


Abstract
Combination antiretroviral drug treatments depend on 3′-deoxy-nucleoside analogs such as 3′-azido-3′-deoxythymidine (AZT) and 2′3′-dideoxyinosine (DDI). Despite being effective in inhibiting human immunodeficiency virus replication, these drugs produce a range of toxicities, including myopathy, pancreatitis, neuropathy, and lactic acidosis, that are generally considered as sequelae to mitochondrial damage. Although cell surface–localized nucleoside transporters, such as human equilibrative nucleoside transporter 2 (hENT2) and human concentrative nucleoside transporter 1 (hCNT1), are known to increase the carrier-mediated uptake of 3′-deoxy-nucleoside analogs into cells, another ubiquitously expressed intracellular nucleoside transporter (namely, hENT3) has been implicated in the mitochondrial transport of 3′-deoxy-nucleoside analogs. Using site-directed mutagenesis, generation of chimeric hENTs, and 3H-permeant flux measurements in mutant/chimeric RNA–injected Xenopus oocytes, here we identified the molecular determinants of hENT3 that dictate membrane translocation of 3′-deoxy-nucleoside analogs. Our findings demonstrated that whereas hENT1 had no significant transport activity toward 3′-deoxy-nucleoside analogs, hENT3 was capable of transporting 3′-deoxy-nucleoside analogs similar to hENT2. Transport analyses of hENT3-hENT1 chimeric constructs demonstrated that the N-terminal half of hENT3 is primarily responsible for the hENT3–3′-deoxy-nucleoside analog interaction. In addition, mutagenic studies identified that 225D and 231L in the N-terminal half of hENT3 partially contribute to the ability of hENT3 to transport AZT and DDI. The identification of the transporter segment and amino acid residues that are important in hENT3 transport of 3′-deoxy-nucleoside analogs may present a possible mechanism for overcoming the adverse toxicities associated with 3′-deoxy-nucleoside analog treatment and may guide rational development of novel nucleoside analogs.
Introduction
Nucleoside analogs are widely used for the prevention and treatment of several viral infections, including human immunodeficiency virus, hepatitis B virus, and herpes infections (Albrecht et al., 1970; Burchenal et al., 1975; Aguirrebengoa et al., 1996; Adams et al., 1997; Bavoux et al., 2000; Quan and Peters, 2004; Anderson, 2008; Angusti et al., 2008; Ambrose et al., 2009; Billioud et al., 2011; Agarwal et al., 2015; Cao et al., 2015; Ehteshami et al., 2017). Due to their hydrophilic nature, nucleoside analogs depend on a carrier-mediated transport process to permeate through host cell membranes before exhibiting their antiviral activities (Yao et al., 1996; Damaraju et al., 2011; Moss et al., 2012). Indeed, numerous studies have suggested nucleoside transporters as one of the critical determinants that contribute to the effectiveness and toxicities of nucleoside analogs (Mangravite et al., 2003; Lang et al., 2004; Koczor et al., 2012). Nucleoside transporters belong to two gene families: namely, the solute carrier SLC28 and SLC29 families, which encode human concentrative and equilibrative nucleoside transporters (hCNTs and hENTs), respectively. The hCNT family has three members (hCNT1, hCNT2, and hCNT3) that are localized at the cell surface, whereas the hENT family is composed of four members (hENT1, hENT2, hENT3, and hENT4) that are either expressed at the cell surface (hENT1, hENT2, and hENT4) and in nuclear membranes (hENT1 and hENT2) or in organelle membranes (hENT3) (Baldwin et al., 2004; Gray et al., 2004; Pastor-Anglada et al., 2008; Young et al., 2008). hENT4 primarily transports monoamines (e.g., serotonin, dopamine) with poor affinity for nucleosides; hence, it has been renamed as the plasma membrane monoamine transporter (Zhou et al., 2007).
