


Abstract
The pivotal role of organic anion-transporting polypeptide 1B1 (OATP1B1) in drug disposition has become clear over the last decade. Therefore, an OATP1B1 inhibition assay suitable for use within early drug discovery was developed and characterized. IC50 estimates for 10 literature compounds using pitavastatin and estradiol-17β-glucuronide as substrates were within 2-fold of each other. In addition, the IC50 estimates using pitavastatin uptake agreed well with literature values (r2 = 0.92, average fold error = 1.3). However, when estrone-3-sulfate was used, OATP1B1 inhibition was underpredicted by as much as 10-fold. A comparison of uptake in human hepatocytes and OATP1B1 inhibition showed a significant correlation (r2 = 0.53, P < 0.001) for more than 40 compounds. These data suggest that, for discrete chemical series, OATP1B1 inhibition data may be used as a surrogate for more costly and time-consuming uptake studies in hepatocytes. OATP1B1 inhibition data, determined for over 260 compounds representing both internal AstraZeneca and literature chemistry, were also used to generate a continuous in silico model. The robustness of the model was demonstrated by accurately predicting OATP1B1 inhibition for external test sets using 50 AstraZeneca compounds (root mean square error = 0.45) and 12 literature drugs (RMSE = 0.32). The most important molecular descriptors for the prediction of OATP1B1 inhibition were maximal hydrogen bonding strength followed by cLogP. This study has shown that a well validated OATP1B1 inhibition assay in conjunction with an in silico approaches has the potential to influence significantly the design-make-test cycle and subsequently reduce the propensity of OATP1B1 ligands.
Introduction
The accurate prediction of human pharmacokinetics has been highlighted as an area that required major improvements to avoid significant attrition in early drug discovery (Prentis et al., 1988). To this end, Drug Metabolism and Pharmacokinetics departments have spent considerable effort in developing suitable strategies to mitigate drug failure attributed to suboptimal human pharmacokinetics (McGinnity et al., 2007; Beaumont and Smith, 2009).
In the past, the majority of new chemical entities (NCEs) have been primarily metabolized by cytochrome P450s (P450s) and particularly by CYP3A4 (Bertz and Granneman, 1997). However, the development of high-throughput in vitro screens (Riley and Grime, 2004) and a greater understanding of the relationship between the physicochemical properties of new chemical entities and their metabolism (van de Waterbeemd et al., 2001; Wenlock et al., 2003) has significantly reduced P450-mediated metabolism. As a consequence, our progress in understanding, predicting, and minimizing P450 metabolic liabilities has arguably resulted in an increase in the relative contribution of phase II metabolism and drug transporters to the clearance of NCEs over the last decade (Soars et al., 2002, 2009; Mizuno et al., 2003; Shitara et al., 2006).
The successful in vitro-in vivo translation of hepatic clearance from both human and preclinical in vitro tools has become commonplace (McGinnity et al., 2004; Ito and Houston, 2005; Riley et al., 2005) for phase I and phase II metabolism. However, a closer inspection of these datasets has highlighted that many of the outliers are known substrates of hepatic uptake transporters (Soars et al., 2007a). As a result, several hepatocyte-based uptake assays have been developed to accurately predict the clearance of this subset of drugs for which traditional approaches fail (Soars et al., 2007b; Paine et al., 2008).
Arguably, the most important superfamily of transporters for the hepatic uptake of anionic drugs is the organic anion-transporting polypeptide (OATP) (Hagenbuch and Meier, 2003, 2004). The expression of individual OATPs in mammalian cell lines and their subsequent characterization against a range of xenobiotics suggests that OATP1B1 and OATP1B3 play a key role in hepatic uptake (Mizuno et al., 2003; Shitara et al., 2006). Furthermore, over the last decade, there has been a significant increase in the number of drug-drug interactions (DDIs) attributed to the inhibition of OATP1B1 (Kalliokoski and Niemi, 2009). Cyclosporine has been shown to increase the plasma concentration of bosentan (Binet et al., 2000), repaglinide (Kalliokoski et al., 2008), and a number of statins, at least in part through an interaction with OATP1B1 (Simonson et al., 2004). The inhibition of OATP1B1 has also been shown to play a role in DDIs involving gemfibrozil and a number of statins, particularly for those that have no CYP2C8 component in their metabolism (Schneck et al., 2004). These clinical data have highlighted the important role that OATP1B1 can play in the disposition of drugs that could be adopted early within drug discovery, which has prompted the pharmaceutical industry and regulators alike to respond by recommending key transporter studies (Giacomini et al., 2010).
To this end, the aims of this study were 3-fold as follows: to develop and characterize an HEK-based cell line that stably expressed OATP1B1; to use literature knowledge to develop an OATP1B1 inhibition assay suitable for screening NCEs early in drug discovery; and to use OATP1B1 inhibition data to generate an in silico model, which could guide future drug discovery programs.
Materials and Methods
Chemicals and Human Hepatocytes.
All chemicals and reagents used were of the highest available grade. Montelukast, bosentan, pravastatin, atorvastatin, and pitavastatin were sourced from Sequoia Research Products Ltd. (Oxford, UK). [3H]Estrone-3-sulfate (specific activity 2120 GBq/mmol) and [3H]estradiol-17β-glucuronide (specific activity 1670 GBq/mmol) were obtained from PerkinElmer Life and Analytical Sciences (Waltham, MA). All other chemicals were purchased from Sigma-Aldrich (Poole, Dorset, UK). AstraZeneca compounds were synthesized at AstraZeneca R&D Charnwood.
Freshly isolated human hepatocytes were obtained from the UK Human Tissue Bank (Leicester, UK) following appropriate consent and ethical approval. Hepatocyte viability was routinely >80%.
Construction of Stably Transfected HEK293 Cells Expressing OATP1B1.
The OATP1B1 cDNA was cloned into the mammalian expression vector pGenIRESneo (a gift from Dr. Hazel Weir, AstraZeneca, Macclesfield, UK) using a SpeI/NotI-digested insert and NheI/NotI-digested vector. The nucleotide sequence of the cloned cDNA was determined using BigDye version 3.1 (Applied Biosystems, Foster City, CA) to confirm that the construct expresses OATP1B1 identical to Swiss-Prot Q9Y6L6. Stable expression of OATP1B1 was achieved by transfecting HEK293 cells (American Type Culture Collection, Manassas, VA), which were maintained in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal calf serum. Transfection was carried out using FuGENE 6 reagent according to the manufacturer's instructions (Roche Diagnostics, Burgess Hill, UK). Forty-eight hours after transfection, the medium was replaced with DMEM/10% fetal calf serum containing 1 mg/ml active Geneticin (G418; Invitrogen, Paisley, UK), and the cells were grown for 2 weeks to select for antibiotic-resistant cells. Monoclonal cells were obtained by limiting dilution cloning.
OATP1B1-HEK Uptake Assays.
OATP1B1-HEK cells and HEK cells transfected with empty vector (pGenIRESneo with no cDNA insert) were grown in DMEM (Invitrogen) supplemented with 10% (v/v) fetal bovine serum (Invitrogen), 4 mM glutamine (Invitrogen) and 1 mg/ml G418 (Invitrogen) at 37°C with 5% CO2 and 95% humidity. Cells were seeded in 12-well plates coated with poly-d-lysine (Becton Dickinson, Oxford, UK) at a density of 0.45 million cells per well, 24 h in advance of any uptake assays. Before assay, cells were washed three times with Krebs-Henseleit buffer prewarmed to 37°C (supplemented with 25 mM HEPES and 2.5 mM CaCl2). After cells were preincubated at 37°C for 30 min in Krebs-Henseleit buffer, uptake assays were initiated with the addition of substrate prepared in Krebs-Henseleit buffer. For initial screening assays pitavastatin and [3H]estradiol-17β-glucuronide were incubated at a final substrate concentration of 1 μM. For kinetic analysis at least seven substrate concentrations were used covering 0.1 to 100 μM. In all cases, the final concentration of dimethyl sulfoxide (DMSO) did not exceed 1% (v/v). Uptake assays were terminated after 1 min (uptake of both pitavastatin and estradiol-17β-glucuronide had been shown to be linear to 1 min previously) via the addition of ice-cold Krebs-Henseleit buffer. Cells were washed two more times with ice-cold Krebs-Henseleit buffer and left dry.
