


Abstract
Lenvatinib is a multityrosine kinase inhibitor that inhibits vascular endothelial growth factor receptors, and is being developed as an anticancer drug. P450s are involved in one of the elimination pathways of lenvatinib, and mono-oxidized metabolites, such as N-oxide (M3) and desmethylated metabolite (M2), form in rats, dogs, monkeys, and humans. Meanwhile, two other oxidative metabolites are produced only in monkey and human liver S9 fractions, and their structures have been identified using high-resolution mass spectrometry as a quinolinone form of lenvatinib (M3′) and a quinolinone form of desmethylated lenvatinib (M2′). The formation of M3′ from lenvatinib occurred independently of NADPH and was effectively inhibited by typical inhibitors of aldehyde oxidase, indicating the involvement of aldehyde oxidase, but not P450s, in this pathway. M2′ was a dioxidized metabolite arising from a combination of mono-oxidation and desmethylation and could only be produced from M2 in a NADPH-independent manner; M2′ could not be generated from M3 or M3′. These results suggested that M2′ is formed from lenvatinib by a unique two-step pathway through M2. Although both lenvatinib and M2 were substrates for aldehyde oxidase, an enzyme kinetic study indicated that M2 was a much more favorable substrate than lenvatinib. No inhibitory activities of lenvatinib, M2′, or M3′ and no significant inhibitory activities of M2 or M3 on aldehyde oxidase were observed, suggesting a low possibility of drug-drug interactions in combination therapy with substrates of aldehyde oxidase.
Introduction
Aldehyde oxidase (AO) is a molybdenum enzyme that catalyzes the oxidation of aldehydes and azaheterocyclic derivatives to corresponding carboxylic acids and lactams, respectively (Kitamura et al., 2006; Pryde et al., 2010; Hutzler et al., 2013). AO transfers the oxygen of water to electrophilic carbon residues in substrates. A marked species-specific distribution of AO has been reported, with high activities reported in monkeys and humans, relatively low activities reported in rats, and no activity reported in dogs (Beedham et al., 1995; Kitamura et al., 2006). Thus, the contribution of AO to the metabolism of drug candidates might become an issue after clinical introduction, with such a contribution possibly resulting in undesirable pharmacokinetic (PK) and toxicity profiles. For example, FK3453 (6-(2-amino-4-phenylpyrimidin-5-yl)-2-isopropylpyridazin-3(2H)-one) showed an unacceptably low plasma concentration after administration to humans even though preferable PK profiles were observed for FK3453 in animals; this result was thought to arise mainly because of the significant contribution of AO metabolism, which occurs only in humans (Akabane et al., 2011). In other cases, SGX523 (6-(6-(1-methyl-1H-pyrazol-4-yl)-[1,2,4]triazolo[4,3-b]pyridazin-3-ylthio)quinoline) is metabolized by AO to a quinolinone derivative, which is more hydrophobic than SGX523. As a result of this reaction, species-specific renal toxicity occurred because of the crystallization of the quinolinone metabolite in monkeys and humans, which is consistent with the observed toxicology and with the lack of formation of the metabolite in rats and dogs (Diamond et al., 2010). In addition, compounds with certain substructures, such as quinolines, are mostly biotransformed by AO in humans, leading to a high plasma clearance (Austin et al., 2001; Akabane et al., 2011). In the most recent decade, drug candidates have been modified to make them resistant to cytochrome P450 (P450)–mediated metabolism when examined using an appropriate microsomal stability assay, so as to avoid clinical failure (Kola and Landis, 2004). Consequently, drug candidates have become less prone to P450-mediated metabolism, but the role of non-P450 metabolism, such as oxidation by AO, has tended to increase.
Thus, the importance of non-P450 metabolism in predicting the intrinsic metabolic fate of drug candidates during the drug-discovery stage is increasing. In addition to the contributions of non-P450 enzymes to drug metabolism, drug-drug interactions (DDIs) related to non-P450 metabolism are also beginning to require attention in drug development because of their increasing contributions to drug metabolism. Zaleplon is a well investigated AO substrate that is metabolized to become 5-oxo-zaleplon (Kawashima et al., 1999). DDIs related to AO inhibition have been reported for zaleplon in combination use with cimetidine (Renwick et al., 2002). Cimetidine has been known as an AO inhibitor, and the coadministration of cimetidine and zaleplon resulted in an increased maximum plasma concentration and area under the curve of zaleplon, compared with the administration of zaleplon alone. Although the frequency of DDIs caused by AO inhibition is relatively low, this mechanism should be considered during drug development.
Lenvatinib mesylate is a multityrosine kinase inhibitor of vascular endothelial growth factor receptors 1-3, platelet-derived growth factor receptor β, rearranged during transfection, and KIT (Matsui et al., 2008a,b; Okamoto et al., 2013). In a phase I study, lenvatinib was shown to have favorable PK properties, and its metabolic fate was investigated (Boss et al., 2012). We previously reported the metabolic pathways of lenvatinib after oral administration in a male cynomolgus monkey (Inoue et al., 2012). Lenvatinib was mainly metabolized to glutathione (GSH)-related derivatives. However, several mono-oxidized metabolites arising from minor oxidative pathways were also observed. To investigate the oxidative metabolic pathways of lenvatinib, liver S9 fractions from animals and humans were used to assess the susceptibility of lenvatinib to P450 and non-P450 metabolism. As a result, P450-mediated metabolism was observed in all species that were tested, whereas the contributions of non-P450 enzymes were observed only in monkeys and humans, as shown by the formation of a quinolinone form of lenvatinib (M3′) and a quinolinone form of the desmethylated form of lenvatinib (M2′). To identify the non-P450 enzymes involved in the oxidative metabolism of lenvatinib, the effects of AO inhibitors (raloxifene and menadione) and a xanthine oxidase (XO) inhibitor (allopurinol) on the formation of M2′ and M3′ were examined. Furthermore, 18O-water and recombinant human AO (rhAO) were used to demonstrate the involvement of AO. In addition, the kinetic parameters of the AO-mediated metabolism of lenvatinib and M2, and the inhibitory activity of lenvatinib and its metabolites on AO were investigated to assess the potential for DDIs related to AO-mediated metabolism in humans.
