


Abstract
Efflux transporters P-glycoprotein (P-gp/ABCB1), multidrug resistance protein 1 (MRP1/ABCC1), and breast cancer resistance protein (BCRP/ABCG2) can affect the efficacy and toxicity of a wide variety of drugs and are implicated in multidrug resistance (MDR). Eight test compounds, recently identified from an intramolecular FRET-based high throughput screening, were characterized for their interaction with MRP1. We report that the active metabolite of vitamin D3, calcitriol, and its analog calcipotriol are selectively cytotoxic to MRP1-overexpressing cells, besides inhibiting transport function of P-gp, MRP1, and BCRP. Calcitriol and calcipotriol consistently displayed a potent inhibitory activity on MRP1-mediated doxorubicin and calcein efflux in MRP1-overexpressing H69AR and HEK293/MRP1 cells. Vesicular transport studies confirmed a strong inhibitory effect of calcitriol and calcipotriol on MRP1-mediated uptake of tritium-labeled estradiol glucuronide and leukotriene C4. In cytotoxicity assays, MRP1-overexpressing cells exhibited hypersensitivity toward calcitriol and calcipotriol. Such collateral sensitivity, however, was not observed in HEK293/P-gp and HEK293/BCRP cells, although the vitamin D3 analogs inhibited calcein efflux in P-gp-overexpressing cells, and mitoxantrone efflux in BCRP-overexpressing cells. The selective cytotoxicity of calcitriol and calpotriol toward MRP1 over-expressing cells can be eliminated with MRP1 inhibitor MK571. Our data indicate a potential role of calcitriol and its analogs in targeting malignancies in which MRP1 expression is prominent and contributes to MDR.
Introduction
The development of multidrug resistance (MDR) remains a major hurdle in chemotherapy, which is presently the standard treatment of many metastatic and leukemic cancers. MDR is characterized by the resistance of malignancies to structurally and mechanistically distinct anti-cancer agents, and can arise from various physiologic changes in the cancer cells (Gottesman et al., 2016). One of the prominent phenotypes of MDR is the overexpression of ATP-binding cassette (ABC) membrane transporters, which mediate MDR by active efflux of its substrate drugs out of cancer cells, leading to sub-therapeutic level of the drugs (Szakács et al., 2006). The main ABC transporters associated with MDR are P-glycoprotein (P-gp/ABCB1), multidrug resistance protein 1 (MRP1/ABCC1), and breast cancer resistance protein (BCRP/ABCG2).
In humans, MRP1 is present in epithelial cells of organs such as the lung, gastrointestinal tract, kidney, and adrenal gland (Flens et al., 1996) and is mainly localized at the basolateral membrane. Consequently, MRP1 plays an important role in the absorption and disposition of a remarkably diverse set of substrates across different organs and physiologic barriers (Leier et al., 1994; Schinkel and Jonker, 2012). What makes MRP1 relevant in MDR, however, is its ability to efflux cytotoxic anti-cancer agents such as doxorubicin, vincristine, and methotrexate (Cole, 2014). Overexpression of MRP1 has been associated with MDR in lung, breast, and prostate cancers, and several types of leukemia (Burger et al., 1994; Nooter et al., 1996; Sullivan et al., 1998; Filipits et al., 2005). In clinics, MRP1 overexpression determines poor prognosis in a number of cancers. In patients with localized high-risk soft tissue sarcoma of limbs and trunk wall treated with anthracycline-based chemotherapy, MRP1 overexpression has been shown to be an independent prognostic factor for relapse-free survival and overall survival (Martin-Broto et al., 2014). Similarly, a large prospective study of primary neuroblastoma has shown that MRP1 overexpression is highly predictive of event-free survival and overall survival (Haber et al., 2006).
Collateral sensitivity is a phenomenon in which the development of resistance toward a cytotoxic agent in the cells simultaneously confers a greater sensitivity to an alternate agent (Szakács et al., 2014). Collateral sensitivity is observed in cell lines overexpressing P-gp, MRP1, and BCRP, and the possibility of exploiting this trait in clinical cancer chemotherapy is being actively explored (Szakács et al., 2014). The underlying mechanisms that mediate collateral sensitivity are yet to be delineated but several putative mechanisms have been proposed, including the generation of reactive oxygen species, change in cellular energy levels, extrusion of essential endogenous substrate, and membrane perturbation in the resistant cells (Pluchino et al., 2012).
Calcitriol (1,25-dihydroxyvitamin D3), the active metabolite of vitamin D3, is a potent hormone that regulates numerous physiologic processes in human body. Traditionally, calcitriol is recognized for its role in bone health through action on calcium and phosphorus absorption (Holick, 2007). In recent years, however, accumulating data has indicated its non-skeletal functions in many chronic diseases (Holick, 2007). In cancer, multiple lines of evidence from epidemiologic and preclinical studies generally suggest a positive role of calcitriol in reducing cancer risk and progression, but evidence from randomized clinical trials has been lacking or inconclusive (Kupferschmidt, 2012; Feldman et al., 2014). Nevertheless, the interest in utilizing calcitriol for the prevention and improvement of cancer and other diseases remains high and a number of large-scale clinical trials are currently underway to determine the effects of calcitriol on these major diseases (Kupferschmidt, 2012).
Previously, we engineered a “two-color MRP1” by fusing green fluorescent protein (GFP) and a red fluorescent protein to the nucleotide-binding domains of MRP1 (Iram et al., 2015). The genetically encoded two-color MRP1 recombinant protein can measure intramolecular fluorescence resonance energy transfer (FRET) efficiency as an index of protein conformational changes upon ligand binding in live cells. Using this approach, a compound library was screened and eight compounds were identified that directly interact, bind and induce a conformational change in the structure of MRP1 (Iram et al., 2015). In the present work, we evaluated the effects of the hit compounds on MRP1 transport activity using different substrates of MRP1 in various cell- and membrane-based assays. We placed our focus on calcitriol and calcipotriol after initial characterization owing to their strong modulatory effects on MRP1 and discovered their selective cytotoxic effect on MRP1-expressing cells.
Materials and Methods
Chemicals.
Calcitriol, calcipotriol, MK571, epigallocatechin gallate (EGCG), hyperoside, LTC4, meropenem, mesalamine, nitazoxanide, and vincristine were purchased from Cayman Chemical (Ann Arbor, MI). AMP, ATP, doxorubicin, estradiol 17-(β-D-glucuronide) (E217βG), mitoxantrone, poly-D-lysine, thiazolyl blue tetrazolium bromide (MTT), and verapamil were procured from Sigma-Aldrich (St. Louis, MO). Apigenin and Ko143 were purchased from Tocris Bioscience (Avonmouth, Bristol, UK). Calcein-AM was obtained from Corning Life Sciences (Corning, NY). [6,7-3H]E217βG (49.9 Ci mmol−1) and [14, 15, 19, 20-3H]LTC4 (177.6 Ci mmol−1) were purchased from PerkinElmer (Waltham, MA).
Cell Lines and Cell Culture.
