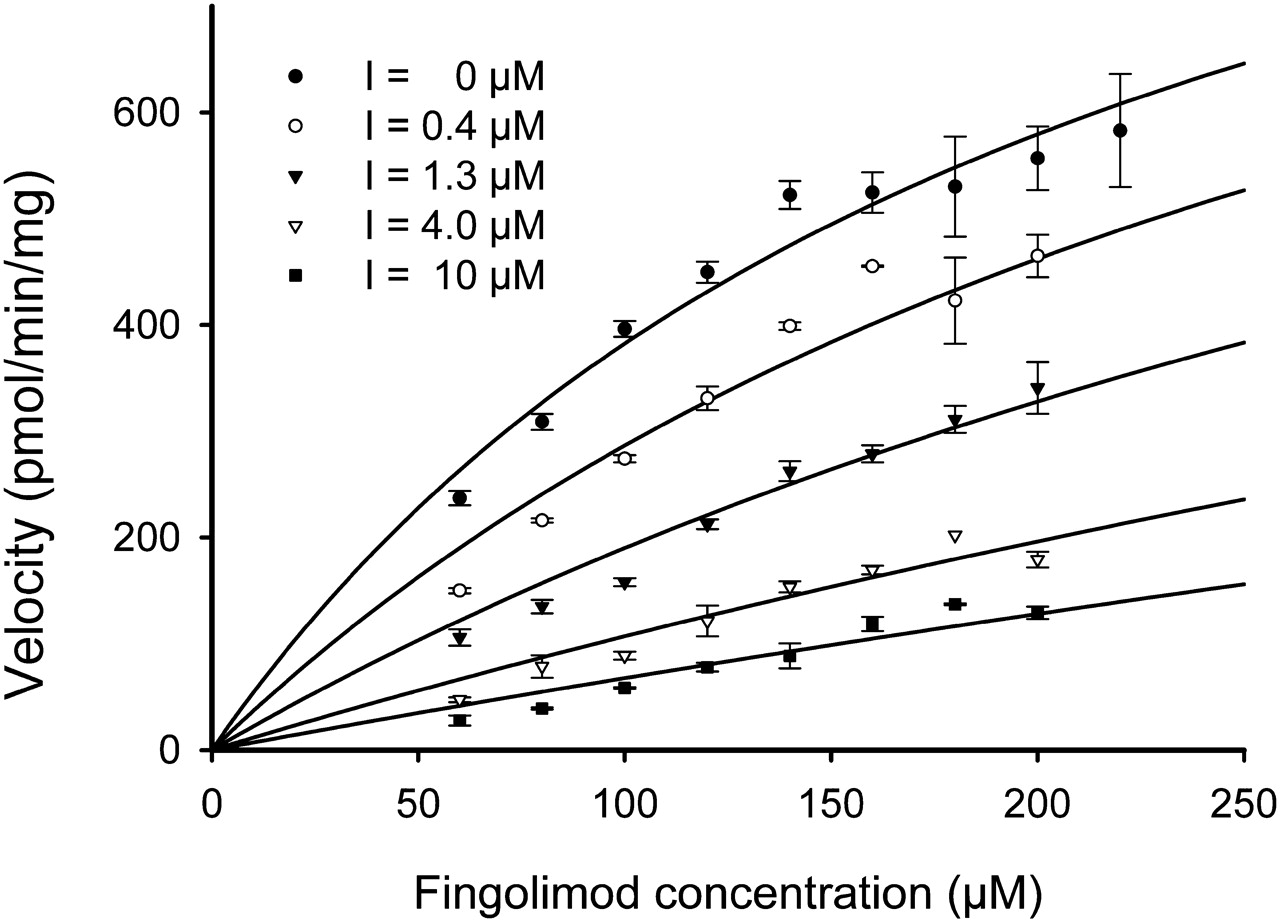
Inhibition of fingolimod metabolism by ketoconazole (Ki determination). The concentration-dependent kinetics of the metabolism of [14C]fingolimod (60–220 μM) by HLM (0.6 mg/ml) were determined in the presence of five different concentrations of the inhibitor (I = 0, 0.4, 1.3, 4, and 10 μM). The incubation times were 20 min. Data are shown as a Michaelis-Menten plot (mean ± S.D. of at least two replicates). The inhibition type was found to be partial competitive, and the Ki value was determined to be 0.74 μM.