Research has shown that 3′-deoxy-nucleoside analogs such as 3′-azido-3′-deoxythymidine (AZT) and 2′3′-dideoxyinosine (DDI) are used in first-line antiretroviral therapy, mostly as combination agents with other protease inhibitors and novel targeted therapies (Maenza and Flexner, 1998). Both rat CNT1 and hENT2 have been shown to be involved in the host cell membrane translocation of 3′-deoxy-nucleoside analogs (Yao et al., 1996, 2001). Unlike the endogenous nucleosides, the lack of the 3′-hydroxyl group in the ribose sugar of 3′-deoxy-nucleoside analogs allows inhibition of viral nucleic acid replication by chain termination subsequent to incorporation into the viral genome (Galmarini et al., 2001). However, this class of nucleoside analogs also produces a range of organ toxicities (e.g., myopathy, pancreatitis, neuropathy, and lactic acidosis) in patients undergoing active antiretroviral therapy, the underlying mechanisms of which are not completely understood (Sanders et al., 1991; Colacino, 1996; Gordon, 1996; Kleiner et al., 1997; Sun et al., 2014). Interestingly, earlier studies have linked 3′-deoxy-nucleoside analog–induced toxicities with mitochondrial toxicities (Colacino et al., 1994; Colacino, 1996; Honkoop et al., 1997). In fact, the mitochondrial DNA polymerase γ is more sensitive to inhibition by 3′-deoxy-nucleoside analogs used for antiviral therapy compared with nuclear DNA, resulting in depletion of mitochondrial DNA in patients undergoing antiretroviral therapy. hENT1 has been shown to be present in mitochondrial membranes and to selectively accumulate nucleoside analogs such as 5-fluorouridine (an investigational anti–hepatitis B agent) into the mitochondria, possibly contributing to the pathogenesis of mitochondrial toxicities (Lee et al., 2006). Although the hENT1 isoform is present in human mitochondrial membranes, this transporter has been found to be incapable of transporting 3′-deoxy-nucleoside analogs leaving the transport mechanisms of 3′-deoxy-nucleoside analogs into the mitochondria a poorly understood process (Yao et al., 2001).
We previously reported the detection of hENT3 in mitochondria of several cultured human cell lines (Govindarajan et al., 2009). Small interfering RNA knockdown of hENT3 reduces mitochondrial import of 3′-deoxy-nucleoside analogs in cultured human placental cells, suggesting a potential role for hENT3 in the mediated uptake of 3′-deoxy-nucleoside analogs into mitochondria (Govindarajan et al., 2009). In addition, the role of ENT3 in maintaining the lysosomal homeostasis of macrophages was demonstrated in mice (Hsu et al., 2012). Numerous independent laboratories are also reporting monogenic mutations in hENT3 that produce a spectrum of human genetic disorders (e.g., H syndrome, familial Rosai-Dorfman disease, pigmented hypertrichotic dermatosis, sinus histiocytosis with massive lymphadenopathy, etc.) to share many clinical resemblances to mitochondrial and lysosomal storage disorders (Molho-Pessach et al., 2008; Cliffe et al., 2009; Kang et al., 2010; Morgan et al., 2010; Campeau et al., 2012). We recently reported the molecular determinants involved in the acidic pH-dependent transport of hENT3, which showed a pH-sensing mechanism favoring the transport-permissible/impermissible conformations of hENT3 (Rahman et al., 2017; Singh and Govindarajan, 2017). Intriguingly, the topological locations of the primary pH-sensing residues of hENT3 facing the opposite sides of the lysosomal or mitochondrial membrane provided a putative mechanism for transport from the lysosome to the cytosol and into mitochondria (Rahman et al., 2017; Singh and Govindarajan, 2017).
Here, we sought to determine the molecular determinants of hENT3 transport of 3′-deoxy-nucleoside analogs. We report the transport features of intracellularly localized hENT3 in handling 3′-deoxy-nucleoside analogs and the putative transporter segment and residues involved in hENT3 transport of 3′-deoxy-nucleoside analogs.
Materials and Methods
Materials and Reagents.
Defolliculated oocytes were obtained from extracted Xenopus laevis ovaries procured from a commercial vendor (NASCO, Fort Atkinson, WI). 3H-radionuclides were obtained from Moravek Radiochemicals (Brea, CA). 4′,6′-diamidino-2-phenylindole, uridine, adenosine, gentamicin, tricaine methanesulfonate, phenylmethanesulfonyl fluoride, and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Collagenase A was purchased from Roche Applied Science (Penzberg, Germany). Taq polymerase, deoxyribonucleotide triphosphates, nuclease-free water, and restriction enzymes were purchased from Promega (Madison, WI). BCA protein assay reagent and SuperSignal West Pico Chemiluminescent substrates were obtained from Thermo Scientific (Waltham, MA).
Plasmid Constructs.
The details of the construction of pOX-Δ36hENT3 were described in a previous study (Govindarajan et al., 2009). The open reading frame cDNA clones of hENT1 and hENT2 were purchased from Dharmacon (Lafayette, CO). The polymerase chain reaction (PCR) products (the coding sequences) of hENT1 and hENT2 were amplified using the primers listed in Table 1 and cloned into a pOX vector using 5′SalI-3′XbaI and 5′HindIII-3′XbaI restriction sites, respectively. A Kozak sequence was introduced before the start codon during amplification. The plasmid constructs of hENT1 and hENT2 were verified by Sanger sequencing by the Ohio State Genomic Facility (Columbus, OH) and the Georgia Genomic Facility (Athens, GA) and were designated pOX-hENT1 and pOX-hENT2, respectively.