Materials and Methods
Lenvatinib (E7080, 4-[3-chloro-4-(N’-cyclopropylureido)phenoxy] 7-methoxy quinoline-6-carboxamide mesylate), M2 (4-{3-chloro-4-[(cyclopropyl-carbamoyl)amino]phenoxy}-7-hydroxyquinoline-6-carboxamide), M3 (6-carbamoyl-4-{3-chloro-4-[(cyclopropylcarbamoyl)amino]phenoxy}-1-hydroxy-7-methoxy-quinolinium), M2′ (4-{3-chloro-4-[(cyclopropylcarbamoyl)amino]phenoxy}-7-hydroxy-2-oxo-1,2-dihydroquinoline-6-carboxamide), and M3′ (4-{3-chloro-4-[(cyclopropylcarbamoyl)amino]phenoxy}-7-methoxy-2-oxo-1,2-dihydroquinoline-6-carboxamide) were synthesized at Eisai Co., Ltd. (Kashima and Tsukuba, Japan). Sprague-Dawley rat liver S9 fraction (male, pool of 400), beagle dog liver S9 fraction (male, pool of 11), cynomolgus monkey liver S9 fraction (male, pool of 10), human liver S9 fraction (mixed gender, pool of 50), human liver cytosol (mixed gender, pool of 50), and rhAO were obtained from Xenotech LLC (Lenexa, KS). Niflumic acid, magnesium chloride hexahydrate, ammonium acetate, and dimethylsulfoxide were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Menadione, allopurinol, phthalazine, phthalazone, 4-phenyl-1-(2H)-phthalazinone, and 18O-water (97 atom %) were obtained from Sigma-Aldrich (St. Louis, MO). Raloxifene hydrochloride and β-NADPH were purchased from Tokyo Chemical Industry (Tokyo, Japan) and Oriental Yeast Co., Ltd. (Tokyo, Japan), respectively. Acetonitrile, methanol, and distilled water were of high-performance liquid chromatography (HPLC) grade. All other reagents used in this study were of reagent grade.
Incubation Conditions.
Lenvatinib and its synthesized metabolites (M2, M3, and M3′, 10 µM each) were individually incubated in reaction solutions consisting of 3 mM magnesium chloride and 2 mg/ml liver S9 fractions with or without 5 mM β-NADPH in 100 mM potassium phosphate buffer (pH 7.4). Experiments were conducted in duplicate. Recombinant human AO (0.5 mg/ml) was incubated with 10 µM lenvatinib or M2 in 100 mM potassium phosphate buffer (pH 7.4). Reactions were kept at 37°C for 2 hours for the incubation of liver S9 fractions and for 22 hours for incubation with rhAO. An incubation sample without the test compounds was used as a control. In the inhibition study using AO and XO inhibitors, raloxifene, menadione, or allopurinol was added at 10, 100, and 100 µM, respectively, and the incubation was conducted without NADPH in liver S9 fractions. The reaction was quenched by the addition of an equal volume of acetonitrile/methanol (2:1, v/v) containing 1 μM niflumic acid as an internal standard (IS). After centrifugation (9000 × g, 10 minutes, 4°C; Centrifuge 5415R; Eppendorf Co., Ltd., Hamburg, Germany), an aliquot of the supernatant of each sample was taken for quantitative analyses of lenvatinib, M2, M3, and M3′ using the Quantum Ultra system (Thermo Fisher Scientific, Waltham, MA). The residual samples were combined, and the mixture was evaporated using a centrifugation evaporator. The obtained residue was reconstituted with acetonitrile/water (3:7, v/v), and the resulting solution was used for a qualitative analysis using LTQ Orbitrap accurate mass spectrometry (Thermo Fisher Scientific).
The incorporation of 18O into lenvatinib and M2 to generate 18O-incorporated M2′ and M3′ was investigated using H218O. The incubation mixture contained 10 μM lenvatinib or M2, and 2 mg/ml human liver cytosol in 100 mM potassium phosphate buffer prepared with H218O. The H218O in the incubation mixture accounted for 69% of the total volume. As a control, lenvatinib or M2 (10 µM) was incubated in H216O-containing mixtures under the same conditions described earlier. The reaction was kept at 37°C for 2 hours and was terminated by the addition of a double volume of acetonitrile/methanol (2:1, v/v). The suspension was centrifuged at 13,000 × g for 10 minutes (Kubota3520; Kubota Corporation, Tokyo, Japan) and the resulting supernatant was evaporated using a centrifugation evaporator (Thermo Fisher Scientific). The residue was reconstituted with acetonitrile/water (3:7, v/v) and then centrifuged to obtain the supernatant, which was used for a qualitative analysis using LTQ Orbitrap accurate mass spectrometry.
Conditions for Quantitative Analysis.