Inhibition of Fingolimod Metabolism in HLM by P450-Specific Inhibitory Antibodies.
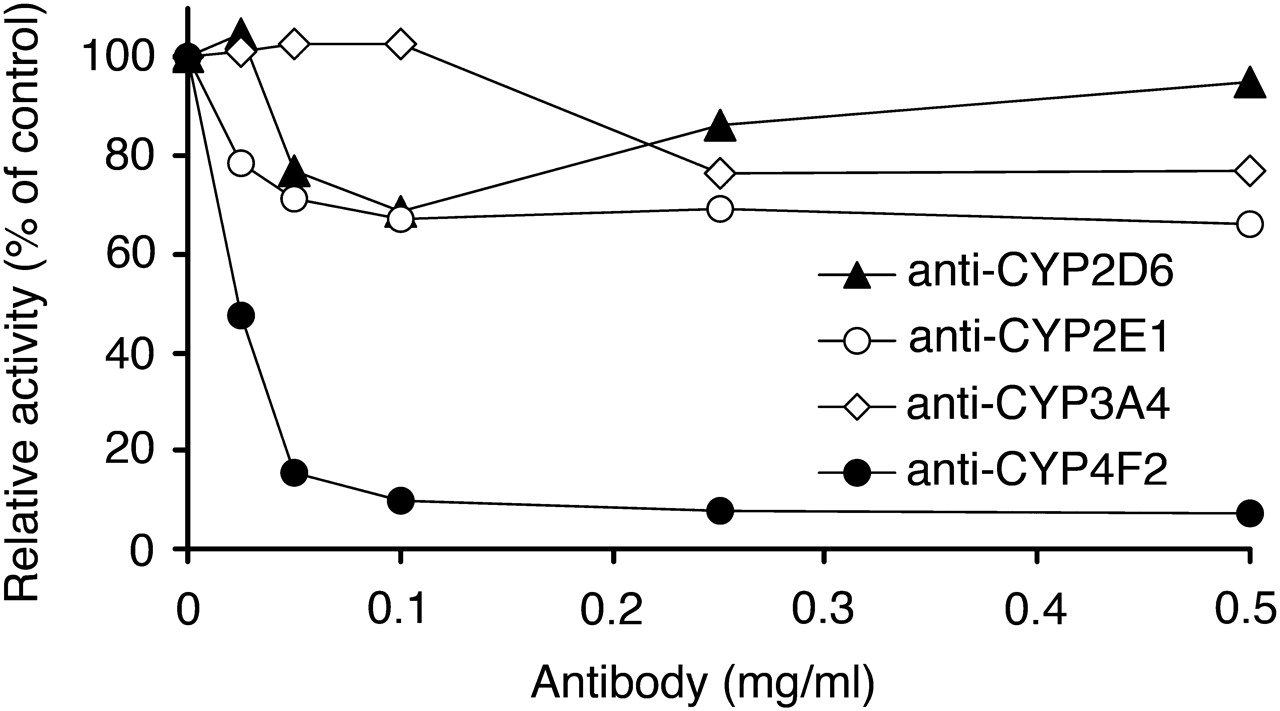
Figure 7 shows the effects of P450-specific inhibitory antibodies on the metabolism of 50 μM fingolimod by HLM (0.5 mg/ml). There were only small changes to the rate of metabolism, or none at all, in the presence of the monoclonal antibodies inhibitory to CYP2D6, CYP2E1, and CYP3A4. In contrast, polyclonal antibodies raised against human liver CYP4F2 were found to be very potent inhibitors of fingolimod metabolism in HLM. The maximum inhibition of total metabolic activity was 93%. In a parallel experiment with recombinant CYP4F2, anti-CYP4F2 showed a similar maximum inhibition (92%) of fingolimod metabolism.