H69 and H69AR cells were procured from ATCC (Manassas, VA). HEK293T cells were a gift from Dr. Adam Hoppe (SDSU, Brookings, SD). HEK293/pcDNA3.1, HEK293/MRP1, HEK293/P-gp, and HEK293/BCRP were kindly gifted by Dr. Suresh V. Ambudkar (NIH, Bethesda, MD) and were maintained as described before (Müller et al., 2002). MDCKII, MDCKII/P-gp, and MDCKII/BCRP cells were kindly provided by Dr. Alfred Schinkel (Netherlands Cancer Institute, Amsterdam, The Netherlands). HEK293 and MDCKII cell lines were grown in DMEM (GE Healthcare, Marlborough, MA) supplemented with 10% FBS. H69 cell lines were cultured in RPMI 1640 (ATCC) media supplemented with 10% FBS. H69AR cells were monthly exposed to 0.8 µM doxorubicin and cultured without drug for 1 week before use in experiments. Cells were cultured at 37°C in a humidified incubator set at 5% CO2.
Western Blot Analysis.
Cell lysates were prepared in RIPA buffer (ThermoFisher Scientific, Waltham, MA) supplemented with 1× Halt Protease Inhibitor Cocktail (ThermoFisher Scientific). Protein concentration was determined using Pierce BCA Protein Assay (ThermoFisher Scientific). Cell lysates (20 µg protein) were electrophoresed on 7.5% Mini-PROTEAN TGX gels (BioRad, Hercules, CA) and transferred onto Immobilon PVDF membranes (EMD Millipore, Burlington, MA). Membranes were blocked for 1 hour at room temperature and incubated overnight at 4°C with monoclonal anti-MRP1 antibody (IU5C1; ThermoFisher Scientific) or anti-α-tubulin antibody (Sigma-Aldrich) at 1:250 and 1:5000 dilutions, respectively. Secondary antibody incubation was performed using horseradish peroxidase-conjugated goat anti-mouse IgG (H + L) (ThermoFisher Scientific) for 1 hour at room temperature. Target proteins were detected using Western Lightning Plus-ECL, Enhanced Chemiluminescence Substrate (PerkinElmer) and an OMEGA LUM G Imaging System (Aplegen, Pleasanton, CA). For protein expression comparison, protein band density was analyzed using the Image Studio Lite (LI-COR Biotechnology, Lincoln, NE) software and corrected for uneven sample loading and transfer using α-tubulin as the loading control.
Calcein, Mitoxantrone and Doxorubicin Accumulation Assay.
Calcein was used as the fluorescent substrate for accumulation assays studying MRP1 and P-gp, whereas mitoxantrone was used for BCRP {Peterson, 2017 #112}. Excitation was performed at 488 nm for calcein and mitoxantrone, with emission bands of 533/30 and >670 nm, respectively. Fluorescence intensity was represented as the mean value of 10,000 events. Experiments were done in duplicate. Doxorubicin accumulation assay was performed as described previously {Peterson, 2017 #112}.
Cytotoxicity Assay.
Cell sensitivity to test compounds was analyzed using the MTT colorimetric assay. HEK293 and H69 cell lines were plated in 96-well plates (CellBIND; Corning) at 5 × 103 and 2.5 × 104 cells per well, respectively, in 100 µl culture medium. The next day, 100 µl of test compound of varying concentrations prepared in culture medium were added to the cells. DMSO concentration was maintained at 0.5%. For drug combination treatment in HEK293/P-gp and HEK293/BCRP cells, 50 µl of calcitriol, calcipotriol, or control compounds were added and incubated for 1 hour before 50 µl of cytotoxic drugs (vincristine or mitoxantrone) were added. Cells were incubated for 72 (HEK293 cell lines) or 96 hour (H69 cell lines). At the end of the incubation period, 100 µl of culture medium was carefully removed and cells were treated with MTT (0.45 mg/ml) for 4 hour. The formazan crystals were dissolved by the addition of 100 µl 15% SDS containing 10 mM HCl and absorbance at 570 nm were recorded using a Hidex Sense Beta Plus plate reader (Turku, Finland).
Membrane Vesicular Transport Assay.
Plasma membrane vesicles were prepared as previously described with modifications {Iram and Cole, 2011; Peterson, 2017 #112 and Loe et al., 1996 #35}. ATP-dependent transport of [3H]E217βG and [3H]LTC4 into the inside-out membrane vesicles was measured by a rapid filtration technique {Loe et al., 1996 #35 and Peterson, 2017 #112}.
RNA Isolation and qPCR Analysis.
Cells treated with calcitriol or calcipotriol for 72 hour were pelleted by centrifugation and stored for RNA isolation. Total RNA was isolated using the Ambion TRIzol plus RNA purification kit (ThermoFisher Scientific). To rid the freshly prepared total RNA of possible DNA contamination, DNase1 enzyme treatment was performed using the Turbo DNA-free kit (ThermoFisher Scientific). The concentration and integrity of RNA was determined using the 260/280 ratios generated by a Nano-drop UV spectrophotometer. cDNA synthesis was done with the recombinant M-MuLV-based ProtoScript II First strand cDNA synthesis kit (New England Biolabs, Ipswich, MA) using 1 µg of the total RNA as starting material. Gene-specific primers were designed using the Primer3 (NIH) and synthesized by Eurofins Scientific (Louisville, KY). Table 1 shows the list of primer sequences. qPCR was performed using Luna Universal Probe qPCR Master Mix (New England Biolabs) according to the manufacturer’s instructions in a QuantStudio3 Real-Time PCR System (Thermo Fisher Scientific). The three-step PCR conditions used are as follows: 95°C denaturation for 1.2 minute, 45 cycles of (annealing at 60°C for 20 second, extension at 72°C for 30 second), and denaturation for 10 second. This was followed by melting curve stage from 60 to 95°C for 20 second while scanning for fluorescence. Relative quantitation was performed using the 2 −∆∆Ct method and data were normalized against housekeeping genes, β-actin and ubiquitin conjugating enzyme (UbCE), of respective samples. The relative messenger RNA (mRNA) expression level was calculated as a fraction of the untreated cells.
Primer sequences
Statistical Analysis.
Statistical analyses were done with IBM SPSS Statistics 19 (IBM, Armonk, NY). The differences between mean values were determined with linear mixed model analysis. For multiple comparisons, Sidak correction was performed.
Results
Effects of Test Compounds on MRP1 Activity in Cell-Based Assays.