For uptake studies using radiolabeled substrates, cells were lysed by incubating with 500 μl of 0.1% (v/v) Triton X-100 for 30 min. After the addition of scintillation cocktail, the amount of radioactivity in the cells was determined using a Packard 2200CA Tri-Carb liquid scintillation counter (Packard Instrument Co., Pangbourne, UK). For uptake studies using pitavastatin as a substrate, cells were lysed by incubating with 500 μl of methanol/acetonitrile (50:50) for 1 min. The samples were placed at −20°C for 1 h and then centrifuged at 2000g for 15 min. The supernatant (200 μl) was transferred to 96-well microtiter plates (Agilent Technologies, Santa Clara, CA) for liquid chromatography-mass spectrometry/mass spectrometry analysis (see below). For protein determinations, unused cells were lysed using 0.1% (v/v) Triton X-100 as stated above with protein concentrations determined using urinary protein as a standard (Randox Laboratories, Crumlin, UK).
OATP1B1-HEK Inhibition Assays.
Inhibition assays using OATP1B1-HEK cells were essentially conducted as for uptake studies (see above), with the exception that in addition to assays containing substrate they also contained varying amounts of inhibitor. Initial substrate and inhibitor stocks were prepared in DMSO at 200× the required concentration so that the final concentration of DMSO did not exceed 1% (v/v). For each IC50 determination, at least six inhibitor concentrations were used. For IC50 determinations of known OATP1B1 inhibitors, the inhibitor concentrations used were selected to span the IC50 for AstraZeneca compounds; the following inhibitor range of 0.1 to 25 μM was used. When pitavastatin or estradiol-17β-glucuronide were used as substrates, the final substrate concentration was 1 μM, whereas a final concentration of 0.1 μM was used for estrone-3-sulfate. The incubation time was 1 min for all substrates.
Determination of Loss from Media CLint,uptake Using Human Hepatocytes.
Loss from media CLint,uptake estimates were determined as stated previously (Soars et al., 2007b). In brief, NCE stocks were prepared in DMSO at 100-fold incubation concentration (100 μM). Of this 100 μM stock, 10 μl was added to a vial containing 490 μl of hepatocyte suspension buffer. A vial containing human hepatocytes at a concentration of 2 million viable cells/ml was preincubated for 5 min in a shaking (80 oscillations/min) water bath at 37°C along with the vial containing the drug/buffer mix. Reactions were initiated by adding 500 μl of hepatocyte suspension to the 500 μl of drug/buffer mix (giving a final substrate concentration of 1 μM at 1% v/v DMSO). Aliquots (80 μl) were removed at 0.5, 1, 2, 4, 6, and 15 min from the incubation and placed into centrifuge tubes. These aliquots were immediately centrifuged at 7000g for 30 s using a MSE MicroCentaur centrifuge (Fisher Scientific, Loughborough, UK), and 40 μl of the supernatant was pipetted into 120 μl of ice-cold methanol. Mock time 0 samples were generated by adding 10 μl of 100 μM stock to 990 μl of hepatocyte suspension buffer from which 40 μl was pipetted into 120 μl of ice-cold methanol. Samples were then frozen for 1 h at −20°C and centrifuged at 2000g for 20 min at 4°C. The supernatants were removed and analyzed as described below.
Analysis of Hepatocyte and OATP1B1 Inhibition Samples Containing Pitavastatin.
Mass spectrometry was conducted on a Micromass Quattro Ultima Platinum triple quadrupole (Waters, Manchester, UK) using a Hewlett Packard 1100 HPLC system (Hewlett Packard, Palo Alto, CA) for separation. Analysis was done by multiple reaction monitoring using either positive or negative ion mode. Cone voltage and collision energy were optimized for each compound.
In these analyses, chromatographic separation was achieved using a Hypersil Gold C18 (4.6 × 50 mm, 3 μm) column obtained from Thermo Electron Corp. (Basingstoke, UK) using 10 μl of each sample. The mobile phase consisted of water with 0.1% (v/v) formic acid with the organic phase being methanol containing 0.1% (v/v) formic acid. All chromatography was performed using a generic gradient (t = 0 min % organic = 5, t = 0.5 min % organic = 5, t = 2 min % organic = 100, t = 3 min % organic = 100, t = 3.1 min % organic 5, total run time = 4 min). The flow rate was set at 1.5 ml/min, which was introduced into the mass spectrometer source at 0.4 ml/min. For kinetics analyses, the amount of each substrate was quantified using authentic standards of known concentration.
Data Analysis.
Kinetic parameters were determined by nonlinear regression using MicroCal Origin 6.0 (OriginLab Corporation, Northampton, MA). The appropriate model (Michaelis-Menten or Hill) used to obtain the kinetic constants was determined by comparing the randomness of the residuals, the size of the sum of the squares of the residuals, and the standard error of the parameter estimates. For each substrate concentration, the initial uptake rate was calculated by subtracting the initial rate determined in HEKs transfected with empty vector from those obtained in HEKs stably expressing OATP1B1.
For IC50 determinations, the initial uptake rate at each inhibitor concentration was calculated by subtracting the initial uptake rate of substrate alone (determined in HEKs transfected with empty vector) from those obtained in HEKs stably expressing OATP1B1. Values were also corrected for any protein differences between HEKs transfected with empty vector or OATP1B1. IC50 values were determined by nonlinear regression analysis (WinNonlin; Pharsight Corporation, Mountain View, CA). IC50 values were converted to Ki estimates for literature comparison by rearranging the eq. 1.
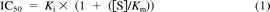 CLint,uptake was estimated from hepatocyte uptake assays using eq. 2:
CLint,uptake was estimated from hepatocyte uptake assays using eq. 2:
 where V is the incubation volume (corrected for nonspecific binding) and k is the elimination rate constant. Nonspecific binding was determined as the difference in drug concentration between the 0- and 0.5-min time point. Therefore, the elimination rate concentration was calculated from the initial linear phase from log concentration-time plots starting from the 0.5-min time point using typically 4 to 5 time points. This method was also used for compounds exhibiting a biphasic profile. Although this represents a potential composite of uptake and metabolism, curve stripping produced similar results for a representative set of compounds (data not shown).
where V is the incubation volume (corrected for nonspecific binding) and k is the elimination rate constant. Nonspecific binding was determined as the difference in drug concentration between the 0- and 0.5-min time point. Therefore, the elimination rate concentration was calculated from the initial linear phase from log concentration-time plots starting from the 0.5-min time point using typically 4 to 5 time points. This method was also used for compounds exhibiting a biphasic profile. Although this represents a potential composite of uptake and metabolism, curve stripping produced similar results for a representative set of compounds (data not shown).
Development of an in Silico Model for the Prediction of OATP1B1 Inhibition.
Dataset.