The TSQ Quantum Ultra/Accela LC/MS/MS system (Thermo Fisher Scientific) was used for a quantitative analysis of lenvatinib and its metabolites, and acquired data were analyzed using Xcalibur software (version 2.0 SR2; Thermo Fisher Scientific). The mobile phases consisted of 10 mM ammonium acetate in water as mobile phase A and methanol as mobile phase B. The analysis was conducted under isocratic conditions with 60% of mobile phase B. The samples (5 µl each) were injected into a Kinetex 2.6u XB-C18 100A, 50 × 2.1–mm column (Phenomenex, Inc., Torrance, CA) at a flow rate of 1 ml/min. The column temperature was kept at 40°C throughout the analysis. The MS/MS analysis was performed in positive ion mode, and the conditions used in this study were as follows: spray voltage, 4000 V; vaporizer temperature, 400°C; and capillary temperature, 300°C. For selected reaction monitoring, the target ions monitored were as follows: for M2, a precursor ion at m/z 413 and product ions at m/z 338.987, 356.001, and 395.988; for M3, a precursor ion at m/z 443 and product ions at m/z 294.929, 342.786, and 385.991; for M3′, a precursor ion at m/z 443 and product ions at m/z 343.105, 369.161, and 426.120; for lenvatinib, a precursor ion at m/z 427 and product ions at m/z 202.038, 311.981, and 370.010; and for niflumic acid (IS), a precursor ion at m/z 283 and a product ion at m/z 265.051.
Qualitative Analytical Conditions.
The LTQ Orbitrap mass spectrometer was coupled with a Shimadzu HPLC (Pump, LC-20AD; degasser, DGU-20A3; column oven, CTO-20AC; autosampler, SIL-20AC; and system controller, CBM-20A; Shimadzu, Kyoto, Japan), and was used for the qualitative analysis of lenvatinib and its metabolites; acquired data were analyzed using Xcalibur software (Thermo Fisher Scientific). Reconstituted samples (10 µl each) were injected into a Synergi 4u Hydro-RP 80A 150 × 4.6–mm column (Phenomenex) at a flow rate of 1 ml/min. The mobile phases consisted of 10 mM ammonium acetate in water as mobile phase A and methanol as mobile phase B. Separation was achieved using a gradient solvent system as follows: 0 minute, 5% B; 0–5 minutes, 5% B; 50 minutes, 80% B; 52 minutes, 90% B; 54.9 minutes, 90% B; 55–60 minutes, 5% B for equilibration. The column temperature and autosampler were kept at 40°C and 15°C, respectively, throughout the analysis. The MS measurement was performed in positive ion mode, and the analytical parameters were as follows: spray voltage, 5 kV; capillary temperature, 360°C; resolution selected, 30,000 for full scan and 7500 for MS/MS scan. The MS/MS analysis was conducted using the collision-induced dissociation mode with a collision energy of 45% for molecular ions at m/z 413.1011, 443.1117, 429.0960, 443.1117, and 427.1168 for M2, M3, M2′, M3′, and lenvatinib, respectively. For 18O-incorporated M2′ and M3′, the MS/MS analysis was conducted using the collision-induced dissociation mode with a collision energy of 45% for molecular ions at m/z 431.1003 and at m/z 445.1159, respectively.
Data Analysis for Metabolic Stability and Inhibitory Effect.
In the Quantum Ultra system, the mass chromatographic and spectral data of lenvatinib, M2, M3, M3′, and IS on selected reaction monitoring were processed using Xcalibur software. The peak area ratios (PARs) of lenvatinib, M2, M3, and M3′ to IS were calculated. Then, the percentages of remaining lenvatinib, M2, M3, and M3′ were calculated using the following equation: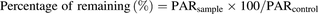 where PARsample represents the mean peak area ratio of lenvatinib, M2, M3, and M3′ to IS after a 2-hour incubation period, and PARcontrol represents the mean peak area ratio of lenvatinib, M2, M3, and M3′ to IS of the control sample.
where PARsample represents the mean peak area ratio of lenvatinib, M2, M3, and M3′ to IS after a 2-hour incubation period, and PARcontrol represents the mean peak area ratio of lenvatinib, M2, M3, and M3′ to IS of the control sample.
In the LTQ Orbitrap system, the MS/MS spectra of lenvatinib, M2, M3, M2′, and M3′ were obtained using Xcalibur software. Peaks consistent with lenvatinib and its synthesized metabolites (M2, M3, M2′, and M3′) were analyzed using MS/MS.
Kinetics of M2′ and M3′ Formation.
For kinetic analysis of the formation of M2′ from M2 and M3′ from lenvatinib, incubation was conducted as follows. Lenvatinib (10–200 µM) or M2 (0.5–50 µM) was incubated with 0.1 mg/ml human liver cytosol in 100 mM potassium phosphate buffer (pH 7.4) at 37°C in triplicate. The incubation time (0–5 minutes) and the protein concentration used provided linear reaction velocity for production of M3′ and M2′ from lenvatinib and M2, respectively. The sampling time points were at 0, 1, 2, 3, and 5 minutes after addition of substrate. The reaction was terminated by the addition of a double volume of acetonitrile/methanol (2:1, v/v) solution containing 1 μM niflumic acid as IS. After centrifugation (1700 × g, 10 minutes, 4°C; Kubota 5930; Kubota Corporation), an aliquot of the supernatant was obtained for analysis using an API5000 (AB Sciex, Framingham, MA)/Shimadzu HPLC system (Pump, LC-20AD; degasser, DGU-20A3; column oven, CTO-20AC; autosampler, SIL-20AC; and system controller, CBM-20A; Shimadzu) equipped with an analytical column (L-column, 2.1 × 100 mm, 5 μm; Chemicals Evaluation and Research Institute, Tokyo, Japan). Separation was achieved using a gradient solvent system with mobile phases of 0.02% formic acid in water (solvent A) and 0.02% formic acid in acetonitrile (solvent B). The initial mobile phase was 5% B, and the mobile phase was linearly increased to 100% B over 3 minutes, then kept at 100% B for the next minute. Equilibration was performed using the initial mobile phase for 2 minutes before the next injection. Quantification was conducted for M2′ and M3′ using multiple reaction monitoring (MRM) with the detection of a product ion at m/z 354.832 from a molecular ion at m/z 428.604 (M2′) and a product ion at m/z 368.949 from a molecular ion at m/z 442.737 (M3′). The obtained data were processed using Analyst software (AB Sciex). Linearity of the reaction velocities (v) for the formations of M2′ from M2 and M3′ from lenvatinib at each substrate concentration within the incubation time (0–5 minutes) was confirmed, so that the v values were calculated by linear regression analysis plotting with the amount of M2′ or M3′ formed and incubation time used. Then, obtained v values of formation of M2′ or M3′ were plotted with substrate concentrations (S; M2 for M2′ and lenvatinib for M3′), and kinetic parameters, Vmax and Km, were obtained by fitting into a substrate inhibition model (Houston and Kenworthy, 2000) using GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA). The equations to calculate kinetic parameters were as follows: where v represents the formation velocity of M2′ and M3′ from M2 and lenvatinib, respectively; Ki represents the inhibition constant; Km represents the Michaelis-Menten constant; S represents the substrate concentration; and Vmax represents the maximum velocity.