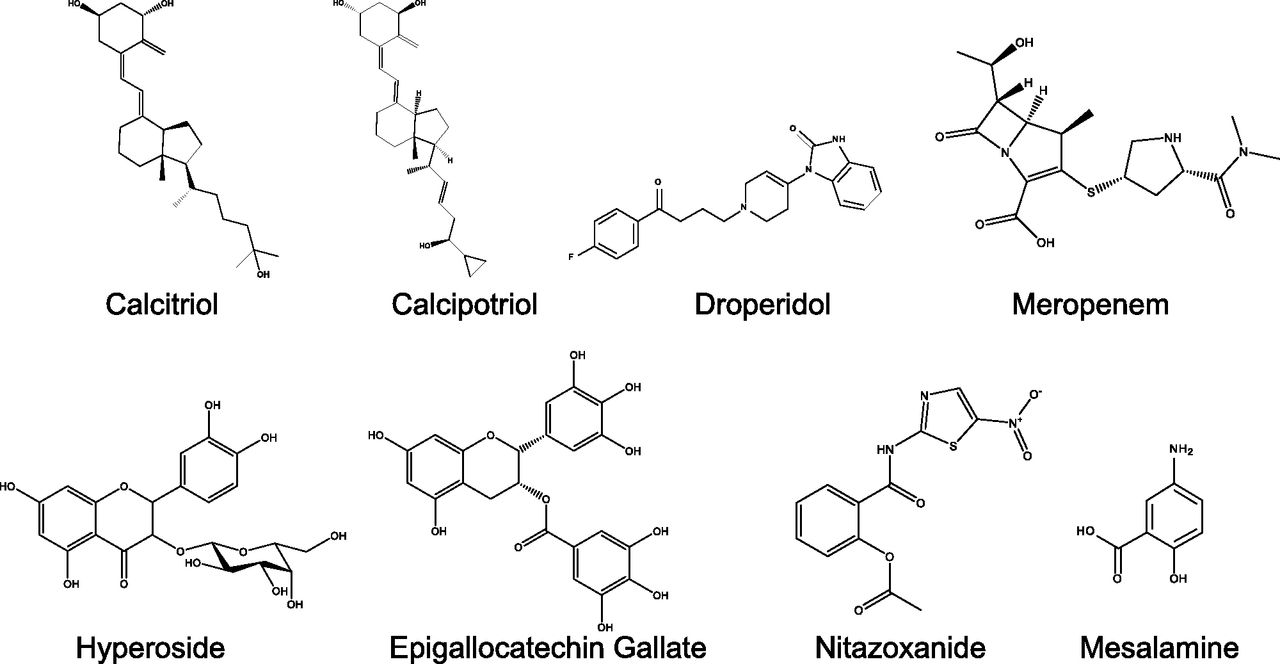
We previously engineered a two-color MRP1 protein that reports intramolecular FRET changes as an index of structural changes in the nucleotide binding domains of MRP1 (Iram et al., 2015). Specifically, a structural change that brings the nucleotide binding domains closer together increases FRET efficiency from a basal to higher level, reflecting the closer proximity of the fluorescent tags. A FRET-based screening of the NIH Clinical Collection compound library identified eight hits that significantly altered the fluorescent lifetime of the donor (Iram et al., 2015). The hit compounds included EGCG (antioxidant), hyperoside (antibiotic and antioxidant), calcitriol and calcipotriol (vitamin D analogs), mesalamine (anti-inflammatory), meropenem (antibiotic), nitazoxanide (antiprotozoal), and droperidol (antipsychotic) (Fig. 1). In the present study, we employed different cell- and membrane-based functional assays to further characterize the effects of these compounds on MRP1 activity.

Chemical structures of the test compounds.
To investigate the interaction of the test compounds with MRP1, we used MRP1-overexpressing cell lines H69AR and HEK293/MRP1. Expression levels of MRP1 in these cell lines as compared with their respective parental cell lines was verified by immunoblot analysis using whole cell lysates as shown in Fig. 2A. As expected, high levels of MRP1 were present in H69AR and HEK293/MRP1 cells, whereas the endogenous levels of MRP1 in the respective parental cell lines were not detectable. Similar levels of α-tubulin in all lysate samples demonstrated equal sample loading. First, we performed calcein efflux assay to determine the inhibitory potency of the test compounds. Calcein-AM, a well-known substrate for P-gp and MRP1, is a cell permeable and non-fluorescent compound that turns into highly fluorescent molecule after the acetoxymethyl ester (AM) moiety is cleaved off by cellular esterases. The intracellular accumulation of fluorescent calcein with or without the presence of test compounds was detected using flow cytometry. As shown in Fig. 2B, accumulation of calcein was enhanced ∼10-fold in H69AR cells with MK571 treatment (50 µM), a widely used MRP1 inhibitor. Calcitriol and calcipotriol (50 µM) increased calcein accumulation to levels comparable to that of MK571. In contrast, droperidol, meropenem, nitazoxanide, hyperoside, EGCG, and mesalamine did not significantly increase calcein accumulation in H69AR cells. On the other hand, no modulatory effect was observed for any of the test compound in the parental H69 cell lines. We also performed the calcein efflux assay using HEK293 cells. In HEK293/MRP1 cells, treatment with MK571 resulted in ∼fourfold increase of calcein accumulation (Fig. 2C). Similar calcein accumulation was observed with calcitriol and calcipotriol treatment, whereas a ∼2.5-fold enhancement in calcein accumulation was seen in treatment with droperidol. Meropenem, nitazoxanide, hyperoside, EGCG, and mesalamine treatments did not exhibit a significant effect on calcein accumulation in HEK293/MRP1 cells. As expected, no or very weak modulatory effect was observed for the test compounds in the control HEK293/pcDNA3.1 cell lines.

Effects of test compounds on MRP1 activity in cell-based assays. (A) Immunoblot analysis of whole cell lysates prepared from indicated cell lines was performed as described in the Materials and Methods section. (B and C) Calcein accumulation assay. Control and MRP1-overexpressing cells were pre-treated with 50 μM MK571 or test compounds for 10 minute before incubation with 25 nM calcein-AM at 37°C for 30 minute. Fluorescence intensity of intracellular calcein was detected using flow cytometry, with excitation and emission wavelengths of 480 and 533/30 nm, respectively. Data are combined from three experiments (performed in duplicate) and presented as mean ± S.E.M. *P < 0.05; ***P < 0.001 compared with control of each cell line, calculated using linear mixed model and Sidak post hoc test. (D) Doxorubicin accumulation assay. HEK293T cells transiently transfected with MRP1-GFP (green) were pre-treated with 50 μM test compounds, before incubation with doxorubicin (red) at 37°C for 1 hour. Images were acquired using confocal microscopy. GFP and doxorubicin were excited at 488 nm, and detected at 475/42 and 605/64 nm, respectively.
Next, the effects of the hit compounds on MRP1 transport activity were evaluated by detecting the accumulation of the fluorescent anti-cancer drug, doxorubicin, a well-known substrate of MRP1. HEK293T cells were transiently transfected with MRP1-GFP vector and confocal microscopy was used to visualize the inhibitory effect of the test compounds on MRP1-mediated doxorubicin efflux in live cells. As shown in Fig. 2D, cells treated with DMSO (vehicle) showed high doxorubicin accumulation in the nuclei of non-transfected cells, while doxorubicin fluorescence was very low or undetectable in cells expressing MRP1-GFP. MRP1-mediated efflux of doxorubicin was blocked by MK571 treatment (50 µM). Among the test compounds (50 µM), calcitriol, calcipotriol, and droperidol treatments exhibited high nuclear accumulation of doxorubicin similar to MK571 treatment. In contrast, treatment with meropenem, nitazoxanide, hyperoside, EGCG, or mesalamine showed very low or undetectable levels of doxorubicin in the nuclei of MRP1-GFP expressing cells, although MRP1 was clearly localized at the plasma membrane.
Effects of Test Compounds on MRP1 Activity in Membrane Vesicle-Based Assays.