Effect of P450-specific inhibitory antibodies on the metabolism of fingolimod by HLM. The total biotransformation rate (formation of M12 plus M15) of fingolimod (50 μM) by HLM (0.5 mg/ml) was investigated after preincubation with antibodies inhibitory to CYP2D6, CYP2E1, CYP3A4, and CYP4F2. The results are expressed as activities relative to the control rates in the absence of antibodies.
Discussion
The in vitro metabolism of fingolimod was investigated in HLM and recombinant human P450s to determine the enzymes involved in the oxidative metabolism of the compound. The biotransformation of fingolimod in HLM was slow with an intrinsic clearance of 10 μl/(min · mg) microsomal protein. This is consistent with the low clearance of the drug found after intravenous administration (6.3 l/h) (Kovarik et al., 2007). In humans in vivo, fingolimod was found to be metabolized by several pathways, including reversible phosphorylation to the pharmacologically active principle. However, the elimination of the compound occurred predominantly by oxidation. The initial step of the oxidative pathway was hydroxylation at the terminal methyl group of the octyl chain (metabolite M12). Because of rapid further oxidation to a carboxylic acid and subsequent β-oxidation, M12 was not detectable in vivo. Yet, in HLM lacking the enzymes catalyzing the further oxidation of the primary hydroxylation product, M12 was the major biotransformation product. In addition, metabolite M15 was observed in HLM as a minor hydroxylation product, and other hydroxylated metabolites (M16–M20) were found in incubates with recombinant P450 enzymes. However, M15 to M20 appeared to play no relevant role because neither these metabolites nor any downstream products could be identified in vivo (Zollinger et al., 2010).
Among the six recombinant P450 enzymes that showed notable metabolism of fingolimod, CYP4F2 and CYP4F3B produced metabolite profiles most similar to those produced by HLM. M12 was the major metabolite formed by CYP4F2 and CYP4F3B (Table 1). The rate of metabolism by CYP4F3B was three times lower compared with that of CYP4F2. In contrast to CYP4F2 and CYP4F3B, CYP2D6, CYP2E1, CYP3A4, and CYP4F12 showed metabolite profiles different from those in HLM. The main metabolites produced by these enzymes (Table 1), all being isomers of M12, were not observed in vivo. This result might be due to differences between recombinant and natural enzymes (Proctor et al., 2004) in their catalytic competency (Ogilvie et al., 2008). Because of the “artificial” nature of expressed enzymes, differences in activity characteristics may exist with respect to P450s in their native membrane environment (Bjornsson et al., 2003). Therefore, CYP2D6, CYP2E1, CYP3A4, and CYP4F12 did not seem to contribute significantly to the metabolism of fingolimod in vivo. Other P450 enzymes and recombinant FMO3 generated only traces of fingolimod metabolites and are therefore also unlikely to contribute.
Abundance values of CYP4F2 in HLM of 142 pmol/mg (Jin et al., 1998) and 155 pmol/mg (Lasker et al., 2000) have been reported previously. A new method using anti-CYP4F2 peptide IgG showed highly variable CYP4F2 contents in HLM with levels ranging between 0 and 80 pmol/mg (Hirani et al., 2008). A recent human liver proteome analysis (Chinese Human Liver Proteome Profiling Consortium, 2010) revealed high protein abundance of CYP4F2 and CYP4F3, representing 42 and 29%, respectively, of the CYP3A4 abundance. J. M. Lasker (personal communication) measured the hepatic CYP4F contents in 29 donors and obtained values ranging from 18 to 128 pmol/mg microsomal protein. According to these results, the hepatic abundance of CYP4F is relatively high. The liver content of CYP4F12 is currently unknown (Chinese Human Liver Proteome Profiling Consortium, 2010). Although mRNA levels of CYP4F2, CYP4F3B, and CYP4F11 were measurable in the liver, the transcript of CYP4F12 was barely expressed, according to a relative abundance study (J. M. Lasker, personal communication). In view of the results with recombinant enzymes, CYP4F2 and, to some extent, CYP4F3B seem to be the major catalysts of fingolimod hydroxylation.