List of primers used in this study
Generation of Mutant Constructs.
Point mutations were introduced into a pOX-Δ36hENT3 construct using a QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA) following the manufacturer’s protocol. The high-performance liquid chromatography–grade mutagenic primers were purchased from Integrated DNA Technologies Inc. (Coralville, IA). The primers that were used to make single point mutations are listed in Table 1. The mutant plasmids were purified using E.Z.N.A.TM Plasmid Mini Prep Kit I (Omega Bio-Tek, Norcross, GA) and were verified by Sanger sequencing by the Ohio State Genomic Facility and Georgia Genomic Facility.
Generation of Chimeric Constructs.
The chimeras containing the N-terminal segment of hENT3 and the C-terminal segment of hENT1 were designated as the (3+1) chimera. The chimeras were generated by overlap extension PCR and were numbered by the position in hENT3 at the sites of the swaps. The sites of the swaps for both hENT3 and hENT1 are presented in Supplemental Fig. 1. The different segments of the chimeras were amplified using pOX-hENT3, pOX-Δ36hENT3, and pOX-hENT1 as templates, which are described above or in previous studies (Rahman et al., 2017). The primers that were used to make the chimeras are listed in Table 1 and were purchased from Integrated DNA Technologies Inc. All PCR amplified chimeras were cloned into the pOX vector using 5′SalI-3′XbaI restriction sites and sequence verified.
In Vitro Transcription and Transport Assays.
The mutant plasmids were linearized by either the NotI or SacI restriction enzyme. The digested products were purified by phenol-chloroform extraction. The cRNAs were synthesized using a T3 mMESSAGE mMACHINE (Ambion, Foster City, CA) transcription kit following the manufacturer’s instructions. The cRNAs were purified by lithium precipitation. Fifty nanoliters (400–800 ng/µl) of equal amounts of cRNAs of various hENTs were injected into oocytes 24 hours after defolliculation. The injected oocytes were incubated at 15°C for 24 hours before transport assays were performed. Uptake of radiolabeled substrates was measured after 30 minutes of incubation in transport buffer (100 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES, pH 7.4) at room temperature. Uptake was terminated by washing the oocytes three times with arrest buffer (100 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 20 mM uridine, pH 7.4). Individual oocytes were shaken overnight in 10% SDS for complete dissolution, and the total radioactivity was then quantified by a Beckman liquid scintillation counter (Beckman Coulter, Brea, CA).
Statistical Analyses.
Statistical analyses were performed with GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA). The t test or one-way analysis of variance was used to identify the significant differences between experimental conditions. Representative experimental results are presented wherein numerous Xenopus oocytes (n = 8–12) were tested for each run. All experiments were repeated at least two to five times. Values of significant correlations are presented with degree of significance indicated as P < 0.05, P < 0.01, P < 0.001, or P < 0.0001.
