


Abstract
Hydrolysis plays an important role in metabolic activation of prodrugs. In the current study, species and in vitro system differences in hepatic and extrahepatic hydrolysis were investigated for 11 prodrugs. Ten prodrugs in the data set are predominantly hydrolyzed by carboxylesterases (CES), whereas olmesartan medoxomil is also metabolized by carboxymethylenebutenolidase (CMBL) and paraoxonase. Metabolic stabilities were assessed in cryopreserved hepatocytes, liver S9 (LS9), intestinal S9 (IS9), kidney S9 (KS9), and plasma from human, monkey, dog, and rat. Of all the preclinical species investigated, monkey intrinsic hydrolysis clearance obtained in hepatocytes (CLint,hepatocytes) were the most comparable to human hepatocyte data. Perindopril and candesartan cilexetil showed the lowest and highest CLint,hepatocytes, respectively, regardless of the species investigated. Scaled intrinsic hydrolysis clearance obtained in LS9 were generally higher than CLint,hepatocytes in all species investigated, with the exception of dog. In the case of human and dog intestinal S9, hydrolysis intrinsic clearance could not be obtained for CES1 substrates, but hydrolysis for CES2 and CMBL substrates was detected in IS9 and KS9 from all species. Pronounced species differences were observed in plasma; hydrolysis of CES substrates was only evident in rat. Predictability of human hepatic intrinsic clearance (CLint,h) was assessed for eight CES1 substrates using hepatocytes and LS9; extrahepatic hydrolysis was not considered due to high stability of these prodrugs in intestinal and kidney S9. On average, predicted oral CLint,h from hepatocyte data represented 20% of the observed value; the underprediction was pronounced for high-clearance prodrugs, consistent with the predictability of cytochrome P450/conjugation clearance from this system. Prediction bias was less apparent with LS9, in particular for high-clearance prodrugs, highlighting the application of this in vitro system for investigation of prodrugs.
Introduction
The prodrug concept has been increasingly used over recent years; 20% of all small molecules approved in the period of 2000–2008 were prodrugs (Stella, 2010; Huttunen et al., 2011; Dahan et al., 2012). Many prodrugs are inactive compounds with carboxyl-ester, thio-ester, or amide (Taketani et al., 2007; Laizure et al., 2013), which are subsequently hydrolyzed to an active drug; the extent of hydrolysis markedly affects the pharmacologic activity and/or toxicity of these compounds. The intentional esterification to a prodrug has been used across different classes of drugs (e.g., antivirals, angiotensin-converting enzyme inhibitors) mostly to improve drug absorption and bioavailability (Imai, 2006; Li et al., 2008). This concept has recently been taken further in a form of carrier-mediated prodrug to target peptide transporter 1 in the small intestine (Gupta et al., 2013).
Carboxylesterases (CES), in particular CES1 and CES2, represent major enzymes involved in the hydrolysis of structurally diverse prodrugs (Satoh and Hosokawa, 1998; Imai, 2006; Hosokawa, 2008). Human CES1 tends to hydrolyze esters with a large acyl moiety relative to the alcohol group (e.g., benazepril), opposite to CES2 substrates with a generally small acyl group, as in the case of candesartan cilexetil (Supplemental Fig. 1) (Taketani et al., 2007; Laizure et al., 2013). Recent studies have highlighted the role of carboxymethylenebutenolidase (CMBL) for the hydrolysis of prodrugs in the liver and intestine (e.g., olmesartan medoxomil) (Ishizuka et al., 2010). CMBL is a relatively novel hydrolysis enzyme, and any potential overlap in its substrates with CES is not well characterized. Cellular localization of these enzymes differs, as CES are present both in the membrane and cytosol (Satoh and Hosokawa, 1998; Fujiyama et al., 2010) whereas CMBL is a predominantly cytosolic enzyme (Ishizuka et al., 2013), emphasizing the need for a careful choice of in vitro system for adequate characterization of hydrolysis. In addition, paraoxonase 1 (PON1), predominantly located in plasma (Bahar et al., 2012), hydrolyzes lactones and aromatic carboxylic acid esters (Fukami and Yokoi, 2012; Ishizuka et al., 2012).
CES1 expression based on mRNA data is highest in the liver, both in human and preclinical species (Supplemental Table 1), suggesting its key role in the hydrolysis of prodrugs (Williams et al., 2010). However, human CES1 is also expressed in the lung, heart, and kidney, though to a lesser extent (Satoh et al., 2002). This dominant CES1 expression relative to CES2 in the liver was also confirmed by proteomic data, with an average protein expression level of 402 and 30 pmol/mg protein reported in human liver microsomes for CES1 and CES2, respectively (Sato et al., 2012). In addition to the small intestine, human CES2 mRNA data have also been reported for the kidney and to a minor extent for the colon and heart (Satoh et al., 2002; Quinney et al., 2005). Analogous to humans, CES2 mRNA has been quantified in the liver and/or intestine of preclinical species, although the data in some species/other tissues are sparse (Supplemental Table 1). The expression data highlight the need for consideration of extrahepatic hydrolysis, analogous to the role of these organs in cytochrome P450 and conjugation metabolism (Galetin and Houston, 2006; Paine et al., 2006; Cubitt et al., 2009; Gertz et al., 2010; Nishimuta et al., 2011; Gill et al., 2012). In addition, significant species differences were observed in CES expression in plasma, with high levels reported for rat, but not human, monkey, or dog plasma (Li et al., 2005; Berry et al., 2009). CMBL is highly expressed in the liver, intestine, and kidney across different species except in the dog intestine (Ishizuka et al., 2013).
Although studies have reported some species differences in CES-mediated hydrolysis of prodrugs (Prueksaritanont et al., 1996; Yoshigae et al., 1998; Williams et al., 2011), the general paucity of data on the activity of CES and/or other hydrolytic enzymes in different systems across species is evident. Therefore, our study investigated species and in vitro system differences in the hydrolysis of 11 selected prodrugs. Most of the prodrugs in the data set are predominantly metabolized by CES1 (8 of 11), whereas candesartan cilexetil and mycophenolate mofetil are substrates for both CES1 and CES2 (Laizure et al., 2013). In addition, olmesartan medoxomil is hydrolyzed by CMBL and PON1, with a minor CES contribution. Intrinsic clearance (CLint) values were obtained in cryopreserved hepatocytes, liver S9 (LS9), intestinal S9 (IS9), and kidney S9 (KS9); the same in vitro systems were used in humans, cynomolgus monkey, beagle dog, and Sprague-Dawley rat. The importance of intestinal and renal hydrolysis relative to hepatic was assessed. Finally, the predictability of in vitro CLint hydrolysis data generated in human hepatocytes and LS9 was investigated for a subset of eight CES1 prodrugs, using the reported in vivo clearance after oral administration.
Materials and Methods
Chemicals.
Benazepril hydrochloride was purchased from Wako Pure Chemical Industries (Osaka, Japan); candesartan cilexetil, perindopril erbumine, quinapril hydrochloride, and ramipril were purchased from LKT Laboratories (St. Paul, MN); cilazapril, trandolapril, temocapril hydrochloride, mycophenolate mofetil, and olmesartan medoxomil were purchased from Toronto Research Chemicals (North York, ON, Canada); oseltamivir phosphate was purchased from Selleck Chemicals (Houston, TX). All other reagents and solvents were of analytic grade and were commercially available.
Subcellular Fractions.