In a panel of individual HLM, no correlation of fingolimod metabolism with CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, CYP4A, and FMO marker activities was found. This finding is in line with CYP4F being the major fingolimod metabolizer.
In agreement with the above results, no significant inhibition of fingolimod metabolism by the chemical P450-selective inhibitors furafylline, paclitaxel, sulfaphenazole, tranylcypromine, quinidine, DETC, and TAO were found. Surprisingly, potent in vitro inhibition by the CYP3A4 inhibitor ketoconazole was observed, in contrast to the absence of an inhibitory effect of TAO, a mechanism-based inhibitor for CYP3A4. However, ketoconazole had also been shown previously to inhibit CYP4F2 (Stresser et al., 2004) although at higher concentrations than for CYP3A4. In line with these results, ketoconazole also inhibited the metabolism of fingolimod by recombinant CYP4F2 (IC50 = 1.6 μM). Wang et al. (2006) found a strong selectivity of ketoconazole for CYP4F2 versus CYP4F3B and proposed this inhibitor as a tool to distinguish the two closely related enzymes. Therefore, the strong effect of ketoconazole on the metabolism of fingolimod in HLM (almost 100% inhibition at 10 μM ketoconazole and 1 μM fingolimod) (Fig. 5) suggests a predominant contribution of CYP4F2 and little involvement of CYP4F3B. Azamulin, a potent and irreversible CYP3A inhibitor, showed only moderate inhibition (by 13–27%) of fingolimod metabolism in HLM at concentrations of 5 and 10 μM, whereas CYP3A activity is strongly inhibited. This finding is consistent with the results of Stresser et al. (2004) who found that azamulin also inhibits CYP4F2 (IC50 = 46 μM). Hence, the effect observed with azamulin was probably caused by a partial inhibition of CYP4F2. Together, the data from chemical inhibitors support the major role of CYP4F2 and little involvement of other P450 enzymes in the hepatic metabolism of fingolimod.
Final confirmation of the major contribution of CYP4F enzymes in fingolimod metabolism was obtained from investigations using P450-specific antibodies. Whereas anti-CYP2D6, anti-CYP2E1, and anti-CYP3A4 showed no or little effect at concentrations that strongly inhibited the metabolism of their known specific substrates (results not shown), an antibody against CYP4F2 was found to be a very potent inhibitor of fingolimod metabolism by HLM, producing a maximum inhibition of 93%.
In the present investigation, four approaches were used to identify the major oxidizing enzymes involved in the biotransformation of fingolimod. Correlation analysis using a panel of HLM from individual donors revealed no correlation of fingolimod metabolism with any of the human P450 and FMO enzymes (which did not include CYP4F) investigated. Recombinant CYP4F2 showed the highest activity in metabolizing fingolimod to M12, the main metabolite in HLM. Experiments with selective chemical inhibitors supported the major role of CYP4F2. The antibody against CYP4F2 inhibited the metabolism of fingolimod in HLM almost completely. All of the four enzyme phenotyping approaches (Ogilvie et al., 2008) were in line with the conclusion that CYP4F enzymes are the major contributors to the hydroxylation of fingolimod. On the basis of our data, CYP4F2 seems to play a major role. However, contributions by other members of the CYP4F subfamily, including CYP4F3B for which only fragmentary data and experimental tools are available at this time, cannot be excluded. The high preference of CYP4 enzymes for ω- over ω-1-hydroxylation of fatty acid chains (Ortiz de Montellano, 2008) is well in line with our results.