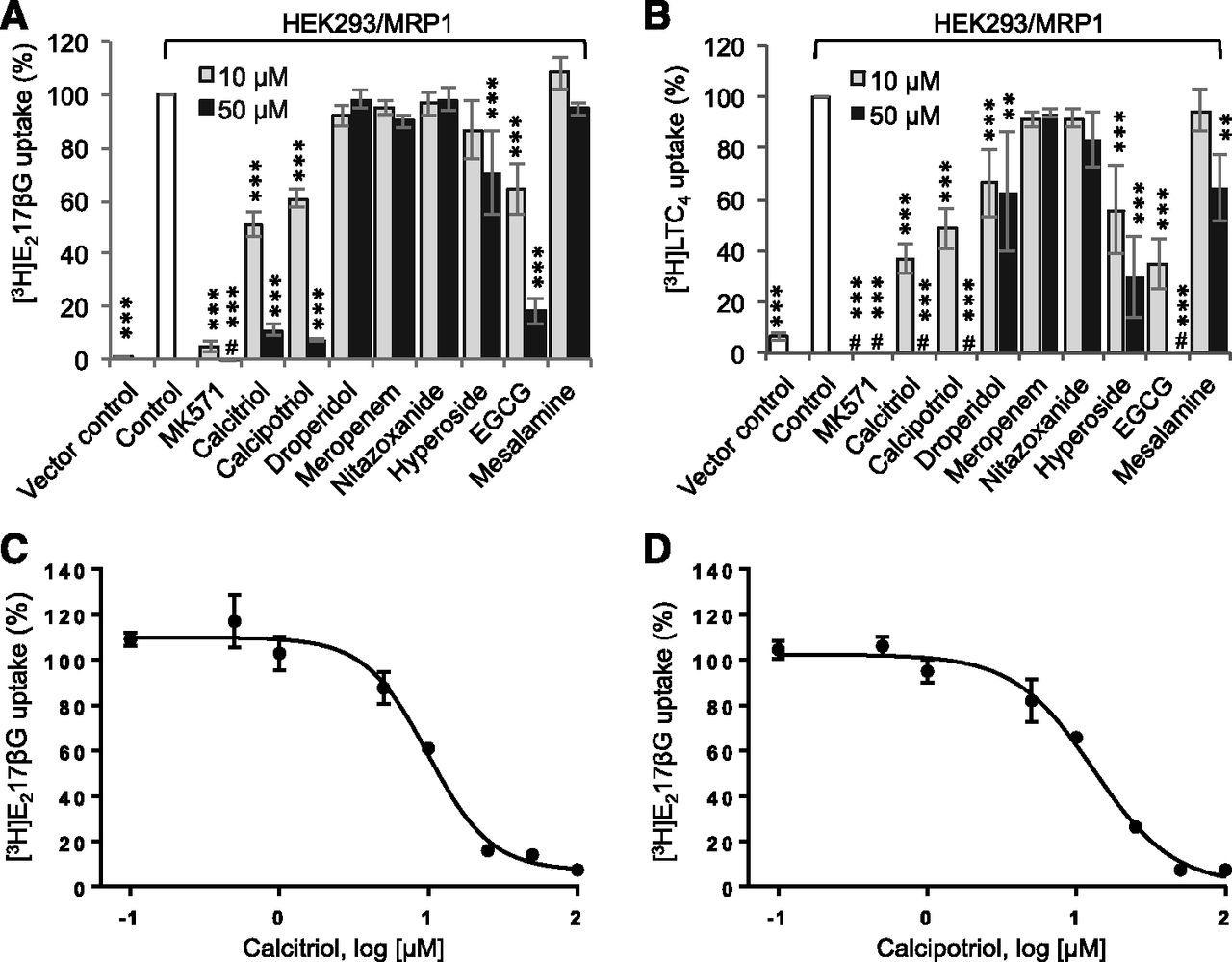
Apart from the abovementioned cell-based assays, we also employed membrane-based vesicular uptake assay to evaluate the effects of test compounds on MRP1-mediated transport of endogenous organic anions. E217βG (a glucuronide conjugate) and LTC4 (a glutathione conjugate) are prototypical substrates of MRP1. Membrane vesicles were prepared from HEK293/pcDNA3.1 and HEK293/MRP1 cells. Transport activities were measured using [3H]E217βG and [3H]LTC4 as MRP1 substrates, and test compound treatment was performed at 10 and 50 µM. As shown in Fig. 3A, transport of [3H]E217βG was reduced by ∼95% with 10 µM MK571 treatment and no active transport was detectable with 50 µM MK571 treatment. Among the test compounds, calcitriol, calcipotriol, and EGCG displayed significant inhibitory activity. For instance, 10 µM treatment with calcitriol, calcipotriol, or EGCG decreased [3H]E217βG transport by ∼50%, 40%, or 35%, respectively (Fig. 3A). In contrast, no significant [3H]E217βG transport inhibition was observed with 10 µM treatments of droperidol, meropenem, nitazoxanide, hyperoside, or mesalamine. Similarly, when MRP1-mediated [3H]LTC4 transport activity was measured, 10 µM treatment with calcitriol, calcipotriol, droperidol, hyperoside, or EGCG decreased transport by ∼65%, 50%, 35%, 45%, or 65%, respectively (Fig. 3B). Among the hit compounds, calcitriol and calcipotriol exhibited strong inhibition for all tested substrates in cell- and membrane-based assays. Therefore, we performed concentration-dependent inhibition experiments to determine their relative potencies. The concentration-response curves generated using [3H]E217βG as substrate showed a typical sigmoidal shape (Fig. 3, C and D), with IC50 values of 8.6 ± 1.4 and 12.2 ± 0.8 µM for calcitriol and calcipotriol, respectively (Table 2).

Effects of test compounds on MRP1 activity in membrane vesicle-based assays. (A and B) Inside-out membrane vesicular uptake assay. Membrane vesicles (2 µg protein) prepared from HEK293/pcDNA3.1 (vector control) and HEK293/MRP1 cells were incubated with 10 or 50 μM test compounds and (A) 400 nM/20 nCi [3H]E217βG or (B) 50 nM/2 nCi [3H]LTC4 at 37°C for 1 minute. MRP1 uptake activity was determined by measuring the radioactivity retained on collected membrane vesicles using liquid scintillation counting. Data are combined from ≥3 experiments (performed in triplicate) and presented as mean ± S.E.M. #, value below background; **P < 0.01; ***P < 0.001 compared with control, calculated using linear mixed model and Sidak post hoc test. Concentration-dependent inhibition of MRP1 activity by calcitriol (C) and calcipotriol (D). MRP1-mediated uptake of [3H]E217βG was performed by incubating HEK293/MRP1 membrane vesicles (2 µg protein) with various concentrations of calcitriol or calcipotriol. Data are representative of three experiments and presented as mean ± S.D. (n = 3).
Reaction parameters derived from the cytotoxicity and membrane vesicular transport assays
Collateral Sensitivity of MRP1-Overexpressing Cells Toward Calcitriol and Calcipotriol.
Based on the strong modulatory effect observed for calcitriol and calcipotriol in both the cell- and membrane-based assays, we decided to focus on these vitamin D3 analogs in our further studies. Additionally, the importance of calcitriol as an essential hormone and the strong interest of applying calcitriol in the clinical settings of various major diseases including cancer prompted us to further investigate their interactions with MRP1. Initially, we intended to test their ability to reverse MRP1-related drug resistance toward anti-cancer agents such as etoposide and vincristine using the MTT cytotoxicity assay. However, in our preliminary studies to determine an appropriate concentration to be used in the resistance reversal assay, we unexpectedly observed a hypersensitivity of MRP1-overexpressing cells toward calcitriol and calcipotriol. Data from the subsequent cytotoxic studies revealed a concentration-dependent cytotoxic effect of calcitriol and calcipotriol, with IC50 values of 11.0 ± 0.7 and 8.5 ± 1.0 µM, respectively, in MRP1 over-expressing H69AR cells (Fig. 4A; Table 2). In contrast, we observed ∼threefold higher IC50 values for calcitriol and calcipotriol in the parental H69 cells. In HEK293/MRP1 cells, calcitriol and calcipotriol also exhibited a concentration-dependent cytotoxic effect, with IC50 values of 8.9 ± 1.3 and 6.0 ± 1.8 µM, respectively, which are ∼fourfold lower than those observed in the HEK293/pcDNA3.1 control cells (Fig. 4B; Table 2). To test whether the observed collateral sensitivity is dependent on MRP1 transport function, we measured the cytotoxicity of calcitriol and calcipotriol treatments in MRP1 over-expressing H69AR and HEK293/MRP1 cells in the presence of 10 and 50 µM MK571 (MRP1 inhibitor). Our results showed that collateral sensitivity of MRP1 over-expressing cell lines toward calcitriol and calcipotriol can be eliminated by MRP1 inhibitor MK571 (Fig. 4; Table 2). By using inhibitor at 10 µM (partial MRP1 inhibition) and 50 µM (complete MRP1 inhibition) the data indicated that collateral sensitivity is proportional to the amount of active MRP1.