Liver S9 from Sprague-Dawley rats (pool of 400 male), beagle dog (pool of 12 male), cynomolgus monkey (pool of 13 male), and human (pool of 50) were purchased from Xenotech (Lenexa, KS). Kidney S9 from Sprague-Dawley rats (pool of 149 male), beagle dog (pool of five male), cynomolgus monkey (pool of five males), and human (pool of four) were purchased from the same supplier. Intestinal S9 from Sprague-Dawley rats (pool of 40 male), beagle dog (individual, male), cynomolgus monkey (individual, male) and human (individual, male) were purchased from the same supplier. To avoid any potential enzyme stability issues, no freeze-thaw cycles were applied during the experiments. Only intestinal S9 prepared in the absence of phenylmethylsulfonyl fluoride (PMSF) was used, considering the reported inhibitory effect of PMSF on CES activity in the process of isolation of enterocytes via the elution method (Kleingeist et al., 1998; Morgan et al., 2008).
Hepatocytes.
Cryopreserved hepatocytes from Sprague-Dawley rat (pool of eight male), beagle dog (pool of three male), cynomolgus monkey (pool of three male), and human (pool of 20, equal number of male and female) were purchased from Xenotech.
Plasma.
Pooled plasma fraction treated by heparin sodium salt from Sprague-Dawley rat, beagle dog, and cynomolgus monkey were purchased from Valley Biomedical Products and Services (Winchester, VA). Pooled plasma fraction treated by heparin sodium salt from human was purchased from Kohjin Bio (Saitama, Japan).
Metabolic Stabilities in Liver, Intestinal, and Kidney S9.
Compounds investigated (200 nM) were incubated at 37°C in 100 μl of 50 mM phosphate buffer (pH 7.4). The same conditions were used for liver, intestinal, and kidney S9 from Sprague-Dawley rats, beagle dogs, cynomolgus monkey, and humans. In the case of the kidney, we conducted preliminary studies only for substrates of CES2 and CMBL, which are highly expressed in the human kidney, namely, candesartan cilexetil, mycophenolate mofetil, and olmesartan medoxomil. Linearity of metabolic activities for S9 concentration (0.01–1 mg S9 protein/ml) and incubation time (5, 15, or 60 minutes) were confirmed, and the optimal reaction conditions were established for each compound. The final concentration of acetonitrile in the incubation mixture was 0.5% (v/v). After preincubation at 37°C for 5 minutes, the reactions were initiated by the addition of the substrates and stopped by addition of 200 μl of ice-cold acetonitrile. Control samples were incubated using the same method as in the absence of substrates; substrates were added after the addition of ice-cold acetonitrile. The reaction mixtures were spiked with 200 μl of acetonitrile containing the internal standard, 200 nM flecainide. The mixtures were centrifuged at 3400g for 10 minutes to remove precipitated protein. The supernatants were then filtrated using 0.45-μm 96-well filter plates (Varian, Palo Alto, CA). The filtrate was diluted 2-fold using distilled water and transferred to 96-well plates, and a 10 μl portion was then injected onto a high-performance liquid chromatography–tandem mass spectrometry (LC-MS/MS) system.
Preparation of Hepatocytes.
The vials of cryopreserved hepatocytes were thawed by immersion for 1.5 minutes in a 37°C water bath. After thawing, the hepatocyte suspension was poured into tube A of the hepatocyte isolation kit (XenoTech) containing supplemented Dulbecco’s modified Eagle’s medium and isotonic Percoll and then centrifuged (70g) for 5 minutes at 25°C. After the supernatant was removed, the cells were resuspended in 5 ml of supplemented Dulbecco’s modified Eagle’s medium in tube B of the hepatocyte isolation kit. The number of viable cells was then determined using trypan blue staining to confirm the viability was >80%. Subsequently, the cells were resuspended in the remaining medium from tube B and then centrifuged (50g) for 3 minutes at 25°C, followed by removal of the supernatant. Finally, the cells were resuspended in the Krebs-Henseleit buffer at a density of 0.1–2.0 × 106 viable cells/ml for the stability experiment.
Metabolic Stabilities in Hepatocytes.
The hepatocyte suspension (150 μl) was distributed in 24-wells plates and incubated in CO2 incubator under 5% CO2 at 37°C for 10 minutes. The reaction was initiated by adding an equal volume (150 μl) of Krebs-Henseleit buffer containing drugs (the final concentration is 200 nM in acetonitrile) to the hepatocyte suspension. The buffer containing drugs was prewarmed at 37°C for 10 minutes before the stability studies. The final concentration of acetonitrile was 0.05% (v/v) in the incubation mixture. After incubation at 37°C for 0, 5, 15, 60, and 120 minutes, 50 μl of the incubation mixture was added into 200 μl of acetonitrile containing the internal standard, 200 nM flecainide. The mixtures were centrifuged at 3400g for 10 minutes to remove precipitated protein. The supernatants were then filtrated using 0.45-μm 96-well filter plates. The filtrate was diluted 2-fold using distilled water and transferred to 96-well plates, and a 10 μl portion was then injected onto the LC-MS/MS system.
Metabolic Stabilities in Plasma.
The compounds investigated were incubated at 37°C in 100 μl of plasma from Sprague-Dawley rat, beagle dog, cynomolgus monkey, or human at a final concentration of 200 nM. The linearity of metabolic activities for incubation time (10 and/or 30 seconds, and/or 1, 2, 5, 15, or 60 minutes) was confirmed in the preliminary studies, and the optimal reaction conditions were established for each compound. The final concentration of acetonitrile in the incubation mixture was 0.5% (v/v). After preincubation at 37°C for 5 minutes, the reactions were initiated by the addition of the substrates and stopped by addition of 200 μl of ice-cold acetonitrile. Control samples were incubated using the same method as in the absence of substrates; substrates were added after the addition of ice-cold acetonitrile. The reaction mixtures were spiked with 200 μl of acetonitrile containing the internal standard, 200 nM flecainide. The mixtures were centrifuged at 3400g for 10 minutes to remove precipitated protein. The supernatants were then filtrated using 0.45-μm 96-well filter plates. The filtrate was diluted 2-fold using distilled water and transferred to 96-well plates, and a 10 μl portion was then injected onto the LC-MS/MS system.
The fraction unbound in human plasma (fu,p) for eight CES1 substrates was determined using a high-throughput dialysis method. The method was considered adequate because of the high stability of these prodrugs in the human plasma samples. Dialysis membranes with a 10 kDa molecular mass cutoff were purchased from Harvard Apparatus (Holliston, MA). Compounds (1 μM, final) with human plasma were added to the acceptor chambers, and phosphate-buffered saline was added to the donor chamber. The dialysis plate was placed in an incubator at 37°C for 22 hours on a plate rotator. After equilibrium had been reached, 10 μl of samples in the acceptor chamber were mixed with 40 μl of phosphate-buffered saline and 40 μl of samples in the donor chamber were mixed with 10 μl of human plasma. These samples were then mixed with 200 μl of methanol containing the internal standard (200 nM flecainide) and centrifuged at 4500 rpm for 10 minutes to remove precipitated protein. The supernatants were filtered using 0.45 μm 96-well filter plates and analyzed using LC-MS/MS.
LC-MS/MS.