where v represents the formation velocity of M2′ and M3′ from M2 and lenvatinib, respectively; Ki represents the inhibition constant; Km represents the Michaelis-Menten constant; S represents the substrate concentration; and Vmax represents the maximum velocity.
Inhibitory Activities for Phthalazine Oxidation to Phthalazone in Human Liver Cytosol.
The incubation mixture consisted of 25 mM phosphate buffer (pH 7.4), 0.1 mM EDTA, 0.1 mg/ml human liver cytosol, and the test compounds (raloxifene, lenvatinib, M2, M3, M2′, or M3′) at the concentrations shown in Table 6. Samples were prepared in triplicate, and the AO reaction was initiated by the addition of phthalazine to obtain a final concentration of 10 μM. Incubation was conducted for 1 minute at 37°C; the reaction was then terminated by the addition of acetonitrile containing 1 M 4-phenyl-1-(2H)-phthalazinone as IS. After centrifugation (9000 × g, 5 minutes, 4°C; Centrifuge 5415R), the supernatant was filtered using an Ultrafree centrifugal filter unit (Durapore-PVDF 0.22 μm; Millipore Corporation, Billerica, MA), and the resulting solution was used for an LC/MS analysis to quantify phthalazone. The LC/MS system consisted of a Shimadzu HPLC system (pump, LC-20AD; degasser, DGU-20A3; column oven, CTO-20AC; autosampler, SIL-20ACHT; and system controller, CBM-20A; Shimadzu) and an API4000 (AB Sciex). Mass spectral data were acquired in positive ion mode, and the analytical conditions were as follows: ion spray voltage, 5500 V; and source temperature, 700°C. Phthalazone was monitored by detecting a product ion at m/z 90.0 generated from a molecular ion at m/z 147 under the MRM scan mode. IS was monitored by detecting a product ion at m/z 130.0 generated from a molecular ion at m/z 223.0 under the MRM scan mode. Samples were injected onto an analytical column (Inertsil ODS-4, 3 μm, φ2.1 mm, 50-mm length; GL Sciences Inc., Tokyo, Japan) equipped with a guard column (Inertsil ODS-4 Guard Column E, 3 μm, φ3 mm, 10-mm length; GL Sciences Inc.). Chromatographic separation was achieved with a gradient solvent system using 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B). The initial mobile phase was 10% B and was linearly increased to 80% B over 2.5 minutes after the injection of the sample. Then, 80% B was maintained for 1.5 minutes, then returned to 10% B for equilibration for 4 minutes. The column temperature and autosampler were kept at 40°C and 5°C, respectively, throughout the analysis. The obtained data were processed using Analyst software (AB Sciex). The remaining activity (percentage of control) was calculated as follows: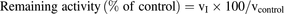 where vI represents the mean of the production rate of phthalazone with the test compound (nmol/min/mg protein of human liver cytosol) and vcontrol represents the mean of the production rate of phthalazone without the test compound (nmol/min/mg protein of human liver cytosol).
where vI represents the mean of the production rate of phthalazone with the test compound (nmol/min/mg protein of human liver cytosol) and vcontrol represents the mean of the production rate of phthalazone without the test compound (nmol/min/mg protein of human liver cytosol).
The remaining activities after incubations with M2 or M3 at 10 µM were around 50%. Thus, IC50 values of M2, M3, and raloxifene were evaluated. Incubation conditions were the same as described earlier. Concentrations of M2, M3, and raloxifene added were as follows: 1, 2, 5, 10, 20, 50, and 100 µM for M2 and M3; 0.0002, 0.0005, 0.001, 0.002, 0.005, 0.01, and 0.02 µM for raloxifene. The IC50 value was calculated using the following equation: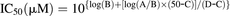 where A and B represent the highest and the lowest concentrations (in micromolar units) to cover an estimated IC50 value, respectively. C represents the remaining activity (percentage of control) at concentration B, and D represents the remaining activity (percentage of control) at concentration A.
where A and B represent the highest and the lowest concentrations (in micromolar units) to cover an estimated IC50 value, respectively. C represents the remaining activity (percentage of control) at concentration B, and D represents the remaining activity (percentage of control) at concentration A.
Results
Structures and Spectra.
The structures and mass spectral data of the authentic standards used in this study are summarized in Table 1. M2, M2′, M3, and M3′ were identified and synthesized as a desmethylated form of lenvatinib, a quinolinone derivative of M2, an N-oxide of lenvatinib, and a quinolinone of lenvatinib, respectively.
Mass spectral data of lenvatinib and metabolites
Metabolism in Liver S9 Fractions.