One of the aims of this study was to develop an in silico method that could be used within the company to assess the propensity for compounds to be OATP1B1 substrates by inference from their ability to act as inhibitors in HEK cells. Data were generated on multiple AstraZeneca sites with compounds from multiple projects that enhanced the chemical diversity of the dataset and increased the applicability of any generated model across the whole company.
The data set in total consisted of 263 compounds, with 201 compounds used in the training set and 50 compounds randomly selected to act as the test set. The test set covered the dynamic range of the dataset that ranged from pIC50 4.3 to 7.0.
A further test set of 12 compounds (losartan, telmisartan, atorvastatin, benazepril, fluvastatin, cerivastatin, irbesartan, valsartan, enalapril, pravastatin, simvastatin, and perindopril) was used to further test the external predictivity of the model (Supplemental Table 2). These 12 compounds, predominantly from the cardiovascular therapeutic area, have been well characterized in the literature and are known to inhibit OATP1B1 to varying degrees.
The properties of the compounds used to generate the in silico model are summarized in Fig. 1. The lipophilicity for these compounds ranged from −0.5 to 7 with a mean lipophilicity of 4.1. The range in molecular weight was from 290 to 760, with a mean of 468, and the polar surface area ranged from 41 to 223, with a mean of 103. This range in physical properties coupled with compounds that were selected from a range of AstraZeneca projects gives a set of compounds with a diverse structural and physical property profile. This dataset was then considered appropriate to build an in silico model with as wide an applicability as was available at this time (Supplemental Table 1).

The frequency distribution of the molecular properties of the compounds used to build the in silico model for OATP1B1 inhibition. The distribution of Clog P (A), molecular weight (B), and polar surface area (PSA) (C).
Modeling.
Molecular Descriptors.
The AstraZeneca in-house descriptor set was used to describe the molecules for the development of the in silico model. This set consisted of 143 descriptors that encode information regarding the topological, geometrical, electronic, and physical properties of each compound. For details of this descriptor set, see Bruneau (2001).
The freely available statistical package R [R Development Core Team 2006, version 2.7.1 (Windows); http://www.r-project.org/index.html] was used to build random forest models using the continuous pIC50 data for OATP1B1 inhibition. The random forest methodology is described in detail by Svetnik et al. (2003).
The random forest algorithm (Breiman et al., 1984) generates a predictive model by use of a set of binary rules to calculate a target value. The algorithm is termed an ensemble method because it combines the results from many (200 in this case) different models to generate a single result. It has been demonstrated that the prediction accuracy from an ensemble model is usually better than the results from one of the individual models (see Dietterich, 2002).
The random forest algorithm was initially provided with the training data, and for each of the models generated, the original training data were split by randomly selecting approximately two thirds of the data with replacement. This process of splitting or sampling the data is termed bootstrapping. The remaining one third of the data, termed the out-of-bag (OOB) data, was used to estimate the error in the model and the importance of each variable toward the overall model.
For each bootstrapped sample (approximately two thirds of the original data), a regression tree was generated with the modification that the best split was chosen using a random selection of the variables rather than the whole descriptor set. The assignment of a single prediction for a given compound was made by taking the average prediction from all of the models generated.
Evaluation of Model Predictivity.
Ideally, the predictivity of any in silico method would be addressed by a large external test set. However, this is an emerging area of science, and a high volume of quality experimental data were not available. In situations such as this, all methods available to these authors were used to generate confidence in the model.
OOB Error.
The OOB error uses the bootstrapping process of tree building and involves sampling of the training data. During this process, some compounds were left out the sample set, whereas others were repeated. Each tree was grown with approximately two thirds of the original data set, whereas one third of the training set could be used in the OOB estimate. Because the OOB samples were not used to construct the tree, they could be used in the estimate of the model prediction accuracy in a similar way as an external test set. An estimate for the mean square error (MSE) of the random forest regression method used in this work is given by eq. 3.
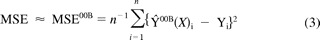 where Ŷ00B(Xi) is the ensemble prediction value for each training sample Xi, n is the number of compounds in the training set, and Yi is the experimentally determined value.
where Ŷ00B(Xi) is the ensemble prediction value for each training sample Xi, n is the number of compounds in the training set, and Yi is the experimentally determined value.
RMSE.
A further estimate of the model predictivity can be assessed by the RMSE of both the external test set consisting of 50 internal AstraZeneca compounds and also the external test set consisting of 12 literature compounds. The RMSE is calculated as in eq. 4.
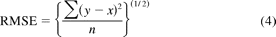
Estimation of Descriptor Importance.
Random forest models are an ensemble of individual models and hence do not produce an explicit model, which can be easily interpreted. Understanding the relationship between the descriptors that contribute toward OATP1B1 inhibition is essential if the model is to be used in the design-make-test cycle of any drug discovery program. However, it is possible to estimate the contribution of each descriptor to the overall model prediction accuracy by replacing each descriptor with random noise. If the model prediction accuracy deteriorated as a consequence of a descriptor being replaced with noise, then that descriptor was contributing significantly to model performance. When the random forest algorithm was used in regression, as was the case in this work, the importance of each descriptor was estimated using the OOB dataset. Each descriptor in the OOB dataset was randomly permuted and then predicted by the tree. The increase in the least squared prediction accuracy was used to estimate the importance of each descriptor.
Results
Characterization of OATP1B1-HEK Substrate Specificity.
Kinetic analyses for the prototypic OATP1B1 substrates pitavastatin and [3H]estradiol-17β-glucuronide were conducted in the OATP1B1-HEK cell line to confirm that uptake was consistent with that obtained in the literature and to determine Km estimates to enable their use as potential substrates in an inhibition assay. The OATP1B1-mediated uptake was determined by subtracting the initial rates (1 min) obtained with empty vector cells from that obtained for HEK cells transfected with OATP1B1 at each substrate concentration. Representative plots for pitavastatin and [3H]estradiol-17β-glucuronide are shown in Fig. 2, A and B, respectively. Figure 2, A and B, shows that the initial rates of uptake for both pitavastatin and estradiol-17β-glucuronide were saturated at concentrations of up to 100 μM and that the data were adequately fitted using a simple Michaelis-Menten model. The Km value of 4.8 ± 0.7 μM obtained for pitavastatin agreed well with that cited previously in the literature (3 μM) (Hirano et al., 2004). The Km obtained for [3H]estradiol-17β-glucuronide (3.6 ± 0.9 μM) was also consistent with the range observed in the literature (3.7–8.2 μM) (König et al., 2000; Cui et al., 2001; Tamai et al., 2001). Because a nonradiolabeled end point is more cost effective and easier to use within early drug discovery, pitavastatin was chosen as a substrate for further inhibition studies.

Determination of pitavastatin (A) and estradiol-17β-glucuronide (B) kinetics using HEK cells stably expressing OATP1B1. At least seven substrate concentrations were used in each kinetic determination. For each substrate concentration, the initial uptake rate was calculated by subtracting the initial rate determined in HEKs transfected with empty vector from those obtained in HEKs stably expressing OATP1B1. Kinetic estimates are the mean ± S.D. of three separate experiments.
Validation of OATP1B1 Inhibition Assay.