Concentrations of compounds in samples were measured using an LC-MS/MS system consisting of an API4000 mass spectrometer (Applied Biosystems, Forester City, CA) with a Shimadzu 20A series high-pressure liquid chromatography (HPLC) system (Shimadzu Corporation, Kyoto, Japan). Chromatography was performed using an Inertsil ODS-3 column (3 μm particle size, 2.1 × 50 mm; GL Science, Tokyo, Japan) warmed to 40°C. The mobile phase consisted of 0.1% formic acid (A) and acetonitrile (B). The flow rate was 0.2 ml/min, and the gradient conditions for elution were as follows: gradient [min, B%] = [0, 10] − [1, 90] − [4, 90] − [4.1, 10] − [7, 10]. Mass spectrometric conditions for individual analytes are given in Table 1.
LC-MS/MS conditions for compounds investigated and internal standards with details on mass transitions
Data Analysis.
The peak area ratios of test compounds to internal standard were used for the calculation in all experiments. The mean value of triplicate determinations was plotted versus the incubation time on a semilogarithmic scale, and the slope was determined by linear-regression analysis as depletion rate constant (k (min−1)). For liver S9, intestinal S9, and hepatocytes, the CLint values were calculated using the following equations: (1)
(1) (2)where V is the initial incubation volume (ml), proteinS9 is the initial amount of S9 protein in incubation (mg), numberhepatocytes is the initial number of hepatocytes in incubation (×106 cells), fu,S9 is fraction unbound in S9 incubation, and fu,hepatocytes is the fraction unbound in hepatocytes incubation. The fu,S9 and fu,hepatocytes values were predicted as reported previously elsewhere (Hallifax and Houston, 2006; Kilford et al., 2008), assuming that binding in S9 was comparable to that in human liver microsomes at the same protein concentrations. Predictive tools were applied to avoid any potential stability issues over prolonged periods in the buffer during the equilibrium dialysis experiments. The protein concentration used in the in vitro experiments was low (whenever possible) to minimize any potential issues associated with nonspecific binding. Most of the prodrugs investigated showed predicted fu,S9 or fu,hepatocytes of >0.8 at the protein/cell concentration used, and the subsequent nonspecific binding correction was not implemented. The only exceptions were candesartan cilexetil (logP = 6.1) (Detroja et al., 2011) and mycophenolate mofetil (logP = 3.0).
(2)where V is the initial incubation volume (ml), proteinS9 is the initial amount of S9 protein in incubation (mg), numberhepatocytes is the initial number of hepatocytes in incubation (×106 cells), fu,S9 is fraction unbound in S9 incubation, and fu,hepatocytes is the fraction unbound in hepatocytes incubation. The fu,S9 and fu,hepatocytes values were predicted as reported previously elsewhere (Hallifax and Houston, 2006; Kilford et al., 2008), assuming that binding in S9 was comparable to that in human liver microsomes at the same protein concentrations. Predictive tools were applied to avoid any potential stability issues over prolonged periods in the buffer during the equilibrium dialysis experiments. The protein concentration used in the in vitro experiments was low (whenever possible) to minimize any potential issues associated with nonspecific binding. Most of the prodrugs investigated showed predicted fu,S9 or fu,hepatocytes of >0.8 at the protein/cell concentration used, and the subsequent nonspecific binding correction was not implemented. The only exceptions were candesartan cilexetil (logP = 6.1) (Detroja et al., 2011) and mycophenolate mofetil (logP = 3.0).
In vitro CLint obtained in hepatocytes (ml/min/106 cells) were scaled by eq. 3 to give an in vivo CLint (ml/min/g liver) using hepatocellularity values of 120 × 106 cells/g liver for human, monkey, and rat, and 240 × 106 cells/g liver for dog (Houston and Galetin, 2008; Hosea et al., 2009):
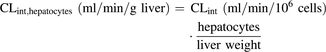 (3)
(3)To allow direct comparison of the intestinal and hepatic S9 data, CLint values (ml/min/mg S9 protein) were scaled to the CLint values to per gram of tissue using eq. 4 and eq. 5 for human LS9 and IS9, respectively. The S9 scaling factors for preclinical species were assumed to be the same as the values reported for humans (Houston and Galetin, 2008; Cubitt et al., 2011).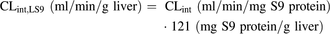 (4)
(4)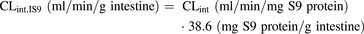 (5)Due to lack of a kidney S9 scaling factor, we used two approaches: using the same scaling factor as for liver S9 [121 mg S9 protein/g liver (Houston and Galetin, 2008)] (scaling factor A) or combining the specific kidney microsomal scaling factor [12.8 mg S9 protein/g kidney (Gill et al., 2012)] with a liver cytosolic scaling factor [80.7 mg S9 protein/g (Houston and Galetin, 2008)], resulting in a value of 93.5 mg S9 protein/g kidney (scaling factor B).
(5)Due to lack of a kidney S9 scaling factor, we used two approaches: using the same scaling factor as for liver S9 [121 mg S9 protein/g liver (Houston and Galetin, 2008)] (scaling factor A) or combining the specific kidney microsomal scaling factor [12.8 mg S9 protein/g kidney (Gill et al., 2012)] with a liver cytosolic scaling factor [80.7 mg S9 protein/g (Houston and Galetin, 2008)], resulting in a value of 93.5 mg S9 protein/g kidney (scaling factor B).
When CLint values could not be estimated because of high stability [>90% of the compound still remaining after 60 minutes (S9) or 120 minutes (hepatocytes) of incubation], a nominal value of 0.1 ml/min/g liver was assigned for these drugs in Figs. 1 and 2; these compounds were not included in the subsequent analysis shown in Figs. 3 and 4. Discrepancies in animal hydrolysis CLint data relative to humans were assessed by geometric fold error (gmfe). This analysis was only performed for hepatocytes because of the high stability of a number of compounds in liver S9 or intestinal S9 from preclinical species.