Collateral sensitivity of MRP1-overexpressing cells toward calcitriol and calcipotriol. H69 and H69AR cells were treated with increasing concentrations of calcitriol or calcipotriol in the absence or presence of MK571 for 96 hour (A). Similar experiments were performed using calcitriol or calcipotriol in HEK293/pcDNA3.1 and HEK293/MRP1 cells in the absence or presence of MK571 for 72 hour (B). Cell viability was evaluated with MTT assay. Data are representative of three experiments and expressed as mean ± S.D. (n = 3).
Effects of Calcitriol and Calcipotriol on MRP1 mRNA and Protein Expression Levels.
To investigate if calcitriol and calcipotriol can affect the mRNA and protein expression levels of MRP1, we performed qPCR and Western blot analysis on HEK293/MRP1 and H69AR treated with calcitriol and calcipotriol at 1 and 10 µM. The mRNA and protein expression levels of MRP1 in both cell lines were not significantly altered by 1 μM calcitriol and calcipotriol (data not shown). However, at 10 µM concentration, calcitriol treatment increased the mRNA levels of MRP1 in HEK293/MRP1 cells but not in H69AR cells (Fig. 5A). In contrast, at 10 µM concentration, calcipotriol treatment increased the mRNA levels of MRP1 in H69AR cells but not in HEK293/MRP1 cells. As shown in Fig. 5C, protein expression levels of MRP1 were decreased significantly in H69AR cells but not in HEK/MRP1 cells with 10 µM calcitriol treatment. However, calcipotriol treatment at 10 µM concentration did not significantly change MRP1 expression levels in both cell lines.
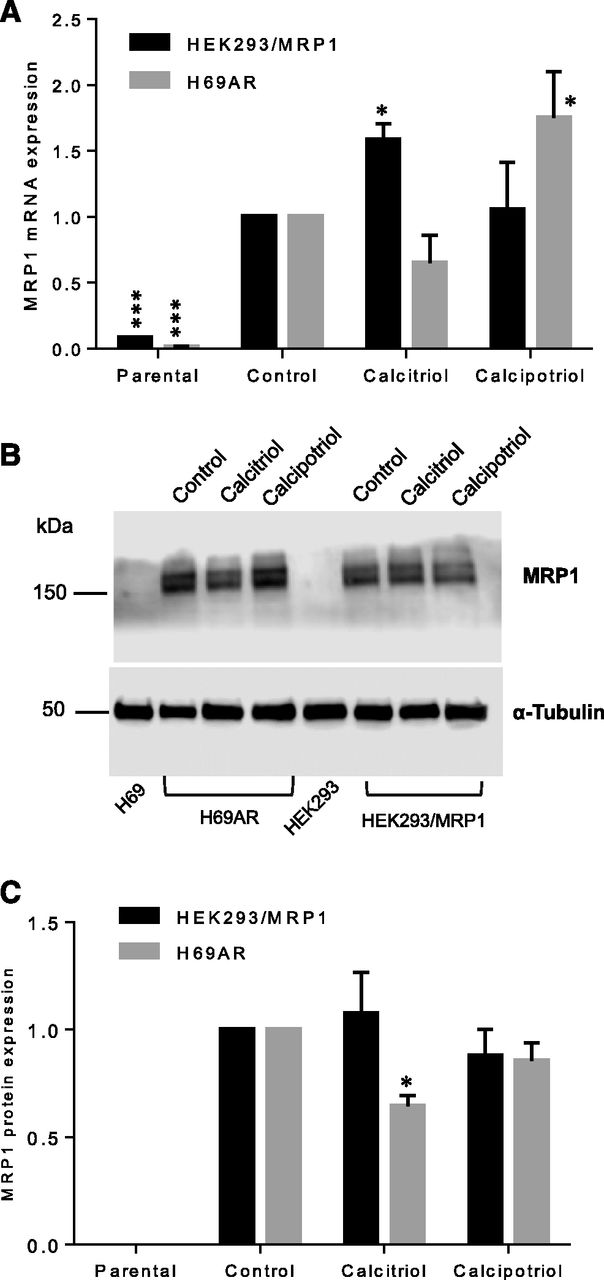
Effects of calcitriol and calcipotriol on the mRNA and protein expression of MRP1. Cells were treated with DMSO, calcitriol, or calcipotriol (10 µM) for 72 hour. (A) mRNA levels of parental (HEK293 and H69) cells and MRP1-overexpressing cells (HEK293/MRP1 and H69AR) were quantified using qPCR as described in the Materials and Methods section. Data are combined from three experiments (each performed in triplicate) and presented as mean ± S.E.M. *P < 0.05; ***P < 0.001 compared with control, calculated using linear mixed model and Sidak post hoc test. (B) Immunoblot analysis of whole cell lysates prepared from indicated cell lines with indicated treatments was performed as described in the Materials and Methods section. Data are representative of three experiments. Mean MRP1 protein expression (±S.E.M., n = 3) shown as a fold change of the DMSO-treated control is shown in (C). Protein band density was analyzed using the Image Studio Lite (LI-COR Biotechnology) software and corrected for uneven sample loading and transfer using α-tubulin as the loading control. *P < 0.05; ***P < 0.001 compared with control, calculated using linear mixed model and Sidak post hoc test.
Inhibition of P-gp and BCRP Transport Activity by Calcitriol and Calcipotriol.
Calcitriol and calcipotriol have not been reported to directly interact with ABC transporters. Therefore, we were interested to investigate if they interact with P-gp and BCRP. We first tested if calcitriol and calcipotriol can modulate the transport function of P-gp and BCRP. To this end, we examined the accumulation of calcein (a P-gp substrate) and mitoxantrone (a BCRP substrate) in P-gp- and BCRP-overexpressing cells, respectively, using flow cytometry. As shown in Fig. 6, A and B, treatment with 25 µM verapamil, a standard P-gp inhibitor, resulted in enhancement of calcein accumulation by 21- and 14-fold in HEK293/P-gp and MDCKII/P-gp cells, respectively. Calcitriol at 10 and 50 µM increased calcein accumulation in HEK293/P-gp cells by 2.3- and 7.8-fold, and in MDCKII/P-gp cells by 2.0- and 7.6-fold, respectively. Similarly, calcipotriol at 10 and 50 µM enhanced calcein accumulation by 2.8- and 7.5-fold in HEK293/P-gp cells, and 3.2- and 7.3-fold in MDCKII/P-gp cells, respectively. On the other hand, no or very weak modulatory effect was observed for calcitriol or calcipotriol in both parental cell lines. Next, the inhibitory effect of calcitriol and calcipotriol on BCRP-mediated efflux was tested. As illustrated in Fig. 6, C and D, treatment with 5 µM Ko143, a widely used BCRP inhibitor, enhanced mitoxantrone accumulation by 5.1- and 5.7-fold in HEK293/BCRP and MDCKII/BCRP cells, respectively. Calcitriol at 10 and 50 µM increased mitoxantrone accumulation in HEK293/BCRP cells by 4.3- and 5.9-fold, and in MDCKII/BCRP cells by 3.0- and 5.3-fold, respectively. Likewise, calcipotriol at 10 and 50 µM resulted in higher mitoxantrone accumulation by 4.6- and 7.0-fold in HEK293/BCRP cells, and 3.4- and 6.2-fold in MDCKII/BCRP cells, respectively. As expected, no or very weak modulatory effect was observed for calcitriol or calcipotriol in both parental cell lines.