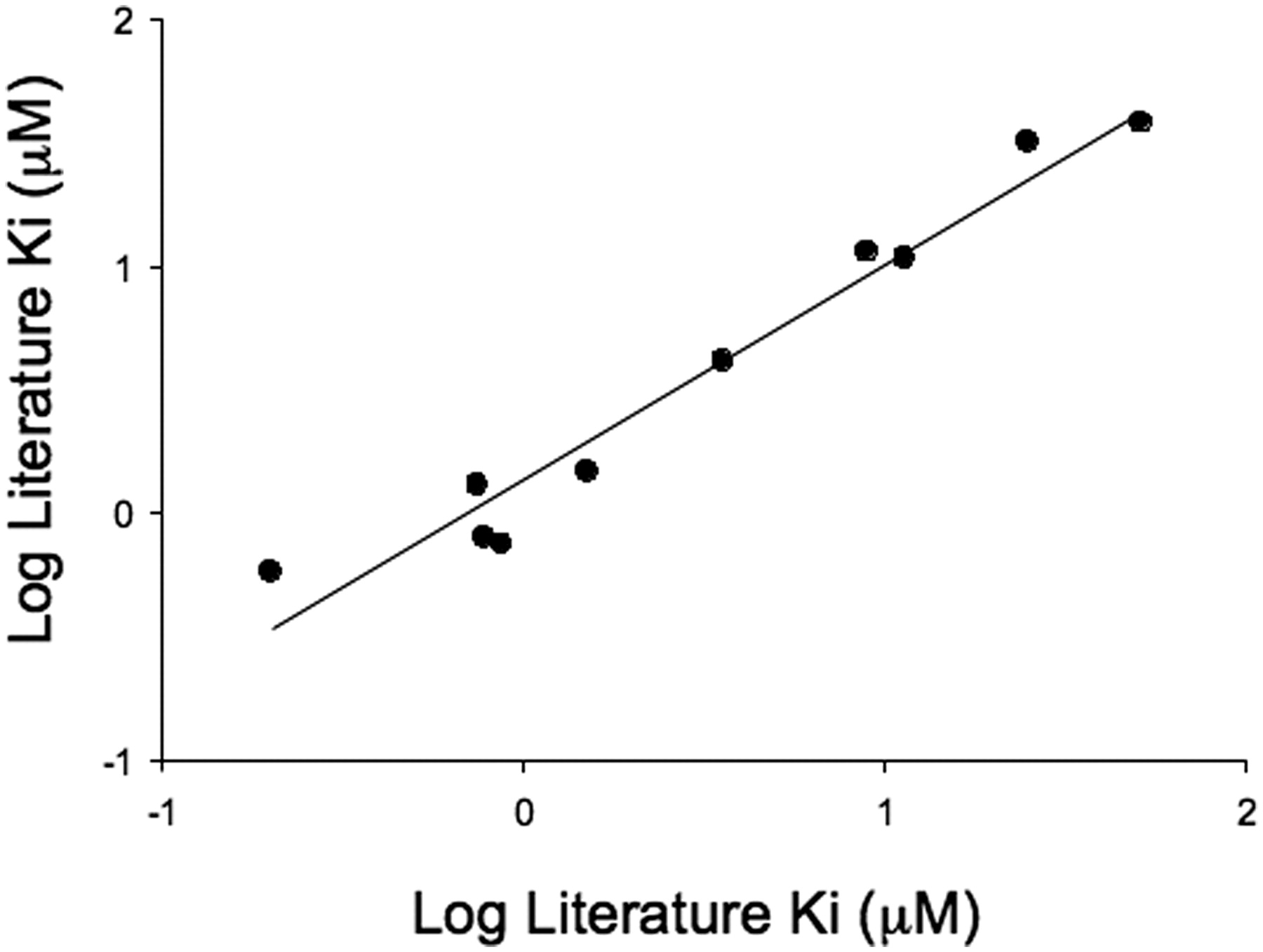
To confirm that pitavastatin was an appropriate substrate to use in an OATP1B1 inhibition assay, a comparison with known OATP1B1 inhibitors was instigated. A literature search highlighted 10 compounds with known Ki values against OATP1B1 ranging from 0.2 μM for cyclosporine A to 52 μM for verapamil. A comparison of the Ki values determined in the literature with those obtained using the AstraZeneca OATP1B1 inhibition assay is shown in Fig. 3. The correlation obtained (r2 = 0.92, average fold error = 1.3) confirms that this cell line can distinguish between weak and potent literature inhibitors of OATP1B1.
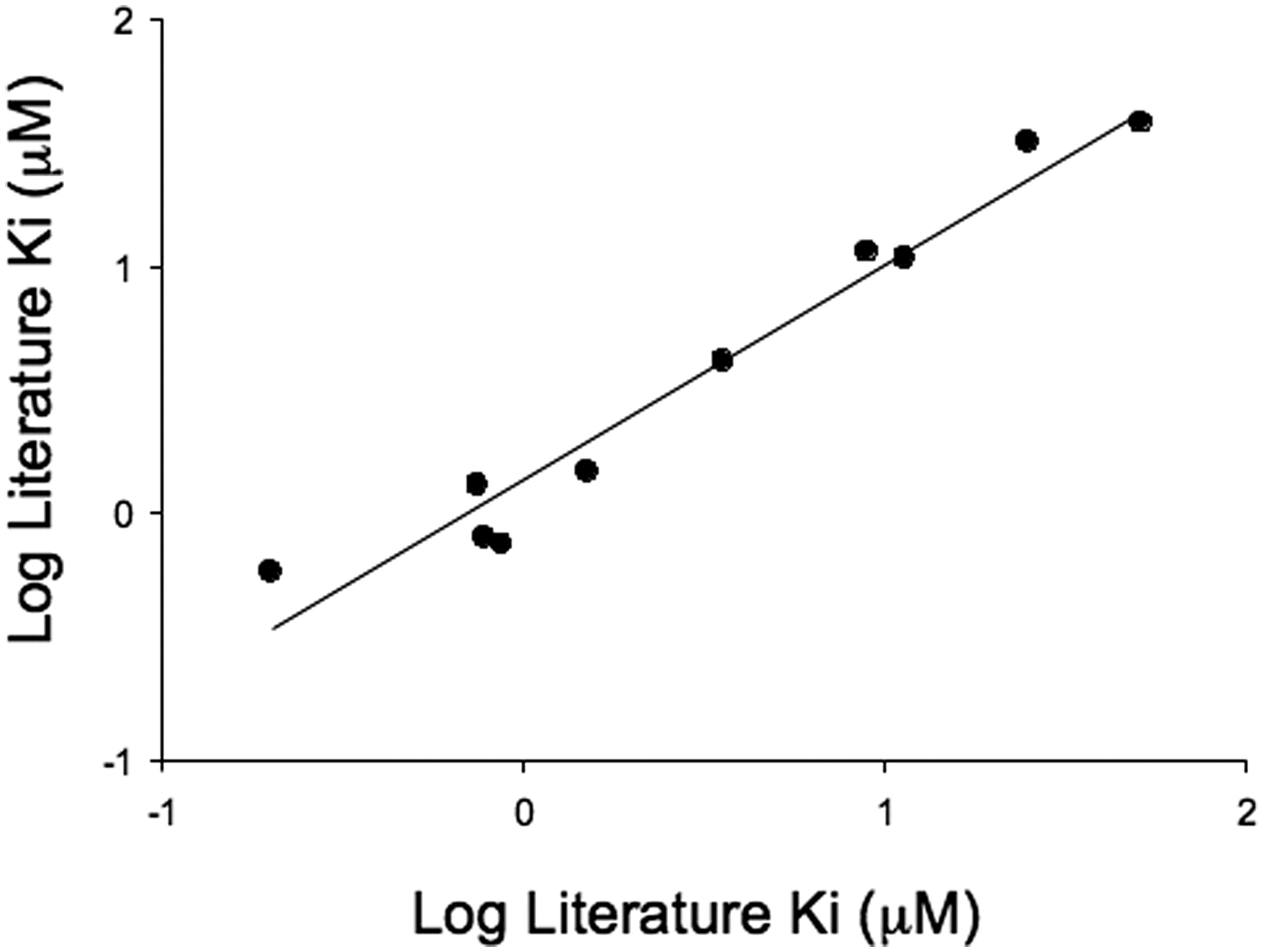
Comparison of Ki estimates determined using OATP1B1-mediated OATP1B1 uptake of pitavastatin with literature values. Ki estimates were determined using the inhibition of OATP1B1-mediated pitavastatin uptake at 1 μM. Six inhibitor concentrations were used in each determination, and each data point represents the mean ± S.D. of at least three experiments. Literature data were sourced from Chen et al. (2005) and Hirano et al. (2006). The solid line represents a regression analysis (line of best fit is given by y = 0.87x + 0.14, r2 = 0.92, average fold error = 1.3).