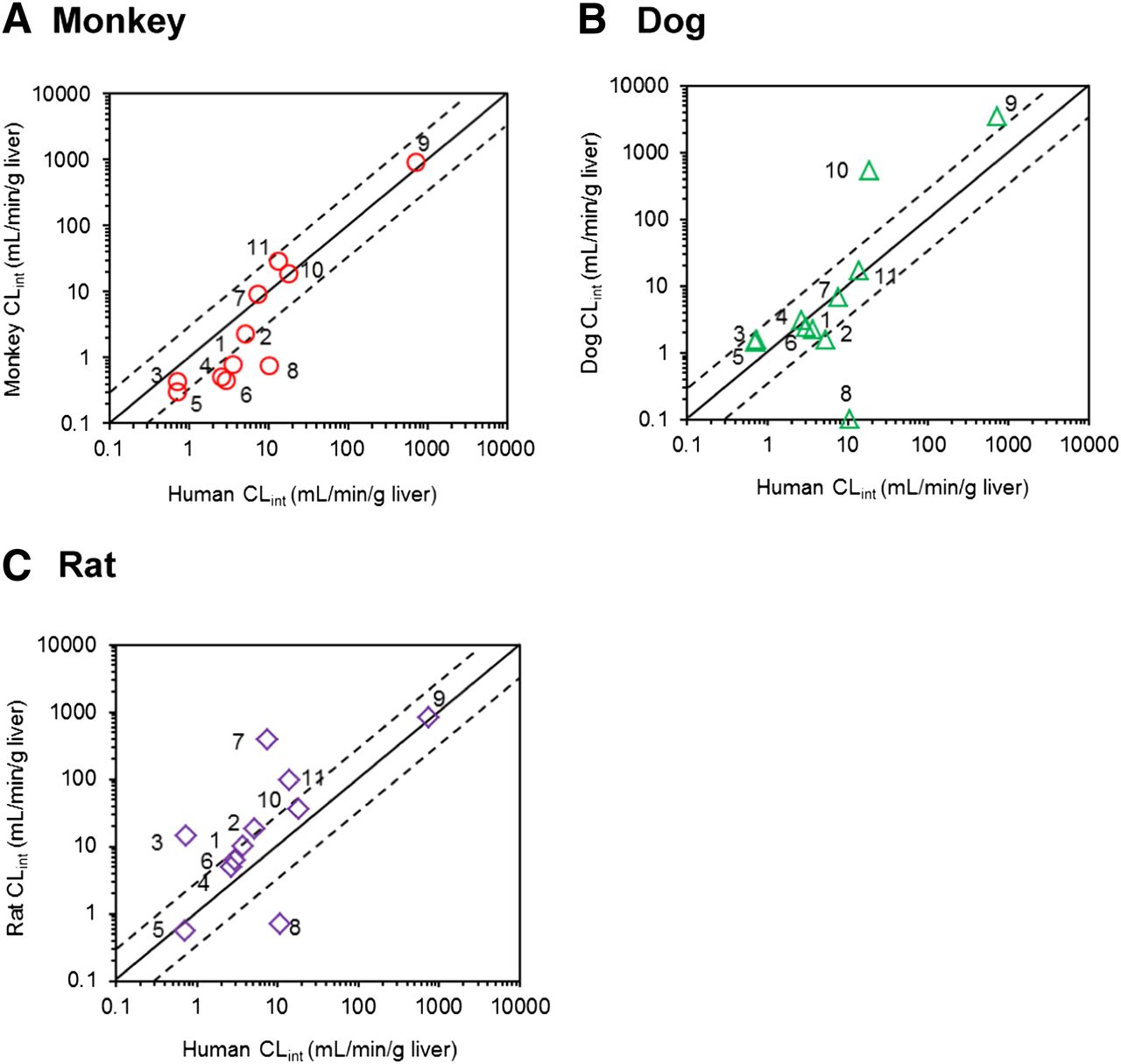
Correlation of the CLint values in hepatocytes between human and monkey (A), dog (B), or rat (C). The numbers allocated per compound are as listed in the Tables. Scaling to CLint (ml/min/g liver) was done using eq. 3. For highly stable compounds (CLint < 0.001 ml/min/106 cells after 120 minutes), the nominal value of 0.1 ml/min/g liver was used. The solid and dashed lines represent line of unity and a 3-fold range, respectively.

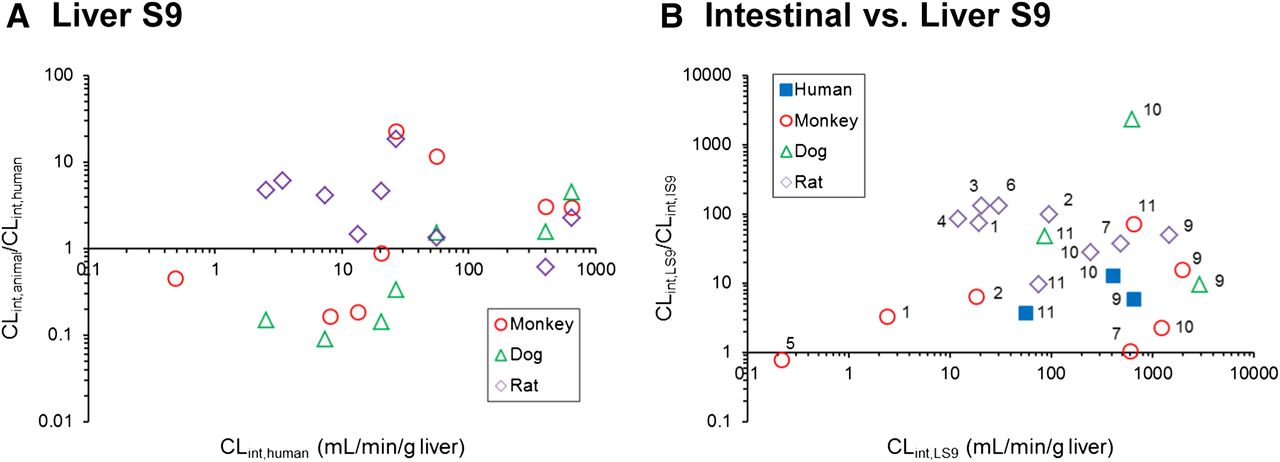
Comparison of scaled animal and human hydrolysis CLint values obtained in LS9 (A). Comparison between the CLint,LS9 values and CLint,LS9/CLint,IS9 values for human, monkey, dog, and rat (B). The numbers allocated per compound are as listed in Tables. Scaling to CLint (ml/min/g liver or ml/min/g intestine) was done using eqs. 4 and 5. For highly stable compounds (CLint <0.002 ml/min/mg S9 protein after 60 minutes), the CLint values could not be obtained and were not considered in this figure.

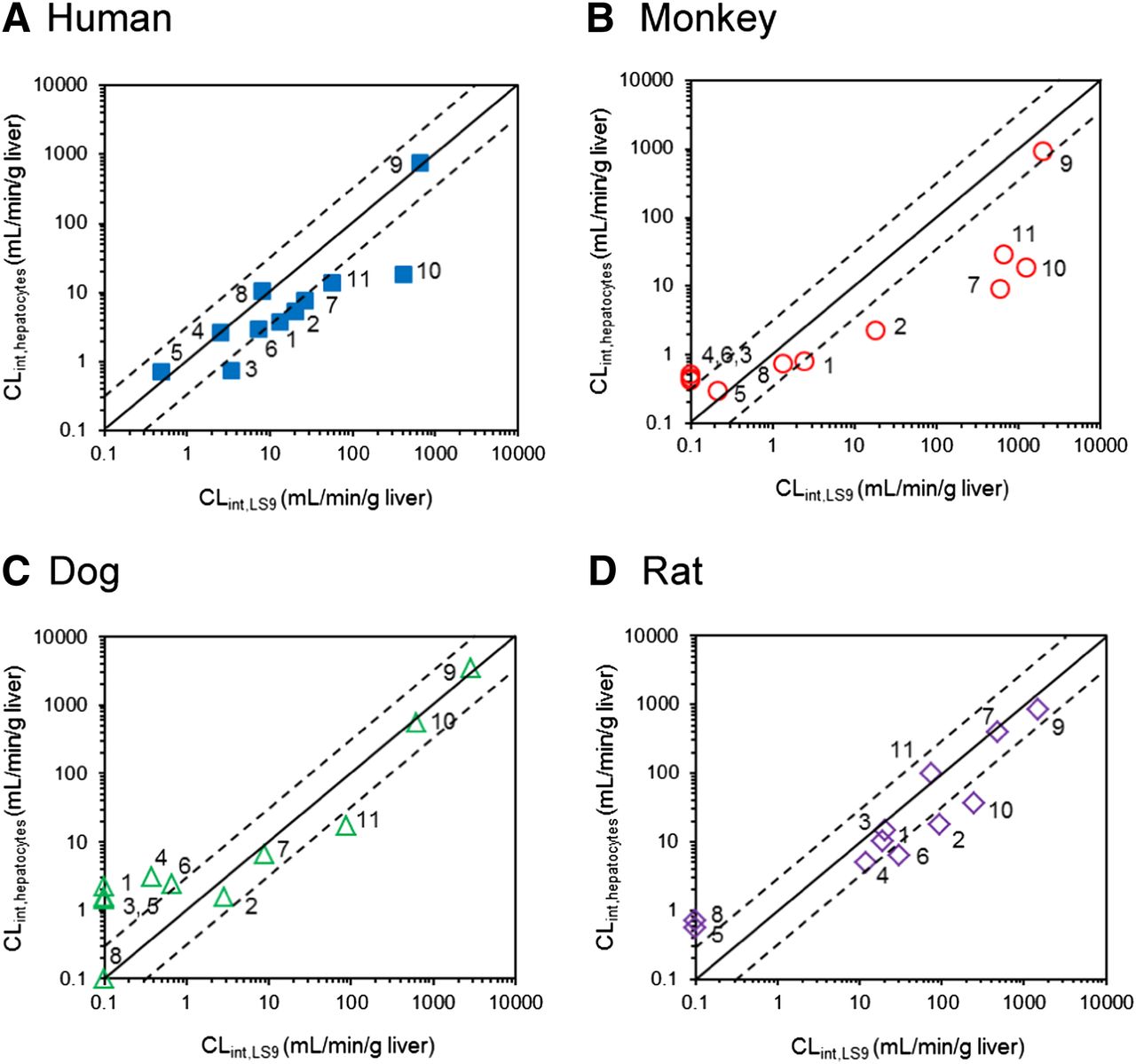
Comparison of the CLint values between liver S9 and hepatocytes for human (A), monkey (B), dog (C), and rat (D). The numbers allocated per compound are as listed in the tables. Scaling to CLint (ml/min per g liver) was done using eqs. 3 and 4. When >90% of the compound still remained after either 60 minutes (S9) or 120 minutes (hepatocytes), the CLint values could not be obtained and were plotted as nominal value of 0.1 ml/min/g liver. The solid and dashed lines represent line of unity and a 3-fold range, respectively.