Calcitriol and calcipotriol inhibit P-gp- and BCRP-mediated efflux. Control, P-gp-overexpressing HEK293 (A) or MDCKII (B), and BCRP-overexpressing HEK293 (C) or MDCKII (D) cells were pre-treated with calcitriol and calcipotriol at 37°C for 10 minute before incubation with 25 nM calcein-AM (A and B) or 5 µM mitoxantrone (C and D) for 30 minute. Verapamil and Ko143 were used as positive controls for P-gp and BCRP inhibition, respectively. Intracellular accumulation of calcein and mitoxantrone was quantified by flow cytometry. Data are combined from three experiments (performed in duplicate) and presented as mean ± S.E.M. *P < 0.05; **P < 0.01; ***P < 0.001 compared with control, calculated using linear mixed model and Sidak post hoc test.
Lack of Collateral Sensitivity in P-gp- and BCRP-Overexpressing Cells Toward Calcitriol and Calcipotriol.
Collateral sensitivity has been observed in cells overexpressing P-gp and BCRP (Szakács et al., 2014). To investigate if the collateral sensitivity effect observed for MRP1-overexpressing cells toward calcitriol and calcipotriol is also inherent in cells overexpressing P-gp and BCRP, two other ABC transporters that are prominently involved in MDR, we examined the cytotoxic effect of calcitriol and calcipotriol on HEK293 cells overexpressing P-gp and BCRP. As shown in Fig. 7, the sensitivity of HEK293/P-gp and HEK293/BCRP cells toward calcitriol and calcipotriol was similar to that of HEK293/pcDNA3.1 cells. The lack of sensitivity toward calcitriol and calcipotriol is also reflected in their relatively close or higher IC50 values as compared with that of HEK293/pcDNA3.1 cells (Table 2).

P-gp- and BCRP-overexpressing cells do not show collateral sensitivity toward calcitriol and calcipotriol. HEK293/pcDNA3.1, HEK293/P-gp, and HEK293/BCRP cells were treated with increasing concentrations of calcitriol (A) or calcipotriol (B) for 72 hour. Cell viability was evaluated with MTT assay. Data are representative of three experiments and expressed as mean ± S.D. (n = 3).
Effects of Calcitriol and Calcipotriol on Drug Sensitivity of HEK293/P-gp and HEK293/BCRP Cells.
Following the observation that calcitriol and calcipotriol do not cause collateral sensitivity in HEK293/P-gp and HEK293/BCRP cells, we investigated if calcitriol and calcipotriol can reverse drug resistance of the cells toward cytotoxic P-gp and BCRP substrates. HEK293/P-gp and HEK293/BCRP cells were treated with anticancer drugs vincristine and mitoxantrone, respectively, in the presence and absence of 10 µM calcitriol and calcipotriol. Verapamil (25 µM) and Ko143 (1 µM) were used as positive controls for P-gp and BCRP, respectively. As shown in Fig. 8, HEK293/P-gp and HEK293/BCRP cells exhibited significant drug resistance as compared with the vector control cell line HEK293/pcDNA3.1. Treatment with verapamil and Ko143 in HEK293/P-gp and HEK293/BCRP cells, respectively, reversed the resistance of the cells toward the anticancer agents. IC50 values of the treatments are shown in Table 3. In both cell lines, co-treatment with 10 µM calcitriol significantly reversed the resistance of the cells to a certain extent. However, no significant reversal was observed in cells co-treated with 10 µM calcipotriol.
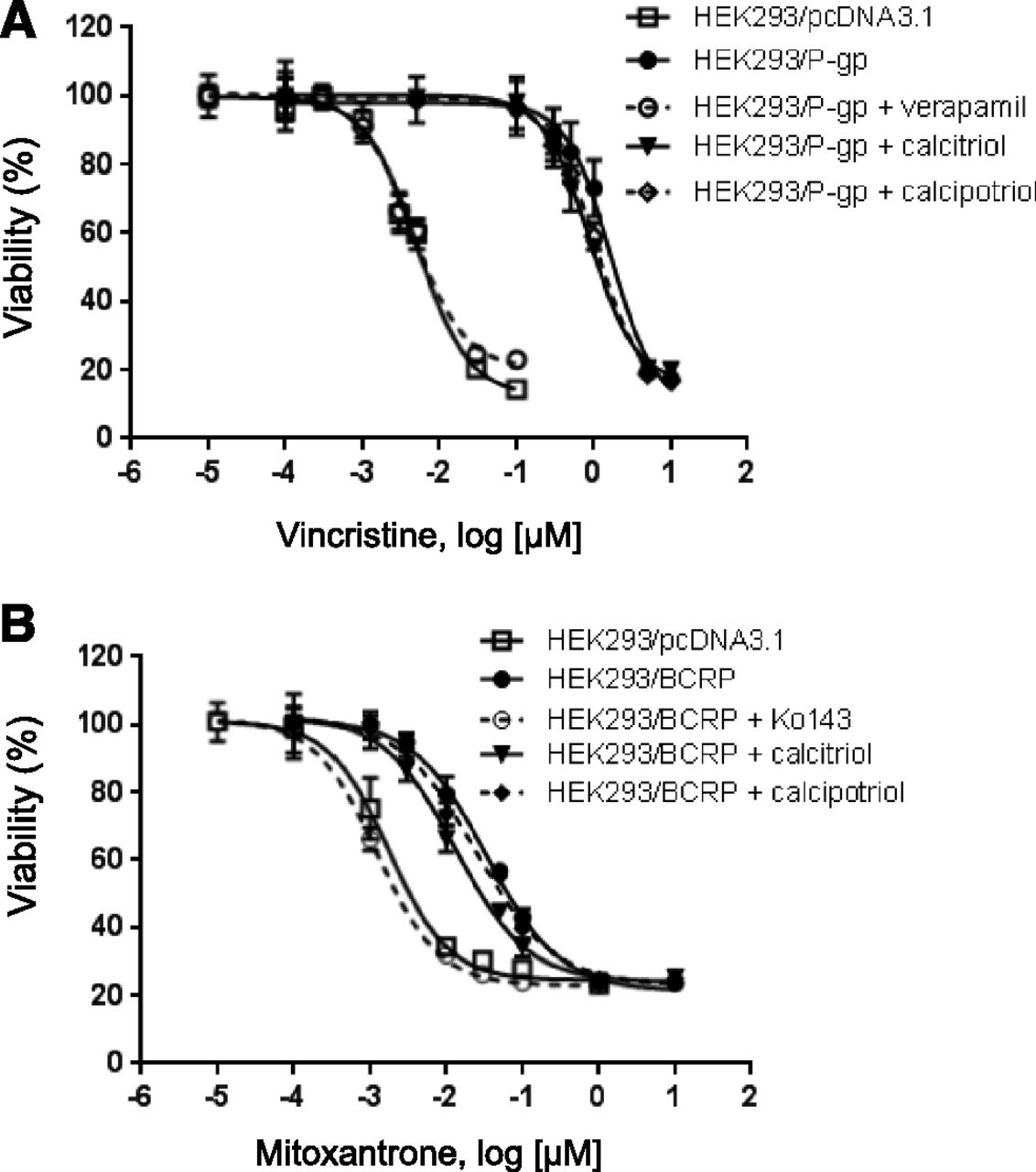
Effects of calcitriol and calcipotriol on drug sensitivity of HEK293/P-gp and HEK293/BCRP cells. HEK293/P-gp (A) and HEK293/BCRP (B) cells were treated with increasing concentrations of vincristine and mitoxantrone, respectively, in the absence and presence of calcitriol or calcipotriol (10 µM) for 72 hour. Verapamil (25 µM) and Ko143 (1 µM) were used as positive controls in HEK293/P-gp and HEK293/BCRP cells, respectively. HEK293/pcDNA cells were included as negative control for drug resistance. Cell viability was evaluated with MTT assay. Data are representative of three experiments and expressed as mean ± S.D. (n = 3).