Lenvatinib was incubated in rat, dog, monkey, and human liver S9 fractions with or without NADPH. As summarized in Table 2, the NADPH-dependent and NADPH-independent metabolic activities for lenvatinib in liver S9 fractions from all the species tested were relatively low. Lenvatinib was metabolized to M2 and M3 only in the presence of NADPH in all species (Table 2). M3′ was formed only in monkey and human liver S9 fractions in an NADPH-independent manner, and its MS/MS analysis showed product ions at m/z 426, 360, and 343, which were identical to those of the synthetic reference for M3′ (Supplemental Fig. 1). As shown in Table 2, a metabolite that was 2 Da heavier than lenvatinib and that showed a molecular ion at m/z 429 was formed in an NADPH-dependent manner only in monkey and human liver S9 fractions; this metabolite was identified as M2′ by comparing the MS/MS spectrum with that of the authentic standard (Supplemental Fig. 2).
Lenvatinib and oxidative metabolites observed in liver S9 fractions
Data represent the mean of duplicate determinations. Numbers in parentheses are the percentage of remaining lenvatinib.
Inhibitory Activities of AO and XO Inhibitors on M3′ Formation.
Since M3′ formation was shown in monkey and human liver S9 fractions in an NADPH-independent manner, the inhibitory activities of AO and XO inhibitors on M3′ formation were investigated in monkey and human liver S9 fractions in the absence of NADPH. The activities were calculated as the percentage of M3′ formation relative to a control sample (100%) incubated without the inhibitors in monkey and human liver S9 fractions. As shown in Table 3, M3′ formation in the human liver S9 fraction was completely inhibited by raloxifene (0% of control, AO inhibitor) and was strongly inhibited by menadione (35% of control, AO inhibitor). In the monkey liver S9 fraction, M3′ formation was strongly inhibited by menadione (12% of control), whereas raloxifene only showed weak inhibitory activity (72% of control). Allopurinol, a selective XO inhibitor, exerted a weak inhibitory activity on M3′ formation in both monkey and human liver S9 fractions (63% and 79% of control, respectively).
Inhibitory activities of AO and XO inhibitors on M3′ formation from lenvatinib and M2 decline incubated without NADPH in monkey and human liver S9 fractions
Data represent the mean of duplicate determinations.
The inhibitory activities of AO and XO inhibitors on the formation of M2′ from M2 were also investigated in those liver S9 fractions; the results are shown in Table 3 as the percentage of decrease of M2 incubated with each inhibitor relative to that in the control sample incubated without inhibitor (100%). In the human liver S9 fraction, raloxifene inhibited the metabolism of M2 (15% of control), whereas menadione moderately inhibited M2 metabolism (77% of control). Allopurinol was not effective for inhibiting M2 metabolism (100% of control). In the monkey liver S9 fraction, menadione inhibited M2 metabolism (54% of control), whereas raloxifene and allopurinol did not have any inhibitory effects (96% and 95% of control, respectively).
Formation of M2′ in Monkey and Human Liver S9 Fractions.
Lenvatinib was metabolized to M2′ in an NADPH-dependent manner only in monkey and human liver S9 fractions. To estimate the formation pathway of M2′, each of M2, M3, and M3′ was incubated with monkey and human liver S9 fractions in the presence or absence of NADPH. As shown in Table 4, M2′ was produced from lenvatinib in the presence of NADPH. Among the metabolites, M2′ was only formed from M2 in an NADPH-independent manner, whereas M3 and M3′ did not yield any metabolites, including M2′.
Formation of M2′ in monkey and human liver S9 fractions incubated with M2, M3, M3′, or lenvatinib
Data represent the mean of duplicate determinations.
Effect of Recombinant Human AO on Formation of M2′ and M3′.
Next, rhAO was incubated with M2 and lenvatinib to demonstrate the involvement of AO in the formation of M2′ and M3′, respectively (Supplemental Fig. 3). M2 and lenvatinib formed an oxidative metabolite that exhibited product ions identical to those of the synthetic standard for M2′ and M3′, respectively (Supplemental Fig. 2 for product ion spectrum of M2′ and Supplemental Fig. 1 for product ion spectrum of M3′).
18O Incorporation Catalyzed by AO.
Lenvatinib and M2 were incubated with human liver cytosol in 18O- and 16O-water containing potassium phosphate buffer. As shown in Fig. 1, A and B for M2′ and Fig. 1, E and F for M3′, 18O-incorporated M2′ and M3′ were observed as molecular ions at m/z 431.0996 (calculated m/z 431.1003 as C20H18ClN4O418O+, −1.6 ppm) and at m/z 445.1155 (calculated m/z 445.1159 as C21H20ClN4O418O+, −0.9 ppm), respectively, after incubation with 18O-water. The product ions that were observed were consistent with the authentic standards (Supplemental Fig. 2 for M2′ and Supplemental Fig. 1 for M3′, respectively). Compared with the MS/MS spectra for 16O- and 18O-labeled M2′ and M3′, +2-Da patterns originating from 18O incorporation were observed, as shown in Fig. 1, C and D for M2′ and Fig. 1, G and H for M3′.

Incorporation of 18O in M2′ and M3′ after incubation with M2 and lenvatinib in 16O-water or 18O-water containing human liver cytosol. MS (A) and MS/MS (C) spectra of 16O-incorporated M2′; MS (B) and MS/MS (D) spectra of 18O-incorporated M2′; MS (E) and MS/MS (G) spectra of 16O-incorporated M3′; and MS (F) and MS/MS (H) spectra of 18O-incorporated M3′.
Kinetic Analysis of M2′ and M3′ Formation in Human Liver Cytosol.