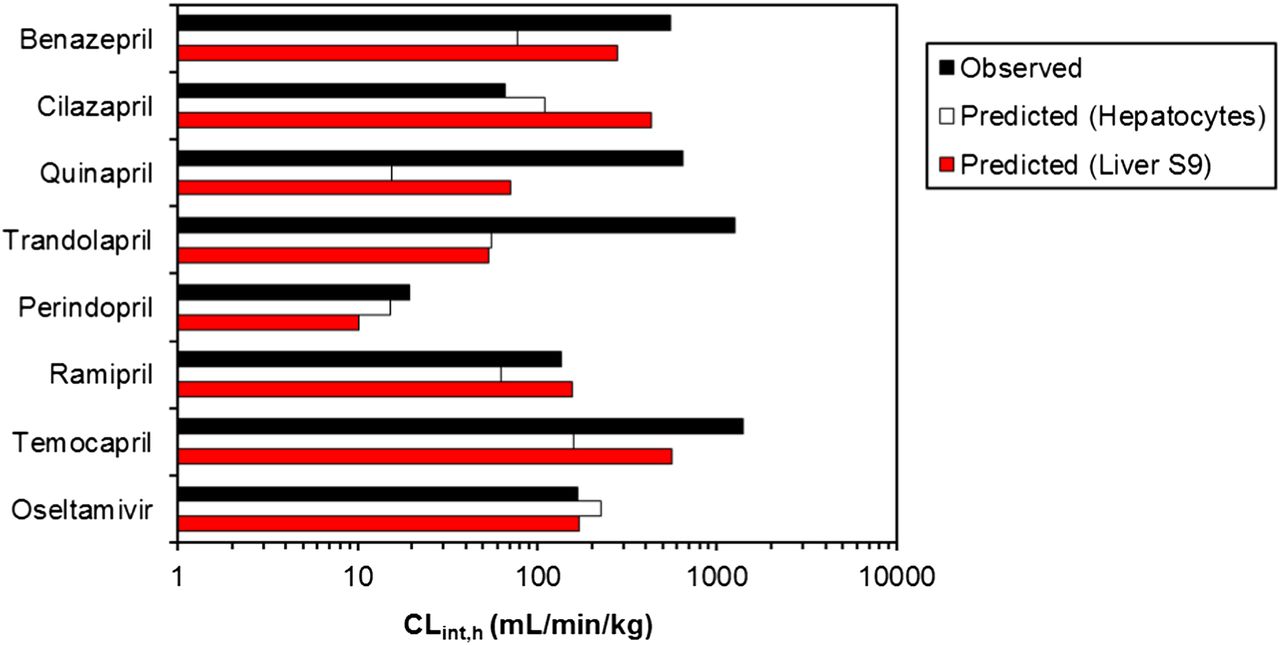
In vitro–in vivo extrapolation of human hydrolysis CLint data for obtained in either human hepatocytes of liver S9. Observed CLint,h were estimated from plasma clearance data after oral administration of prodrugs, assuming negligible contribution of intestinal hydrolysis and complete absorption.
In Vitro–In Vivo Extrapolation of Human Prodrug Clearance.
Because of the lack of intravenous clearance data reported for prodrugs, the predictability of human hydrolysis CLint data was assessed against oral clearances. The hepatic intrinsic clearance (CLint,h) values after oral drug administration were estimated using eq. 6 for a subset of eight CES1 substrates (Pang and Rowland, 1977; Gertz et al., 2010)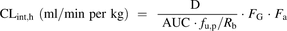 (6)where D/AUC represents the hepatic blood clearance obtained from mean plasma data after correcting for renal excretion (where applicable) and the blood to plasma distribution ratio (Rb). Renal excretion was minimal for most of the prodrugs (<1%) with the exception of quinapril, perindopril, and oseltamivir (3–14%) (Supplemental Table 4). The fu,p represents the fraction unbound in plasma, D represents the oral drug dose (mg/kg), AUC represents the area under the drug concentration-time curve (mg/min per ml), FG is intestinal availability, and Fa is the fraction absorbed. The oral clearance data for eight CES1 substrates and the Rb for oseltamivir were collated from the literature (Supplemental Table 4). The Rb value for the seven remaining prodrugs was assumed to be 0.55, an assumption used previously for acidic drugs (Kilford et al., 2009). The FG values for all eight prodrugs were assumed to be 1, considering their high stability in intestinal S9 (see Results). In addition, the absorption of these drugs was assumed to be complete (Fa = 1) because of their high solubility (>1 mg/ml in buffer and in fasted-state-simulated intestinal fluid (FaSSIF) for all eight prodrugs, in-house data) and low dose number (<0.3 for all the prodrugs in the data set). The lowest parallel artificial membrane permeability assay estimate in this data set was obtained for perindopril, which translated to effective permeability of 1.34 × 10−4 cm/s, confirming the validity of Fa = 1 assumption (Lennernas, 2014). The unbound CLint,h from human hepatocytes or liver S9 (eqs. 3 and 4, respectively) was scaled using an average liver weight of 21.4 g of liver/kg.