To investigate any potential intersubstrate differences in OATP1B1 inhibition, IC50 estimates were obtained for eight known OATP1B1 inhibitors using pitavastatin, estradiol-17β-glucuronide, and estrone-3-sulfate as substrates (see Table 1). The IC50 determinations obtained using pitavastatin and estradiol-17β-glucuronide as substrates were within 2-fold of each other for the majority of compounds investigated. When estone-3-sulfate was used as a substrate, the results were more variable (see Table 1). The IC50 values obtained for the potent OATP1B1 inhibitor cyclosporine A were comparable using either of the three substrates; however, the IC50 estimates determined for ritonavir and glibenclamide were up to ∼5- to 10-fold higher than those obtained using either pitavastatin or estradiol-17β-glucuronide as substrates.
Effect of substrate on OATP1B1 inhibition
Data represent individual experiments or mean ± S.D. of three experiments
Utility of OATP1B1 Inhibition as a Surrogate for Human Hepatic Uptake Assays.
A set of AstraZeneca and literature compounds shown previously to be uptake substrates in human hepatocytes (see Soars et al., 2007b) was investigated in the OATP1B1 inhibition assay (see Fig. 4). For several discrete series of AstraZeneca compounds, there was a statistically significant relationship between hepatic uptake rate assessed by human hepatocytes and OATP1B1 inhibition. A similar trend was also apparent for the more limited set of literature compounds (n = 4), which had been included in these studies. Although for each of the three datasets hepatic uptake increased (lower 1/CLint,uptake), with an increase in OATP1B1 inhibition (lower IC50), the relationship for the literature compounds investigated was offset from that observed for the AstraZeneca chemistries. Essentially, for a given CLint,uptake, the OATP1B1 IC50 was on average 5-fold lower for the literature compounds than for each of the AstraZeneca series, which may indicate differential selectivity for OATPs and/or passive permeability.

Relationship between the inhibition of OATP1B1-mediated uptake and CLint,uptake determined in human hepatocytes for several chemically distinct series of AstraZeneca compounds (closed symbols) and four literature compounds (open symbols). IC50 estimates were determined using the inhibition of OATP1B1-mediated pitavastatin uptake at 1 μM. Six inhibitor concentrations were used in each determination. CLint,uptake was determined using human hepatocytes via a media loss incubation. Each data point is the mean of at least two separate experiments. The solid lines represent a regression analysis (equations given by y = 0.78x − 1.82, r2 = 0.95, P < 0.05 for literature compounds and y = 0.85x − 2.60, r2 = 0.53, P < 0.01 for AstraZeneca compounds).
Development and Characterization of an OATP1B1 Inhibition in Silico Model.
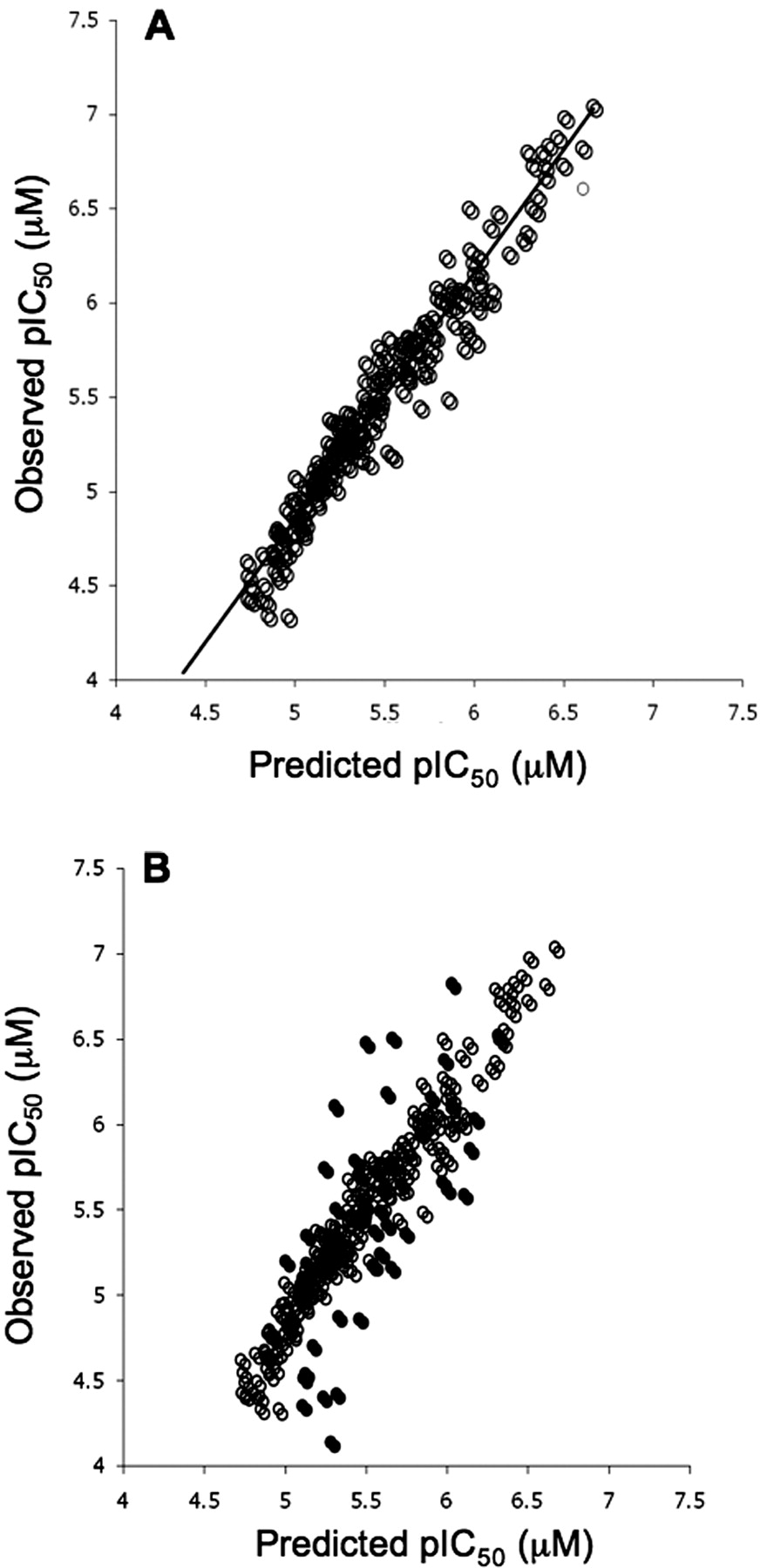
A single computational model was developed using the random forest algorithm and a descriptor set, which covered topological, electronic, geometric, and the physical properties of each compound. Figure 5A shows the correlation between experimentally determined inhibition of OATP1B1 and the predicted inhibition based on the random forest model. The statistics of the model in training are good, as is expected given the nature of the random forest algorithm, and result in an RMSE of 0.2. An assessment of the external predictivity of the model was obtained by calculating OATP1B1 inhibition estimates for a further set of 50 in house AstraZeneca compounds (overlaid in Fig. 5B). For this external test set, the model performed well over a pIC50 range from 4.1 to 6.8, producing an RMSE of 0.45. Although the model was built using AstraZeneca compounds, Fig. 6 shows the capability to predict accurately (RMSE = 0.32) the inhibition of OATP1B1 for a series of 12 literature compounds with pIC50 values ranging between 4.5 and 6.3. This series of compounds represents a further external estimate of the accuracy in predicting OATP1B1 inhibition using the random forest model.