Effect of calcitriol and calcipotriol (10 µM) on IC50 of vincristine and mitoxantrone in HEK293/P-gp and HEK293/BCRP, respectively
Verapamil (25 µM) and Ko143 (1 µM) are used as positive controls.
Discussion
In the present study, we report for the first time that the active metabolite of vitamin D3, calcitriol, and its analog calcipotriol, cause selective cytotoxicity in MRP1-overexpressing, but not P-gp- and BCRP-overexpressing cell lines. In addition, we demonstrate an inhibitory activity of calcitriol and calcipotriol on the transport function of MRP1, P-gp, and BCRP, which are prominently involved in MDR and drug disposition. Our results show that collateral sensitivity of MRP1 over-expressing cell lines toward calcitriol and calcipotriol can be eliminated by MRP1 inhibitor MK571. We initiated the present work as a follow up of our previous study, which used a genetically-engineered two-color MRP1 for high-throughput identification of potential MRP1 substrates and modulators (Iram et al., 2015). The two-color MRP1 reports ligand-induced structural changes through changes in intramolecular FRET efficiency. Such structural approach has also been successfully applied for the discovery of activator/inhibitor candidates of the P-type sarco/endoplasmic reticulum calcium ATPase (Gruber et al., 2014).
High throughput screening of the NIH Clinical Collection compound library using the MRP1 FRET-based reporter identified eight compounds that directly interact, bind and induce a conformational change in the structure of MRP1 (Iram et al., 2015). However, this FRET-based screening does not provide any information about the biochemical nature of interaction of the hit molecules with MRP1. The identified compounds from this screen could be a substrate, inhibitor, activator or a molecule that simply interacts with MRP1 to stimulate transport of a substrate or binds with the transporter for unknown reasons or could be a false hit due to non-specific interaction with MRP1. In the present work, we evaluated the effects of the hit compounds on MRP1 transport activity using different substrates of MRP1 in various cell- and membrane-based assays. Based on our results, we identified calcitriol and calcipotriol as inhibitors of MRP1. Both agents showed strong inhibition of MRP1 activity for all substrates tested. We also discovered that calcitriol and calcipotriol inhibit transport activity of P-gp and BCRP. However, further work is needed to determine whether calcitriol and calcipotriol are also substrates of these transporters. Droperidol exhibited both substrate selective and cell line specific inhibition of MRP1. It showed no inhibition of E217βG and LTC4 by MRP1 while strong inhibition was observed for the anti-cancer drug doxorubicin. There is plenty of data to indicate that MRP1 has multiple distinct substrate binding sites. Hydrophobic drugs are predicted to bind on the top half of the binding pocket while the anionic compounds are predicted to bind in the lower half closer to the membrane interface. Substrate selective inhibition of MRP1 by Droperidol is of high interest because it can block the efflux of cytotoxic drug doxorubicin to improve its efficacy while keeping the transport of important physiologic substrates (E217βG and LTC4) unaltered. Droperidol showed moderate inhibition of calcein efflux in HEK/MRP1 cells while no inhibition was observed for calcein efflux in H69AR cells. It is not uncommon to observe cell line specific effects of a drug. H69AR was derived by long-term exposure to a drug while HEK/MRP1 was developed by transfection and they can be very different in terms of their molecular profile and membrane dynamics. The flavonoids EGCG and hyperoside showed inhibition of E217βG and LTC4 transport by MRP1 while no inhibition was observed for calcein and doxorubicin (Figs. 2 and 3). EGCG is a known substrate of MRP1 (Hong et al., 2003). The inhibition of endogenous organic anion substrates E217βG and LTC4 by EGCG and hyperoside is probably due to transport competition when the two substrates compete for the same binding site or if the binding sites are overlapping. Mesalamine only showed weak inhibition for LTC4. Two of the hit compounds, nitazoxanide and meropenem, which did not show inhibition on any tested substrates, are anticipated to be substrates of MRP1 with a distinct substrate binding site. They could also be false hits from the FRET-based screening. Further work is needed to verify their interaction with MRP1.
Generally, among ABC drug transporters, P-gp is considered most relevant for clinical MDR. The traditional approach in targeting ABC transporter as a strategy for overcoming MDR in chemotherapy involves treatment with a combination of a cytotoxic anti-cancer agent and an inhibitor of P-gp as drug sensitizer. However, clinical trials conducted with P-gp inhibitors continued to yield negative outcomes, which lead to a general skepticism on the feasibility of the MDR reversal approach (Libby and Hromas, 2010; Tamaki et al., 2011). More recently, there is a strong interest in exploiting collateral sensitivity in cancer cells, a phenomenon in which drug resistant cells concomitantly develop hypersensitivity to alternate drugs (Szakács et al., 2014; Genoux-Bastide et al., 2011). Recent advances in the field have shown that collateral sensitivity in tumors can be temporal and/or persistent due to the dynamic nature of tumor clonal evolution and the sequential use of drugs targeting dynamic tumor vulnerabilities can, therefore, be a promising approach toward eliminating MDR (Colmegna et al., 2015; Zhao et al., 2016). In the present study, we show that MRP1-overexpressing H69AR and HEK293/MRP1 cells exhibit collateral sensitivity toward calcitriol and calcipotriol. Although both H69AR and HEK293/MRP1 cell lines over-express MRP1, the former was obtained under doxorubicin selection and may have other physiologic changes as well, whereas the later was obtained through stable transfection and likely purely expressing the transporter without other physiologic changes. Interestingly, this phenomenon appears to be specific to MRP1-overexpressing cells as the sensitivity was not observed in HEK293/P-gp or HEK293/BCRP cells. Furthermore, we found that this collateral sensitivity can be eliminated by MRP1 inhibitor MK571. There could be various possibilities how calcitriol and calcipotriol can be selectively cytotoxic to MRP1 over-expressing cells including stimulation of GSH efflux, up-regulation of apoptosis in a MRP1-dependant manner and assuming if these molecules are indeed substrates of MRP1 then they may bind something which is essential for the cell and the co-transport is toxic to cells (Laberge et al., 2007; Lorendeau et al., 2014; Trompier et al., 2004). These data provide a framework for further investigation into the use of vitamin D analogs in clinical chemotherapy, especially in cancers with prominent MRP1 expression, such as primary neuroblastoma (Haber et al., 2006; Yan and Nuriding, 2014).