(6)where D/AUC represents the hepatic blood clearance obtained from mean plasma data after correcting for renal excretion (where applicable) and the blood to plasma distribution ratio (Rb). Renal excretion was minimal for most of the prodrugs (<1%) with the exception of quinapril, perindopril, and oseltamivir (3–14%) (Supplemental Table 4). The fu,p represents the fraction unbound in plasma, D represents the oral drug dose (mg/kg), AUC represents the area under the drug concentration-time curve (mg/min per ml), FG is intestinal availability, and Fa is the fraction absorbed. The oral clearance data for eight CES1 substrates and the Rb for oseltamivir were collated from the literature (Supplemental Table 4). The Rb value for the seven remaining prodrugs was assumed to be 0.55, an assumption used previously for acidic drugs (Kilford et al., 2009). The FG values for all eight prodrugs were assumed to be 1, considering their high stability in intestinal S9 (see Results). In addition, the absorption of these drugs was assumed to be complete (Fa = 1) because of their high solubility (>1 mg/ml in buffer and in fasted-state-simulated intestinal fluid (FaSSIF) for all eight prodrugs, in-house data) and low dose number (<0.3 for all the prodrugs in the data set). The lowest parallel artificial membrane permeability assay estimate in this data set was obtained for perindopril, which translated to effective permeability of 1.34 × 10−4 cm/s, confirming the validity of Fa = 1 assumption (Lennernas, 2014). The unbound CLint,h from human hepatocytes or liver S9 (eqs. 3 and 4, respectively) was scaled using an average liver weight of 21.4 g of liver/kg.
Results
CLint values were obtained for 11 prodrugs in four different species across a range of in vitro systems: cryopreserved hepatocytes, LS9, IS9, and KS9 using the substrate depletion approach. The CLint values were corrected for nonspecific binding and scaled to CLint per gram of tissue to allow comparison among different tissues and/or species. In addition, the stability of prodrugs in plasma samples from each species was investigated. The predictability of human hydrolysis CLint data was assessed for a subset of eight prodrugs with reported in vivo data.
Assessment of Hydrolysis Activity in Hepatocytes across Species.
The individual CLint values (ml/min/106 cells) obtained for 11 prodrugs in human, monkey, dog, and rat hepatocytes are listed in Table 2. The correlation between CLint (ml/min/g liver) scaled from the human and animal hepatocytes data for all the compounds investigated is shown in Fig. 1.
CLint values obtained in hepatocytes of human, monkey, dog, and rat
Each value presents the mean ± S.D. (n = 3).
The human intrinsic hydrolysis clearance obtained in hepatocytes (CLint,hepatocytes) ranged three orders of magnitude from 0.73–740 ml/min/g liver for perindopril and candesartan cilexetil, respectively. Similarly, the monkey CLint,hepatocytes ranged from 0.30 to 930 ml/min/g liver for perindopril and candesartan cilexetil, respectively (Fig. 1A). The monkey CLint values for seven drugs were within 3-fold of the human CLint values, with an excellent agreement in the CLint values in the case of candesartan cilexetil, mycophenolate mofetil, and temocapril. The monkey CLint,hepatocytes showed a 2.7-fold deviation from human hydrolysis CLint data obtained in the same cellular system. The most pronounced outlier was oseltamivir, where the monkey CLint,hepatocytes value represented only 6% of the corresponding value in human hepatocytes.
Analogous to monkeys and humans, dog CLint,hepatocytes showed a wide range (1.4–3400 ml/min/g liver), with perindopril and candesartan cilexetil being the drugs with lowest and highest CLint values (Fig. 1B). The dog hydrolysis CLint were within 3-fold of the human hepatocyte values for seven compounds; however, an increased divergence from human CLint,hepatocyte data was apparent (gmfe of 3.6). The largest disagreement between dog and human hepatocyte data was seen for mycophenolate mofetil and oseltamivir. The mycophenolate mofetil dog CLint,hepatocytes value was 29-fold higher than the human CLint value, whereas oseltamivir was highly stable in dog hepatocytes (CLint < 0.1 ml/min per g liver) relative to human cells (11 ml/min per g liver; Table 2).
Of all the preclinical species investigated, rat hydrolysis CLint,hepatocytes showed the most pronounced disagreement with human data (gmfe = 4.4; Fig. 1C). In general, most of the CLint values obtained in rat hepatocytes were higher than the human data obtained in the same in vitro system; the difference was up to 52-fold in the case of temocapril (Supplemental Fig. 2). Analogous to the trends seen in the monkey and dog CLint,hepatocytes data, oseltamivir was the outlier, with the rat CLint value representing <10% of the human CLint.
Assessment of Hydrolysis Activity in LS9 across Species.
Individual CLint values (ml/min/mg S9 protein) obtained in human, monkey, dog, and rat liver S9 are shown in Table 3. A three orders of magnitude range in human intrinsic hydrolysis clearance obtained in liver S9 (CLint,LS9) was observed (0.48–650 ml/min/g liver); perindopril and candesartan cilexetil were extremes, as seen in hepatocyte CLint data (Fig. 2). Analogous to hepatocytes, oseltamivir CLint data in monkey LS9 were 6-fold lower than in humans. In contrast, monkey CLint,LS9 was higher than in human for candesartan cilexetil, mycophenolate mofetil, olmesartan medoxomil, and temocapril (up to 23-fold). In general, the rat CLint,LS9 values for CES1 substrates were higher than the human values, whereas an opposite trend was seen for dog CLint,LS9 data relative to human (candesartan cilexetil exception). Differences in CLint,LS9 values across three preclinical species were not pronounced for candesartan cilexetil, mycophenolate mofetil, or perindopril (up to 5-fold), in contrast to trandolapril, quinapril, and ramipril, where a >100-fold difference in scaled CLint,LS9 was observed (Fig. 2A).
CLint values in liver S9 of human, monkey, dog, and rat
Comparison of CLint,hepatocytes and CLint,LS9.
On average, the CLint,LS9 values were higher than CLint,hepatocytes values in all species other than dogs. Human CLint,LS9 data for trandolapril, perindopril, oseltamivir, and candesartan cilexetil were in excellent agreement with CLint,hepatocytes, whereas the CLint,LS9 value for all the remaining prodrugs was higher than in human hepatocytes (up to 20-fold in the case of mycophenolate mofetil) (Fig. 3A).
In contrast to human LS9, a number of compounds showed high stability in animal LS9, and CLint could not be obtained (Fig. 3, B–D). In particular, this was evident for perindopril, oseltamivir, and quinapril where the CLint data were not determined in two out of three preclinical species investigated (Table 3). Analogous to human, mycophenolate mofetil monkey CLint data showed the most pronounced system differences (50-fold higher value obtained in monkey LS9 compared with hepatocytes). This trend was consistent across all preclinical species, as the CLint,LS9 values for this prodrug were higher than the CLint,hepatocytes. The number of stable prodrugs in dog LS9 was the largest among the species investigated, and the CLint was not determined for 4 of 11 prodrugs (Fig. 3C). The range of scaled dog CLint,LS9 for the remaining drugs was 0.38–2900 ml/min/g liver; as in previous cases, candesartan cilexetil showed the highest CLint.
Assessment of Hydrolysis Activity in Intestinal S9 across Species.
CLint values (ml/min per mg S9 protein) obtained in human, monkey, dog, and rat intestinal S9 fractions are listed in Table 4. In the case of human intestine, the CLint for CES1 substrates (drugs 1–8 in each table) was not detected (intrinsic hydrolysis clearance obtained in intestinal S9 [CLint,IS9] <0.002 ml/min per mg S9 protein). In contrast, the CLint,IS9 values for CES2 and CMBL substrates ranged from 0.4 to 2.9 ml/min per mg S9 protein for olmesartan medoxomil and candesartan cilexetil, respectively. Unlike in humans, the CLint was detected for CES1 substrates in monkey and rat IS9. However, the intestinal hydrolysis clearance values in both of these preclinical species were generally low for these drugs (CLint <0.1 ml/min per mg S9 protein for all the drugs investigated, with the exception of temocapril). The monkey CLint,IS9 values for CES2 and CMBL substrates were either comparable (candesartan cilexetil and olmesartan medoxomil) or up to 17-fold higher than for human IS9 CLint (mycophenolate mofetil). Analogous to humans, dog CLint,IS9 for eight CES1 substrates was not detected in dog intestine (Table 4). For the remaining CES2 and CMBL substrates, the trends were inconsistent with either human or monkey IS9 data; this discrepancy was particularly evident in the case of mycophenolate mofetil, where the CLint was <0.1% of the human IS9 estimate. The rat CLint,IS9 values for CES2 and CMBL substrates were on average 30% of the human CLint,IS9 (Table 4).