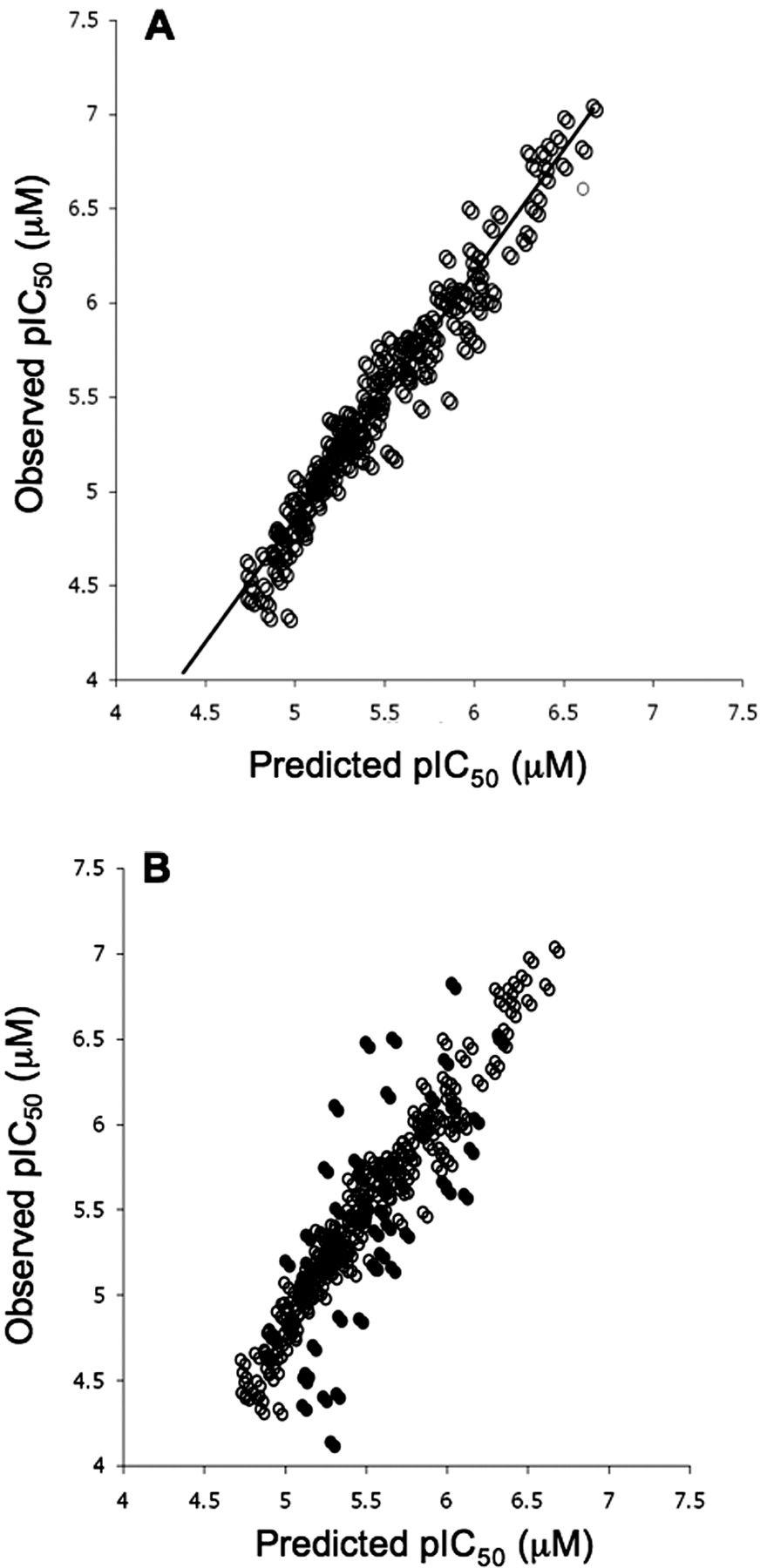
Relationship between the predicted inhibition of OATP1B1-mediated uptake of pitavastatin (A) and observed inhibition based on random forest model external test set overlaid on training set (B).
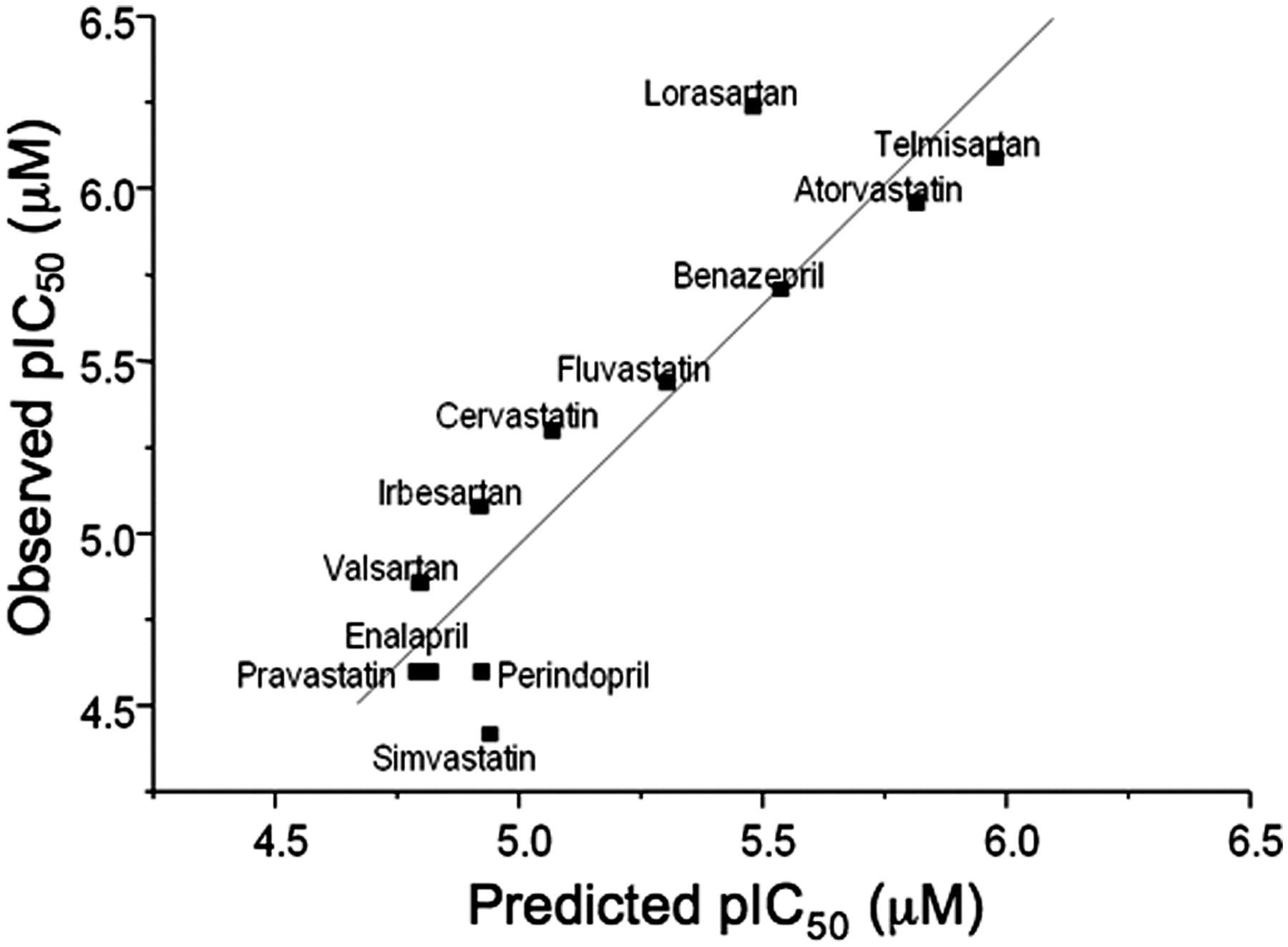
Relationship between the predicted inhibition of OATP1B1-mediated uptake of pitavastatin and observed inhibition based on random forest model for a series of literature compounds.