Calcitriol is a multifunctional hormone that exerts its activity through the nuclear vitamin D receptor (VDR), which is a member of the nuclear receptor superfamily of ligand-activated transcription factors. Recent evidence has linked VDR to the upregulation of ABC transporter expression. In Caco-2 cells, which are commonly used as a physiologic barrier model for the small intestine, treatment with calcitriol activates the expression of P-gp, MRP2, and MRP4 through VDR (Fan et al., 2009). A study performed in conscious rats also reported that calcitriol-induced VDR activation increases cerebral P-gp expression and restricts brain distribution of P-gp substrates (Durk et al., 2015). While the longer treatment (typically 2 to 3 days) with calcitriol appears to upregulate the expression levels of ABC transporters and thus increase the overall efflux of substrate drugs, the immediate effect of calcitriol on the transport function of ABC transporter has not been reported. In the present study, we observed no significant alteration in MRP1 mRNA and protein expression with 2-day calcitriol or calcipotriol treatment at 1 µM concentration. However, at 10 µM concentration, we observed increased mRNA levels for MRP1 in HEK293/MRP1 cells, although the changes in protein levels were not significantly different. On the contrary, calcitriol treatment at 10 µM did not significant affect the mRNA levels but significantly reduced MRP1 protein levels in H69AR cells. Our results demonstrate a clear inhibitory effect of calcitriol and calcipotriol on the efflux activity of P-gp, MRP1, and BCRP. However, it is uncertain whether the observed inhibitory activity is implicated in pharmacokinetic interactions with substrate drugs of P-gp, MRP1, and BCRP in vivo. A phase I study using intravenous calcitriol in combination with oral dexamethasone and gefitinib in patients with advanced solid tumors reported calcitriol serum concentrations of 4–13 ng/ml (1–30 nM) when calcitriol was administered at doses in the range of 57–163 µg (Muindi et al., 2009). As we observed inhibitory effect of calcitriol and calcipotriol on P-gp, MRP1, and BCRP in the micromolar range, it appears unlikely for clinically administered calcitriol to exert significant inhibition on drug efflux transporters. Indeed, phase II and III clinical trials using high-dose concentrated calcitriol (DN-101) in combination with docetaxel, a known P-gp substrate, for patients with castration-resistant prostate cancer did not observe an increase in docetaxel toxicity (Beer et al., 2007; Scher et al., 2011). However, in these clinical trials, calcitriol was administered 1 day before docetaxel, which might have evaded P-gp-mediated drug-drug interaction. Therefore, the inhibitory effect of calcitriol on drug efflux transporters should be considered if calcitriol is co-administered at high doses with substrates of ABC transporters, as interference on transporter function remains possible.
Calcipotriol, a less calcemic analog of calcitriol, is a widely used first-line topical ointment for the treatment of psoriasis, a common and chronic immune-mediated skin disease (Menter et al., 2009; Dubertret et al., 1992). The mode of action of calcipotriol in alleviating psoriasis is not fully understood, but current knowledge has suggested that VDR agonists, like calcitriol, drive T cell population toward a T helper (Th) type 2 profile while inhibiting the Th1 and Th17 phenotypes, which are associated with the pathogenesis of psoriasis (Boonstra et al., 2001; Nestle et al., 2009). Th1 and Th17 cells are stimulated by proinflammatory cytokines including tumor necrosis factor-α, interleukin (IL)-12, and IL-23. Once activated, Th1 and Th17 cells secrete mediators such as tumor necrosis factor-α, IL-17A, and IL-22, which trigger the proliferation of keratinocytes and other cellular events leading to psoriasis (Nestle et al., 2009). Interestingly, keratinocyte proliferation is also stimulated by LTC4, a high affinity substrate of MRP1, in a mouse model of atopic dermatitis to promote hyperkeratosis and fibrosis (Oyoshi et al., 2012). It will be of interest to investigate the probable involvement of MRP1 in the pathogenesis of psoriasis, as inhibition of MRP1-mediated efflux of inflammatory mediators could be a mechanism by which calcipotriol exert its therapeutic effect. In our study calcipotriol treatment at 1 and 10 µM did not affect the MRP1 protein levels in HEK293/MRP1 and H69AR cells whereas our results demonstrate a clear inhibitory effect of calcipotriol on the efflux activity of MRP1, P-gp and BCRP.
In conclusion, this study brings a new perspective to the application of calcitriol and its analogs in cancer treatment. Specifically, our work suggests the potential use of calcitriol and its analogs in selectively targeting tumors with the MDR phenotype conferred by MRP1 overexpression and provides the rational for sequential use of calcitriol and other anti-cancer agents to circumvent the development of MRP1-mediated MDR in clinical chemotherapy. Future work should be focused on designing calcitriol analogs with improved potency and validating the feasibility of applying the MRP1-selective effect of calcitriol in vivo.
Acknowledgments
We thank Dr. Suresh V. Ambudkar (NIH), Dr. Alfred Schinkel, Piet Borst (The Netherlands Cancer Institute, the Netherlands), and Dr. Adam Hoppe (South Dakota State University, SD) for providing the cell lines used in this study. We also thank Dr. Adam Hoppe and members of his laboratory for assistance with confocal microscope and flow cytometer. S.H.I. is a member of BioSNTR.
Authorship Contributions
Participated in research design: Tan, Osa-Andrews, Iram.
Conducted experiments: Tan, Sampson, Osa-Andrews.
Contributed new reagents or analytic tools: Iram.
Performed data analysis: Tan, Sampson, Osa-Andrews.
Wrote or contributed to the writing of the manuscript: Tan, Sampson, Osa-Andrews, Iram.
Footnotes
- Received March 22, 2018.
- Accepted September 10, 2018.
This work is supported by National Science Foundation/EPSCoR Award No. IIA-1355423, by the state of South Dakota’s Governor’s Office of Economic Development as a South Dakota Research Innovation Center (SDRIC), and with financial/match commitment from all participating institutions. This work was also supported by Research/Scholarship Support Fund (SDSU), Scholarly Excellence Funds (SDSU), and Laboratory Start-up funds to S.H.I. S.H.I. is a member of BioSNTR.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Abbreviations
- ABC
- ATP-binding cassette
- BCRP
- breast cancer resistance protein
- E217βG
- 17β-estradiol-17-β-(D-glucuronide)
- EGCG
- epigallocatechin gallate
- FRET
- fluorescence resonance energy transfer
- GFP
- green fluorescent protein
- HEK293
- Human embryonic kidney 293
- IC50
- half-maximal inhibitory concentration
- LTC4
- cysteinyl leukotriene
- MDCKII
- Madin-Darby canine kidney II
- MDR
- multidrug resistance
- MRP1
- multidrug resistance protein 1
- MTT
- thiazolyl blue tetrazolium bromide
- P-gp
- P-glycoprotein
- Th
- T helper
- VDR
- vitamin D receptor
- Copyright © 2018 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}