A kinetic study examining the formations of M2′ and M3′ was conducted using human liver cytosol. As shown in Fig. 2, the formation velocities of M2′ from M2 and M3′ from lenvatinib were plotted against the substrate concentrations. At higher substrate concentrations, the enzymatic activity was slightly decreased; thus, the kinetic parameters for the formation of M2′ and M3′ by AO were calculated using nonlinear regression fitting in the substrate inhibition model. The kinetic parameters are summarized in Table 5. M2 showed a much higher affinity for AO than lenvatinib (Km for M2 was 1.7 ± 0.2 μM, whereas that for lenvatinib was 161.5 ± 62.5 μM). The maximum velocity of reaction (Vmax) for the formation of M2′ from M2 was 269.8 ± 10.1 pmol/min/mg of cytosolic protein, whereas that for the formation of M3′ from lenvatinib was slower (73.0 ± 22.2 pmol/min/mg of cytosolic protein). The intrinsic clearance values calculated from the parameters were 2.2 ml/min/mg of cytosolic protein for lenvatinib metabolism and 163.9 ml/min/mg of cytosolic protein for M2 metabolism.

Enzymatic kinetic plots for formation of M3′ from lenvatinib (A), and that of M2′ from M2 (B), respectively. The initial velocity for the formations of M3′ and M2′ was plotted versus substrate concentrations. The experiments were conducted in triplicate, and the error bars in the panels indicate S.D. A substrate inhibition model with nonlinear regression was used.
Kinetic parameters of lenvatinib and M2 metabolized by AO in human liver cytosol
Data represent the mean ± S.D. of triplicate determinations. Kinetic parameters are calculated using the substrate inhibition model.
Inhibitory Activities of Lenvatinib and Its Metabolites on the Oxidation of Phthalazine to Phthalazone.
The inhibition of AO activity against a typical substrate, phthalazine, was investigated using lenvatinib and its metabolites (M2, M3, M2′, and M3′). The apparent Km value of phthalazine for human liver cytosol used was 24.9 µM, and phthalazine was incubated at 10 µM with raloxifene, lenvatinib, M2, M3, M2′, and M3′ as inhibitors. Raloxifene, a typical AO inhibitor, showed a potent inhibitory activity, with an IC50 value of 0.0035 µM (Table 6). Lenvatinib, M2′, and M3′ showed almost no inhibitory effects on AO activity, even when administered at a concentration of 50 (M3′) or 100 μM (lenvatinib and M2′) as shown in Table 6. Meanwhile, M2 and M3 at a concentration of 10 μM exerted inhibitory activities, and the IC50 values for M2 and M3 were estimated to be 11.6 and 30.8 µM, respectively (Table 6).
Inhibitory activities of lenvatinib and metabolites on the phthalazine oxidation activity of AO
Data represent the mean of triplicate determinations.
Discussion
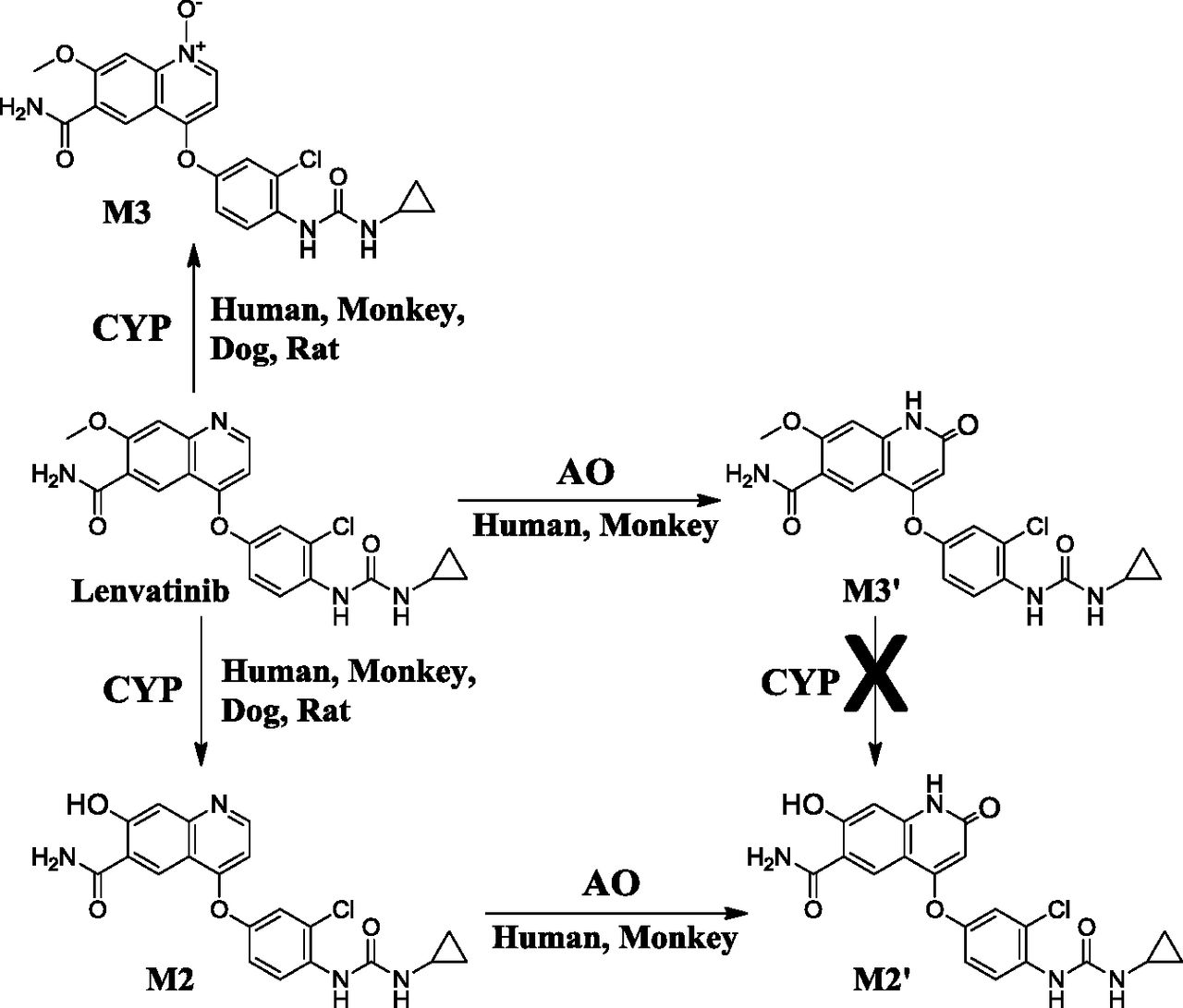
Lenvatinib is known to be nonenzymatically transformed to a GSH conjugate and its related metabolites in cynomolgus monkeys after oral administrations of radiolabeled lenvatinib (Inoue et al., 2012). Oxidative metabolites found in monkeys have been identified as M2 (desmethylated metabolite), M2′ (desmethylation + quinolinone), M3 (N-oxide), and M3′ (quinolinone), shown in Table 1. In rat, dog, monkey, and human liver S9 fractions, M2 and M3 were generated in an NADPH-dependent manner in all species. As shown in Table 2, the percentage of remaining lenvatinib was around 83–97% in liver S9 fractions with or without NADPH, indicating that lenvatinib is modestly metabolized in vitro. M3′ was produced only in monkey and human liver S9 fractions in an NADPH-independent manner, suggesting that the formation of M3′ would be mediated by species-specific non-P450 enzyme(s) (Table 2). Lenvatinib has a quinoline moiety in its structure that is known to be a typical substructure susceptible to metabolism by AO (Kitamura et al., 2006; Pryde et al., 2010; Hutzler et al., 2013). AO belongs to a molybdenum-containing enzyme subfamily that includes XO, and XO exhibits a substrate preference similar to AO based on a reaction mechanism that transfers electrons from water to the electrophilic carbon residues of xenobiotics (Kitamura et al., 2006; Pryde et al., 2010). Thus, to confirm the enzyme responsible for M3′ formation, the contributions of AO and XO were investigated using selective