Ketoconazole inhibited the biotransformation of fingolimod by HLM with a Ki of 0.74 μM. This value was in line with the IC50 value determined with recombinant CYP4F2 (1.6 μM). The in vitro data suggest that ketoconazole may have the potential to inhibit the metabolic clearance of fingolimod. Therapeutic plasma concentrations of ketoconazole are in the range of 2 to 12 μM (Schulz and Schmoldt, 2003). These values are 3- to 16-fold greater than the Ki value. Therefore, ketoconazole has a potential to inhibit the metabolic clearance of fingolimod (Bjornsson et al., 2003). In a clinical study, ketoconazole was used as a putative CYP4F2 perpetrator to quantify its influence on fingolimod pharmacokinetics in healthy subjects. A modest average 1.7-fold increase of fingolimod exposure (area under the blood concentration time curve) was found (Kovarik et al., 2009). The in vivo data confirmed the clinical relevance of CYP4F2 inhibition by ketoconazole. A Ki value for the effect of ketoconazole on CYP4F2 has not been published. The value determined in this work may be applicable for predicting the effects of ketoconazole on metabolism of CYP4F2 substrates with the I/Ki approach (Bjornsson et al., 2003) or using modeling and simulation tools such as DDI Predict and Simcyp (Rostami-Hodjegan and Tucker, 2007).
A recent genome-wide association study revealed that CYP4F2 was one of three principal genetic determinants of warfarin dose, the other two being CYP2C9 and vitamin K epoxide reductase complex, subunit 1 (Takeuchi et al., 2009). Ward et al. (2008) demonstrated a significant association of the CYP4F2 GA/AA genotype with elevated systolic blood pressure and significantly increased urinary 20-HETE excretion. Hence, a genetic polymorphism of CYP4F2 might possibly contribute to the interindividual variability in the metabolism of fingolimod. However, functional consequences of the CYP4F2 polymorphism were found to be substrate-dependent (Stec et al., 2007). The amino acid change at position 433 (V433M) decreased 20-HETE production by 34 to 44% but had no effect on ω-hydroxylation of leukotriene B4. A preliminary pharmacogenetic analysis indicated no significant effect of CYP4F2 genetic polymorphism on the pharmacokinetics of fingolimod (unpublished Novartis internal data).
In conclusion, CYP4F enzymes, in particular CYP4F2, were identified as the major catalysts of fingolimod oxidation. The lack of involvement of the common drug-metabolizing enzymes in fingolimod clearance suggests a reduced drug interaction potential. This provides an advantage regarding the safety of this novel oral therapy for relapsing multiple sclerosis.
Authorship Contributions
Participated in research design: Jin, Zimmerlin, and Patten.
Conducted experiments: Jin, Zollinger, Borell, Zimmerlin, and Patten.
Performed data analysis: Jin, Zollinger, Borell, Zimmerlin, and Patten.
Wrote or contributed to the writing of the manuscript: Jin, Zollinger, Zimmerlin, and Patten.
Acknowledgments
We thank Dr. Rhys Salter (Isotope Laboratory, Novartis) for the synthesis of the radiolabeled drug and Claudia Sayer for technical support in the LC-MS analysis. We also thank Dr. Jerome Lasker (Puracyp Inc., Carlsbad, CA) for helpful discussion and for providing unpublished information on CYP4F enzymes, Drs. Shivani Mittra and Rama Sivasubramanian (Integrated Information Sciences, Novartis Healthcare Pvt. Ltd.) for assistance in preparing the manuscript, and Drs. Gian Camenisch, Heike Gutmann, Pieter J. Swart, and Olivier Kretz for critical review of the article.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.035378.
-
ABBREVIATIONS:
- fingolimod (FTY720)
- 2-amino-2-[2-(4-octylphenyl)ethyl]-1,3-propanediol
- P450
- cytochrome P450
- 20-HETE
- 20-hydroxyeicosatetranoic acid
- HLM
- human liver microsomes
- DETC
- sodium diethyldithiocarbamate
- TAO
- troleandomycin
- HPLC
- high-performance liquid chromatography
- FMO
- flavin-containing monooxygenase
- HPLC-RA
- high-performance liquid chromatography with radiodetection
- LC-MS
- liquid chromatography-mass spectrometry.
- Received August 16, 2010.
- Accepted November 2, 2010.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}