A summary of the statistics for this model together, with an assessment of the accuracy in predicting OATP1B1 inhibition, is given in Table 2. In addition to using an RMSE for both the in-house and literature external datasets (see Table 2), an OOB error, which is a form of cross-validation available within the R software, was also used resulting in an RMSE of 0.23. The consistently low error rate obtained using each of these three approaches adds confidence in the robustness of the OATP1B1 inhibition model.
Data set information and model summary statistics
Key Descriptors in the OATP1B1 in Silico Model.
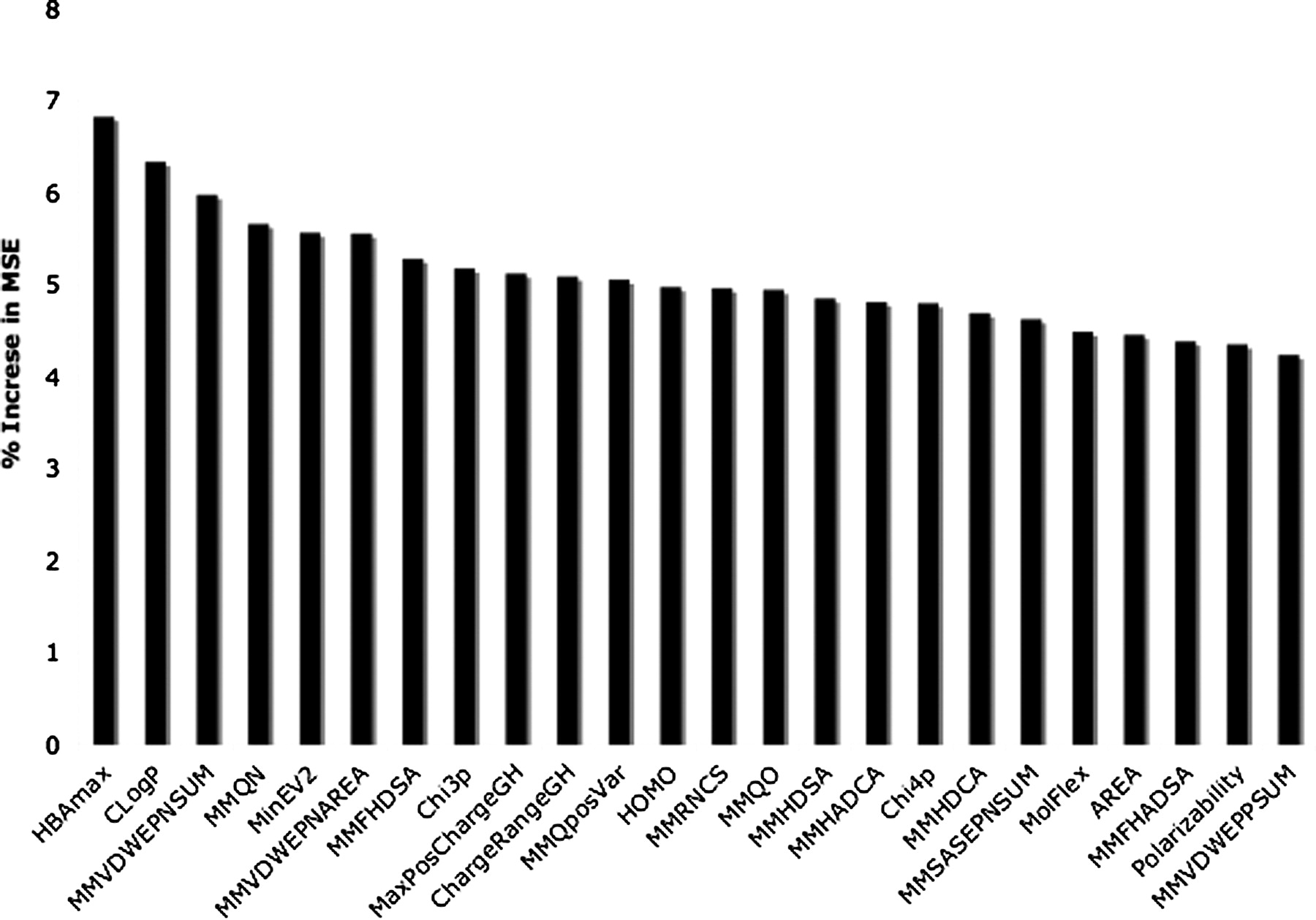
Figure 7 shows the 24 most important descriptors contributing to the random forest model as assessed by the increase in MSE after randomizing each descriptor. The most important descriptor is the maximal hydrogen bonding strength followed by the cLogP of the compounds. The next five descriptors are the molecular mechanics descriptors relating to the general hydrogen-bonding potential of the compound and further strengthen the importance of this property in the determination of affinity at OATP1B1.