inhibitors. Raloxifene, known to act as a potent human AO inhibitor (Obach, 2004), inhibited M3′ formation, although menadione, another selective AO inhibitor, showed a moderate inhibitory effect in human liver S9 fractions (Table 3). In monkeys, M3′ formation was strongly inhibited by menadione, but not by raloxifene. Species differences in the inhibitory effects of AO inhibitors on AO activity have been reported, and the differences observed in the extents of inhibition by raloxifene and menadione between humans and monkeys were consistent with the results of previous reports (Obach, 2004; Sahi et al., 2008). Allopurinol, an XO inhibitor, slightly inhibited M3′ formation in both humans and monkeys, but the inhibitory effect was much smaller than those of the AO inhibitors described earlier. These results indicated that M3′ formation from lenvatinib was mainly catalyzed by AO. Regarding species differences, AO activity is known to be relatively high in monkeys and humans, relatively weak in rats, and absent in dogs (Kitamura et al., 2006). In rats and dogs, M3′ was not generated from lenvatinib, supporting the contribution of AO to the metabolism of lenvatinib.
A secondary oxidative metabolite corresponding to M2′ was detected as a product of a minor pathway in monkeys and was also detected in the human mass balance study. This metabolite was assumed to be a further oxidative metabolite generated from the desmethylated metabolite, M2. Lenvatinib generated M2′ only in the presence of NADPH in monkey and human liver S9 fractions (Table 2), suggesting that P450s would contribute to the formation of M2′ from lenvatinib. Synthesized standards of M2, M3, and M3′ were then incubated to assess the formation pathway of M2′ in monkey and human liver S9 fractions with or without NADPH. As shown in Table 4, only M2 was metabolized to M2′ in both liver S9 fractions in an NADPH-independent manner, and M3 and M3′ did not yield M2′. These results suggest that M2 is likely to be a precursor of M2′. M2 formation from lenvatinib was mediated by P450s in all species tested (Table 2, +NADPH), although M2′ was found only in primates. Thus, we hypothesized that M2′ formation was also mediated by AO as a secondary oxidative metabolite of M2. To assess the contribution of AO, the effects of AO and XO inhibitors on M2′ formation were assessed. As shown in Table 3, the inhibition profile on M2 metabolism was similar to that of M3′ formation from lenvatinib, implying that AO would also be responsible for M2 metabolism. The contributions of AO to the formations of M2′ and M3′ were confirmed using rhAO, and M2 and lenvatinib generated M2′ and M3′ in rhAO, respectively (Supplemental Fig. 3). Additionally, lenvatinib and M2 were incubated in human liver cytosol and 18O-water, and the incorporation of 18O in structures of M2′ and M3′ was observed (Fig. 1). These results indicated a role of AO in M2′ and M3′ formation from M2 and lenvatinib, respectively. Based on these facts, mechanisms for the formations of M2, M3, M2′, and M3′ were proposed (Fig. 3). In humans, as well as monkeys, lenvatinib was oxidized by P450s to form M2 and M3 and by AO to form M3′. For the formation of M2′, two sequential steps of enzyme-mediated reactions were demonstrated: M2 formation via a P450-mediated pathway, followed by the further oxidation of M2 by AO. In humans, the majority of M2′ and M3′ was observed in feces (data not shown). A kinetic analysis of the AO-mediated metabolism of lenvatinib and M2 revealed that lenvatinib did not have a high affinity toward AO. The apparent Km values of lenvatinib and M2 for human AO were 161.5 and 1.7 μM, respectively (Table 5). Ghafourian and Rashidi (2001) reported that substrate preference of AO toward phthalazine derivatives could be altered by a variety of substituent groups on the structure that affected electron deficiency on the metabolic site. In the case of lenvatinib, AO favored metabolism of M2 rather than lenvatinib, and it indicated that desmethylation of the methoxy group to form a hydroxyl group on the quinoline structure may affect the chemical property of lenvatinib such as the electron deficiency or ability for formation of a hydrogen bond with AO, although further investigation would be needed.