CLint values in intestinal S9 of human, monkey, dog, and rat
Each value presents the mean ± S.D. (n = 3).
Assessment of Intestinal Hydrolysis Relative to Hepatic.
The comparison of CLint values obtained in LS9 relative to IS9 in each individual species is shown in Fig. 2B. It is evident that the hepatic hydrolysis CLint was higher than the intestinal, regardless of the species investigated. The data set was reduced in human and dog S9 data because of the high stability of CES1 substrates in the corresponding IS9. The ratio of monkey CLint,LS9/CLint,IS9 for CES1 substrates ranged from 0.8 to 6.0 for perindopril and cilazapril, respectively. Across all species, the difference in hepatic hydrolysis CES1 CLint values relative to intestine was the most pronounced in the rat, with on average 92-fold higher CLint in LS9 (37- to 131-fold for temocapril and ramipril, respectively).
For CES2 and CMBL substrates, the ratio of human CLint,LS9/CLint,IS9 ranged from 4 to 13 for olmesartan medoxomil and candesartan cilexetil, respectively. Data in preclinical species reflected this trend of higher CLint values in liver hydrolysis relative to intestine. However, species differences were evident, as illustrated nicely in the case of mycophenolate mofetil. Intestinal and hepatic hydrolysis were comparable in cynomolgus monkeys (within 2-fold), in contrast to the >2000 CLint,LS9/CLint,IS9 ratio seen in dog, illustrating a very different contribution of intestinal hydrolysis to the first-pass effect and bioavailability of this prodrug across preclinical species.
Assessment of Hydrolysis Activity in Kidney S9.
CLint values (ml/min/mg S9 protein) in KS9 of each species were only obtained for candesartan cilexetil, mycophenolate mofetil, and olmesartan medoxomil (Table 5); hydrolysis was detected in KS9 from all species investigated. For candesartan cilexetil and mycophenolate mofetil, the CLint,KS9 (ml/min/mg S9 protein) values in preclinical species were higher than those in humans (on average 5.4- to 33-fold, respectively), with the most pronounced differences seen in dog CLint,KS9 data relative to human. In the case of olmesartan medoxomil, the CLint,KS9 (ml/min/mg S9 protein) values among all four species were within 7-fold. The scaled CLint,KS9 (ml/min/g kidney) value was calculated using two alternative kidney S9 scaling factors; details are shown in Supplemental Table 3. Human scaled CLint,KS9 (ml/min/g kidney) values for candesartan cilexetil and mycophenolate mofetil (CES2 substrates) were lower than the CLint,IS9 (ml/min/g intestine) value, in contrast to olmesartan medoxomil where they were comparable.
CLint values in kidney S9 of human, monkey, dog, and rat
Assessment of Hydrolysis Activity in Plasma.
CES substrates showed high stability in human, monkey, and dog plasma as >90% of the drug remained after 60 minutes of incubation. The only exception was rat plasma, where the hydrolysis was evident, even in the case of CES substrates, with the half-life (t1/2) values ranging from 0.11 to 5.2 minutes for trandolapril and oseltamivir, respectively (Supplemental Table 2). In contrast, olmesartan medoxomil hydrolysis was seen in plasma from both human and preclinical species (t1/2 < 0.1 minutes, in contrast to t1/2 of 2.3 minutes in rat plasma).
In Vitro–In Vivo Extrapolation of Hydrolysis Clearance for CES1 Prodrugs.
The ability of in vitro CLint hydrolysis data obtained in human hepatocytes and LS9 to predict oral clearance for eight CES1 substrates was investigated; no intravenous clearance data were available for any of the prodrugs in the data set. Hepatic hydrolysis was assumed to be the only contributor to the oral clearance, considering the high stability of these prodrugs in intestinal S9. In vitro–in vivo extrapolation was limited to human because of the limited availability of data in preclinical species; in this case, oral clearances could not be considered because of the intestinal hydrolysis observed in some preclinical species.
Hepatocyte data predicted CLint,h for four of eight prodrugs within 3-fold of the observed data. However, an underprediction trend was particularly evident for high clearance prodrugs (CLint,h >550 ml/min per kg), as the predicted clearance represented only 2 to 14% of the observed value for quinapril and benazepril, respectively. This trend was not as apparent when LS9 data were used, as temocapril and benazepril clearances were predicted relatively well (up to 50% of observed); in contrast, the prediction of trandolapril and quinapril clearance was comparable to hepatocytes (4 and 11% of the observed, respectively). Prediction bias was less apparent with LS9 (gmfe of 3.2 versus 5 seen in the case of hepatocytes), highlighting the utility of this subcellular fraction for the prediction of human hydrolysis clearance.
Discussion
The current study represents the most comprehensive analysis to date of the species and in vitro system differences (hepatocytes versus S9) in hydrolysis CLint generated under standardized conditions. In addition, the importance of extrahepatic hydrolysis (intestine and kidney) relative to hepatic has been assessed. Quantitative prediction of in vivo hydrolysis clearance focused on human data only; inconsistent or unavailable in vivo data in preclinical species limited our attempts to assess species differences in the predictability of hydrolysis clearance.
Species Differences in Hepatic Hydrolysis Activity.
The hydrolysis CLint could be obtained in hepatocytes for all the drugs investigated in both human and preclinical species. Regardless of the species, perindopril and candesartan cilexetil were the prodrugs with the lowest and highest CLint, whereas the rank order of other compounds varied.
Across all species investigated, the monkey hepatocyte data showed the most comparable hydrolysis CLint relative to human hepatocytes (evident in particular for candesartan cilexetil, mycophenolate mofetil, and temocapril, Fig. 1A), in agreement with previous studies in recombinant CES (Williams et al., 2011) and high homology of amino acid sequence reported between cynomolgus monkeys and human CES1 and CES2 (Taketani et al., 2007; Hosokawa, 2008; Imai and Ohura, 2010). The most pronounced differences in hydrolysis CLint,hepatocytes in preclinical species relative to human were seen in rat (Fig. 1C); the esterase family in this preclinical species includes multiple enzymes with 67–77% homology with human CES1 (Taketani et al., 2007; Hosokawa, 2008). Dog hepatocytes data showed a comparable wide range in hydrolysis CLint to other species and good agreement with human CLint, hepatocyte data for a number of low- and high-clearance prodrugs (e.g., temocapril and candesartan cilexetil); however, a discrepancy compared with human data (gmfe = 3.6) was more pronounced relative to monkey CLint,hepatocytes. Oseltamivir was generally more stable in all animal hepatocytes relative to humans; this trend was particularly evident in dog, as >90% of the drug was stable after 120 minutes. Dog CES1 shows lower homology with human CES1 (80%) compared with monkeys (Taketani et al., 2007; Hosokawa, 2008), which may rationalize some species difference in substrate recognition, especially considering structure similarities of CES1 substrates investigated (with the exception of oseltamivir; Supplemental Fig. 1). The homology of the amino acid sequence between human and dog CES2 is currently unknown.