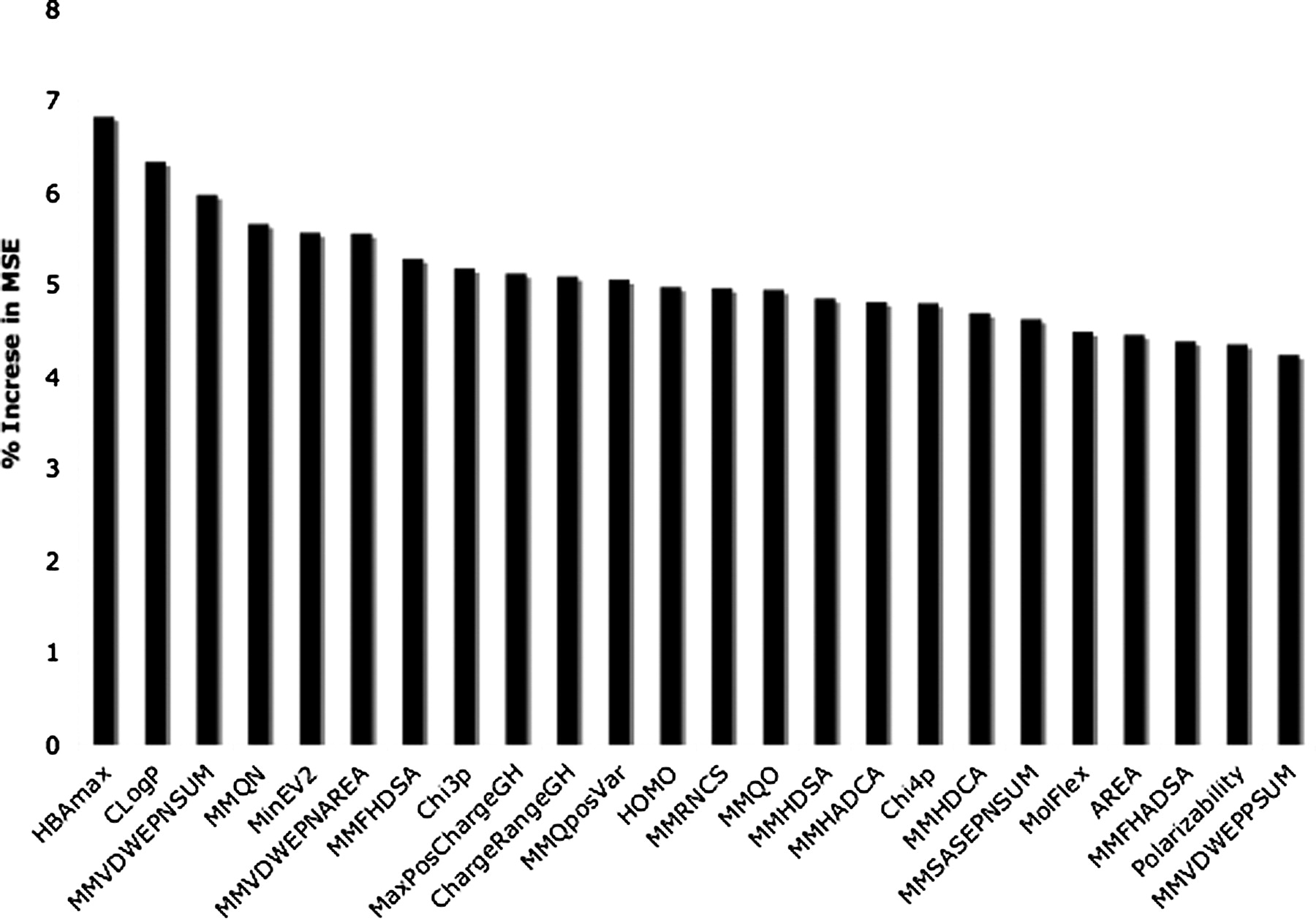
Key properties of random forest model. Descriptor importance based on the increase in means square error when the descriptor is replaced with random noise.
Discussion
Over the last 10 years, the number of DDIs involving drug transporters has increased significantly (Kindla et al., 2009). This has prompted the drug industry and regulators alike to highlight how in vitro transport assays could be used to mitigate and/or form clinical DDI studies (Giacomini et al., 2010). Therefore, the development and characterization of robust transport assays, such as the OATP1B1 inhibition assay described here, are pivotal in providing early assessments of DDI potential.
The importance of using more than one substrate in inhibition assays to get an accurate assessment of inhibitory potential has been highlighted for enzymes that have multiple binding sites or modes such as CYP3A4 (Kenworthy et al., 1999). The differences in IC50 values observed in this study when using estone-3-sulfate as a substrate compared with pitavastatin/estradiol-17β-glucuronide (see Table 1) suggest that care should be taken when selecting a probe for OATP1B1 inhibition assays. An underprediction in the inhibition of OATP1B1 with gemfibrozil has been observed previously by Noé et al. (2007) when estone-3-sulfate was used as a substrate. Subsequent analysis showed that estrone-3-sulfate kinetics in OATP1B1 cells were most accurately described by a two-site model, and this was attributed to OATP1B1 containing at least two binding sites. Further evidence for multiple binding sites has been obtained with mutation studies (Miyagawa et al., 2009). The relevance of OATP1B1 inhibition data generated using pitavastatin as a substrate has been highlighted by Hirano et al. (2006) who showed that these data could be used to predict in vivo interactions mediated via OATP1B1. Whereas pitavastatin has been selected as an easy-to-use and cost-effective probe for OATP1B1 inhibition studies in early drug discovery, it should be noted that [3H]estradiol-17β-glucuronide has also been used successfully in a drug development setting (Sharma et al., 2010).
The emerging relationship between hepatic uptake in human hepatocytes and OATP1B1 inhibition for several series of AstraZeneca compounds (see Fig. 4) suggests a further potential application of OATP1B1 inhibition data. It is noteworthy that such a relationship implies that the compounds investigated in this study interact competitively at a single OATP1B1 binding site. Exceptions may be anticipated for compounds that bind noncompetitively or demonstrate mixed type inhibition. Nevertheless, the potential exists to facilitate rapid decision-making in drug discovery using such an early indirect assessment to prioritize those compounds studied in a more time-consuming and expensive, functional hepatic uptake assay as initially recommended for other drug transporters, e.g., P-glycoprotein/multidrug resistance 1 (Schwab et al., 2003). Further work is required in this area to build on these findings.
The current OATP1B1 inhibition in silico model has been developed using a set of descriptors that describe the topological, electronic, geometric, and physical properties of the test compounds. The ability of the model to discriminate between active and inactive compounds plays a critical part in the design-make-test cycle guiding design teams to optimize the interaction between NCEs and OATP1B1.
The model performs well in predicting an external test set of 50 in-house AstraZeneca compounds where the RMSE in prediction is 0.45 together with literature test set of 12 compounds for which the RMSE is 0.30. The key descriptors for increasing OATP1B1 inhibition as determined from this in silico model are to increase the hydrogen bond-accepting strength and increase the lipophilicity. The key molecular features identified in this two-dimensional quantitative structure activity relationship approach are consistent with the work of Chang et al. (2005) who by use of a three-dimensional pharmacophore approach also identified hydrogen bond accepting and hydrophobicity as key features in OATP1B1 inhibition. These key interactions are further substantiated by Gui et al. (2009) who by use of a comparative molecular field analysis approach identified hydrophobicity and basicity of the side chain residues as being critical for OATP1B1 inhibition. Badolo et al. (2010) investigated the ability of 179 compounds to inhibit the uptake of estradiol-17β-glucuronide in human hepatocytes to develop a structure-activity relationship for OATP1B1/1B3. Lipophilicity, polarity, pKa, and the number of hydrogen bond donors and acceptors again were shown to play a critical role in determining the molecular interactions with the OATP1B1/1B3. More recently, the work of Karlgren et al. (2011) showed that OATP1B1 inhibitors tend to be more lipophilic and larger and display a larger polar surface area than noninhibitors.
This report has highlighted the importance of robustly characterizing a cell line (in this case OATP1B1) before it is used as a primary inhibition screen. By analyzing over 250 compounds in an OATP1B1 inhibition assay, it has been possible to develop the first continuous quantitative structure activity relationship model that can accurately predict OATP1B1 inhibition for internal and literature compounds alike. Future efforts to build robust in silico models for key transporter isoforms will further enhance our ability to optimize transporter interactions in early drug discovery.
Authorship Contributions
Participated in research design: Soars, Barton, Ismair, and Riley.
Conducted experiments: Soars and Ismair.
Contributed new reagents or analytic tools: Jupp.
Performed data analysis: Barton.
Wrote or contributed to the writing of the manuscript: Soars, Barton, Jupp, and Riley.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.ABBREVIATIONS:
- NCE
- new chemical entity
- P450
- cytochrome P450
- OATP
- organic anion-transporting polypeptide
- DDI
- drug-drug interaction
- HEK
- human embryonic kidney
- DMEM
- Dulbecco's modified Eagle's medium
- DMSO
- dimethyl sulfoxide
- OOB
- out-of-bag
- MSE
- mean square error
- pIC50
- negative log of the IC50 estimate
- RMSE
- root mean square error.
- Received August 25, 2011.
- Accepted May 14, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}