Proposed formation pathway of M2, M3, M2′, and M3′ mediated by P450 and AO.
At the drug-discovery stage, P450-dependent metabolism is generally assessed to identify promising candidates with acceptable clearances for drug development. This approach sometimes results in a higher contribution of non-P450 enzymes, including AO, that may exhibit significant species differences. In some cases, AO metabolism causes an unexpectedly high clearance in human clinical studies, occasionally leading to the postponement or termination of drug development (Austin et al., 2001; Akabane et al., 2011). In the case of lenvatinib, AO was shown to be involved in its oxidative metabolism. However, lenvatinib had a low affinity toward AO. Although M2 had a high affinity to AO, metabolic clearance of lenvatinib to form P450-mediated oxidative metabolites including M2 is limited, resulting in a low exposure of M2 in humans. Based on these in vitro data, lenvatinib was thought to have an acceptable clearance through oxidative metabolism by P450s and AO in humans, and a favorable PK profile of lenvatinib has been confirmed in humans in a phase I study (Boss et al., 2012).
In terms of the DDI risk for AO-mediated metabolism, the inhibitory effects of lenvatinib and its metabolites on AO-mediated metabolism were considered. AO activity measured as the oxidation of phthalazine to phthalazone was not inhibited by lenvatinib, M2′, or M3′ in human liver cytosol (IC50 value >50 μM). M2 and M3 showed moderate inhibitory effects, with IC50 values of 11.6 and 30.8 μM, respectively (Table 6). At the maximum tolerable dose in humans, the Cmax of lenvatinib at steady state was 1.4 μM after multiple doses (Boss et al., 2012), and plasma concentrations of metabolites such as M2, M3, M2′, and M3′ were low (data not shown). Thus, the possibility of clinical DDIs via AO inhibition by lenvatinib administration in humans is likely to be low. To estimate the risk for victim DDIs, estimation of responsible enzymes or other pathways for the elimination of a new drug is important; however, complete understanding of this is very complicated for drugs with multiple clearance pathways. The finding of this study will contribute to a more accurate estimation of responsible enzymes of lenvatinib, since the result of this study clearly shows that M2, M3, and their sequential metabolites including M2′ are results of P450-dependent metabolism, and M3′ is of AO-dependent metabolism. In terms of contribution of other metabolic enzymes, recombinant human flavin-containing monooxygenase (FMO) isoforms (FMO1, FMO3, and FMO5) were incubated with lenvatinib, and none of them were involved in metabolism of lenvatinib (data not shown).
In conclusion, lenvatinib was metabolized to M2, M3, and other oxidative metabolites via P450s in rat, dog, monkey, and human liver S9 fractions. AO contributes to the formations of M2′ and M3′ from M2 and lenvatinib, respectively, in monkeys and humans, but not in rats and dogs. M2′ was formed by a unique two-step pathway through M2. A kinetic analysis revealed a low affinity of lenvatinib and a relatively higher affinity of M2 toward AO, suggesting lenvatinib would be stable against AO metabolism unless it would be transformed to M2 by P450. In addition, lenvatinib and its metabolites did not exert significant AO inhibition, and low possibility of clinical DDIs caused by lenvatinib related to AO metabolism was expected. Lenvatinib can be eliminated by multiple pathways mediated by P450, AO, and conjugation with GSH from the body, suggesting that the risk of clinical DDIs of lenvatinib as a victim in its metabolism would be attenuated.
Acknowledgments
The authors thank Dr. George Lai for scientific suggestions in this work and editorial assistance with this manuscript.
Authorship Contributions
Participated in research design: Inoue, Mizuo.
Conducted experiments: Inoue, Mizuo.
Performed data analysis: Inoue, Mizuo, Kawaguchi.
Wrote or contributed to the writing of the manuscript: Inoue, Mizuo, Kawaguchi, Fukuda, Kusano, Yoshimura.
Footnotes
- Received March 19, 2014.
- Accepted June 5, 2014.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AO
- aldehyde oxidase
- DDI
- drug-drug interaction
- FMO
- flavin-containing monooxygenase
- GSH
- glutathione
- HPLC
- high-performance liquid chromatography
- IS
- internal standard
- LC
- liquid chromatography
- Lenvatinib
- (4-[3-chloro-4-(N’-cyclopropylureido)phenoxy] 7-methoxyquinoline-6-carboxamide mesylate); M2, (4-{3-chloro-4-[(cyclopropyl-carbamoyl)amino]phenoxy}-7-hydroxyquinoline-6-carboxamide); M2′, (4-{3-chloro-4-[(cyclopropylcarbamoyl)amino]phenoxy}-7-hydroxy-2-oxo-1,2-dihydroquinoline-6-carboxamide); M3, (6-carbamoyl-4-{3-chloro-4-[(cyclopropylcarbamoyl)amino]phenoxy}-1-hydroxy-7-methoxy-quinolinium); M3′, (4-{3-chloro-4-[(cyclopropylcarbamoyl)amino]phenoxy}-7-methoxy-2-oxo-1,2-dihydroquinoline-6-carboxamide)
- MRM
- multiple reaction monitoring
- MS
- mass spectrometry
- P450
- cytochrome P450
- PAR
- peak area ratio
- PK
- pharmacokinetic
- rhAO
- recombinant human AO
- XO
- xanthine oxidase
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}