Comparison of CLint,LS9 and CLint,hepatocytes across Species.
In the current analysis of 11 prodrugs, the scaled CLint,hepatocytes values were generally lower than the CLint,LS9 values in all species, with the exception of dog (Figs. 2 and 3). In humans, the fold range in CLint,LS9 was analogous to hepatocytes, highlighting the utility of LS9 for the characterization of prodrugs in humans. However, it is important to note that some of the low-clearance compounds in animal hepatocytes (e.g., perindopril, oseltamivir, and quinapril) were more metabolically stable in animal LS9 (under the conditions selected) and thus CLint data could not be determined in all the preclinical species investigated. This stability could not be associated with any methodological differences, as all LS9 fractions were obtained from the same supplier.
Subsequently, in vitro–in vivo extrapolation of hydrolysis CLint data was performed using data obtained in human hepatocytes and LS9. A subset of eight CES1 substrates with available clearance data after prodrug oral administration was selected, as plasma concentrations of prodrugs after intravenous dosing are often very low and/or not reported, with the most data available for the active metabolite. The data set covered a 75-fold range in observed CLint,h, with perindopril and temocapril on the lower and higher end, respectively. Use of hydrolysis hepatocytes data resulted in reasonably good prediction of low clearance prodrugs (50% within 3-fold). However, an overall underprediction trend was evident (5-fold bias), in agreement with trends reported previously for cytochrome P450 substrates (Hallifax et al., 2010). This trend was particularly apparent for high clearance prodrugs, as the predicted clearance was <15% of the observed value. For certain prodrugs in this range (e.g., trandolapril), the extent of underprediction was comparable between hepatocytes and LS9 (the predicted CLint was approximately 4% of the in vivo value regardless of the system). However, this underprediction trend was not evident for all high clearance prodrugs when LS9 data were used, emphasizing the promising application of this subcellular in vitro system for the prediction of human hydrolysis clearance. Unfortunately, predictability of in vitro hydrolysis CLint from individual preclinical species was not possible; this kind of analysis would increase the confidence for the subsequent predictive use of human hydrolytic data obtained in the same cellular system/subcellular fraction.
Species Differences in Intestinal and Kidney Hydrolysis Activity.
The metabolic activities for CES1 substrates were not detected in human intestine, in contrast to CES2 and CMBL substrates (Fig. 2B; Table 4); the findings were generally in agreement with expression data reported for these enzymes in human intestine (Hosokawa, 2008; Ishizuka et al., 2013). The scaled human CLint,IS9 values were lower than the CLint,LS9 values for CES2 and CMBL substrates (Fig. 2B), suggesting lower expression levels per gram of tissue of these hydrolases in the intestine. Analogous to humans, CES1 substrates were also metabolically stable in dog intestinal S9 data, whereas opposite trends were seen in the case of monkey and rat IS9 (Table 4). Surprisingly, the metabolic activities for two CES2 substrates were detected in dog IS9; although the CLint,IS9 value for mycophenolate mofetil was low, in particular relative to dog liver S9 data, candesartan cilexetil CLint,IS9 was the highest across all species (Table 4). This result was in contrast to the reported lack of expression of both CES1 and CES2 in dog intestine (Hosokawa, 2008), indicating a potential contribution of additional hydrolases. The latter is most likely the reason for observed hydrolysis of CES1 substrate temocapril in rat IS9 (Table 4), the opposite of the CES expression reported in this species. In the case of olmesartan medoxomil, the lowest CLint obtained in dog IS9 supported the minimal CMBL expression levels reported in dog intestine relative to other species (Ishizuka et al., 2013). There was no consistency in differences seen in animal intestinal hydrolysis CLint for CES2 and CMBL substrates relative to human IS9. In addition, differences in the contribution of intestinal hydrolysis to the first-pass effect and bioavailability were evident across preclinical species relative to human (e.g., mycophenolate mofetil).
An important consideration is that the human, monkey, and dog intestinal S9 were prepared from a single individual, which may bias the analysis; however, pooled samples prepared without the use of PMSF (CES inhibitor) during the isolation process were not available. In addition, owing to a lack of data, the intestinal and liver S9 scaling factors for preclinical species were assumed to be the same as human. However, a number of issues have already been highlighted (Cubitt et al., 2011) with respect to the value of the human intestinal cytosolic scaling factor (obtained from limited number of samples prepared by mucosal scraping method, no information on the variability), which also propagate here for the scaling of hydrolysis data. In addition, a single intestinal S9 scaling factor was used, and no potential regional differences were accounted for, which all may impact the analysis of the relevance of intestinal hydrolysis relative to hepatic. Similar problems occurred in the case of kidney data, where the general lack of cytosolic/S9 scaling factors led to the use of indirect values (liver), emphasizing the need for more work in this area—in particular if the data were to be used in physiologically based models to simulate concentration-time profiles of prodrugs and their active metabolites in relevant tissues of interest (for safety concerns or to assess potential drug-drug interaction risks). Hydrolysis activities were detected in kidney S9 for CES2 and CMBL substrates only (Table 5), in agreement with the expression data reported for these enzymes in the kidney across species (Supplemental Table 1).
Assessment of metabolic stability of prodrugs in plasma highlighted very clear species differences, with fast hydrolysis observed for CES substrates only in rat plasma. For olmesartan medoxomil, a PON1 substrate, the metabolic activity in human plasma was comparable to the monkey and dog, and in all cases was significantly more pronounced than in the rat, emphasizing the need for consideration of these species differences when performing in vivo studies in preclinical species.
In summary, the systematic analysis of species and system differences was performed for 11 prodrugs. Hydrolysis CLint data obtained in monkey hepatocytes were the most comparable to human for the current data set. The importance of intestinal hydrolysis and species difference in its potential contribution to the overall bioavailability has been illustrated for CES2 and CMBL substrates. Uncertainty in scaling factors, in particular highlighted in the case of kidney and intestinal S9 data, may impact the analysis of the relevance of extrahepatic hydrolysis relative to hepatic. For the first time, the predictive utility of hydrolysis CLint obtained in human hepatocytes and liver S9 was assessed. Reduced prediction bias observed with liver S9 data, in particular for high clearance prodrugs, highlights the application of this subcellular fraction for the characterization of hydrolysis clearance and its prediction. Further evaluation of this in vitro system using the extended data set of prodrugs is needed.
Authorship Contributions
Participated in research design: Nishimuta, Galetin.
Conducted experiments: Nishimuta.
Performed data analysis: Nishimuta, Galetin.
Wrote or contributed to the writing of the manuscript: Nishimuta, Houston, Galetin.
Footnotes
- Received January 28, 2014.
- Accepted July 2, 2014.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- CES
- carboxylesterase
- CLint
- intrinsic clearance
- CLint,h
- hepatic intrinsic clearance
- CLint,hepatocytes
- intrinsic hydrolysis clearance obtained in hepatocytes
- CLint,LS9
- intrinsic hydrolysis clearance obtained in liver S9
- CLint,IS9
- intrinsic hydrolysis clearance obtained in intestinal S9
- CMBL
- carboxymethylenebutenolidase
- fu
- fraction unbound
- gmfe
- geometric fold error
- IS9
- intestinal S9
- KS9
- kidney S9
- LS9
- liver S9
- LC-MS/MS
- high-performance liquid chromatography–mass spectrometry
- PMSF
- phenylmethylsulfonyl fluoride
- PON1
- paraoxonase 1
- t1/2
- half-life
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}