Results
hENT3 Transports 3′-Deoxy-Nucleoside Analogs.
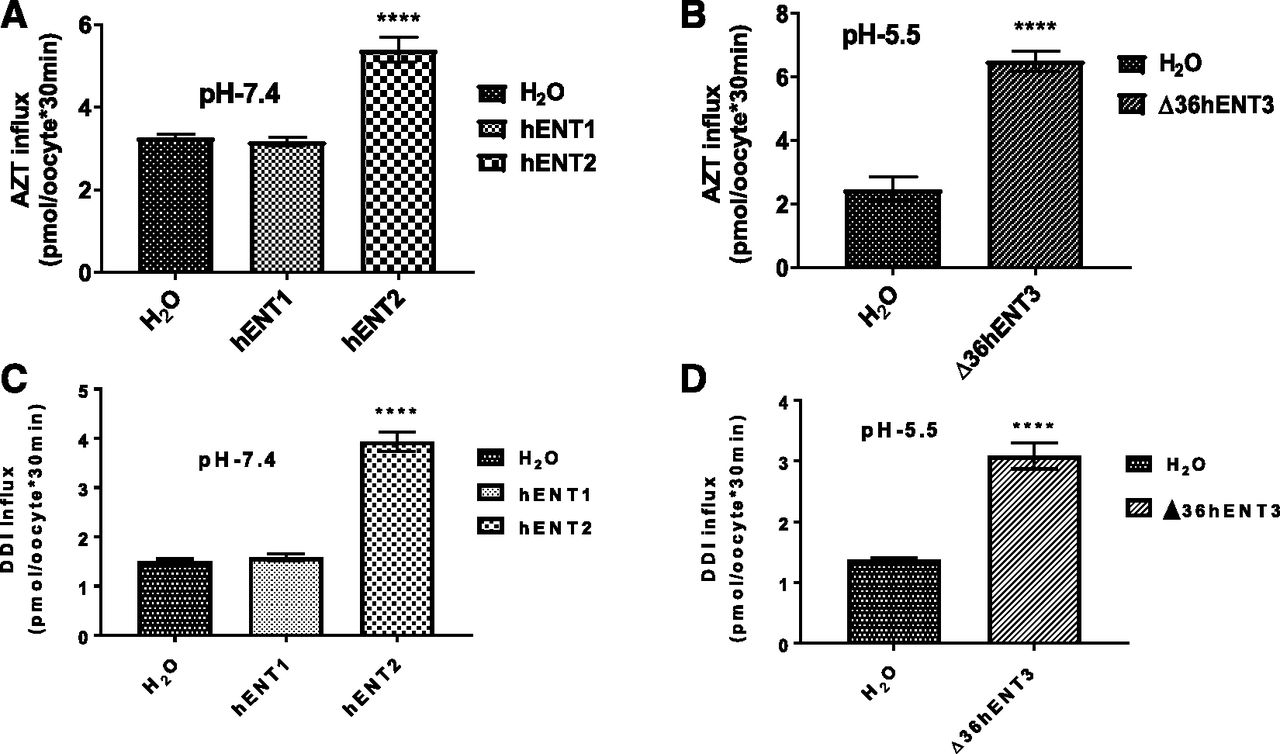
To identify the 3′-dideoxy-nucleoside analog transport capabilities of hENTs, we expressed individual hENTs (hENT1, hENT2, and hENT3) in Xenopus oocytes and analyzed the mediated flux of a pyrimidine-based and a purine-based 3′-dideoxy-nucleoside analog (i.e., 3H-AZT and 3H-DDI, respectively). 3H-adenosine was used as a common control substrate, which is transported by all ENTs. Since hENT3 harbors intracellular targeting signals at the beginning of its N terminus, we used an N-terminal 36 amino acid truncated hENT3 (designated ∆36hENT3) for analysis (Baldwin et al., 2005; Govindarajan et al., 2009; Kang et al., 2010; Rahman et al., 2017). After measuring transport levels of hENT cRNA-injected Xenopus oocytes using extracellular solutions buffered at various pHs (pH 7.4 for hENT1 and hENT2 and pH 5.5 for ∆36ENT3), hENT1 demonstrated no significant transport activity for AZT compared with H2O-injected oocytes (Fig. 1A). However, hENT2 showed a 1.65-fold (P < 0.0001) increased transport activity in transporting AZT compared with H2O-injected oocytes (Fig. 1A). Interestingly, ∆36ENT3 showed a 2.63-fold (P < 0.0001) increase in mediated AZT transport compared with H2O-injected oocytes (Fig. 1B). Next, we investigated the capability of hENTs in transporting DDI. As shown in Fig. 1, C and D, hENT2 showed a 2.62-fold (P < 0.0001) increased transport activity, consistent with its higher apparent affinity for its natural nucleoside counterpart (inosine), and hENT3 showed a 2.23-fold (P < 0.0001) increased transport activity compared with H2O-injected oocytes in transporting DDI. Again, hENT1 showed no or poor transport activity for DDI (Fig. 1C). Together, these results identify that, unlike hENT1, hENT3 is capable of transporting both AZT and DDI, similar to hENT2.

Determination of the differential 3′-deoxy-nucleoside analog (AZT and DDI) transport by hENTs. (A) Transport activities of 3H-AZT (20 µM) were measured and plotted at pH 7.4 of oocytes at 25°C 24 hours after injection of H2O, hENT1, or hENT2 transcripts. (B) Transport activities of 3H-AZT (20 µM) at pH 5.5 after injection of oocytes with H2O or ∆36hENT3 transcripts. (C) Transport activities of 3H-DDI (20 µM) were measured and plotted at pH 7.4 of oocytes at 37°C 24 hours after injection of H2O, hENT1, or hENT2 transcripts. (D) Transport activities of 3H-DDI at pH 5.5, with H2O or ∆36hENT3 transcript injection. ****P < 0.0001 (one-way analysis of variance/t test with hENTs compared with H2O). Bars represent the average ± S.E. (n = 8–12 oocytes).
Generation and Characterization of hENT3-hENT1 Chimeric Transporters.
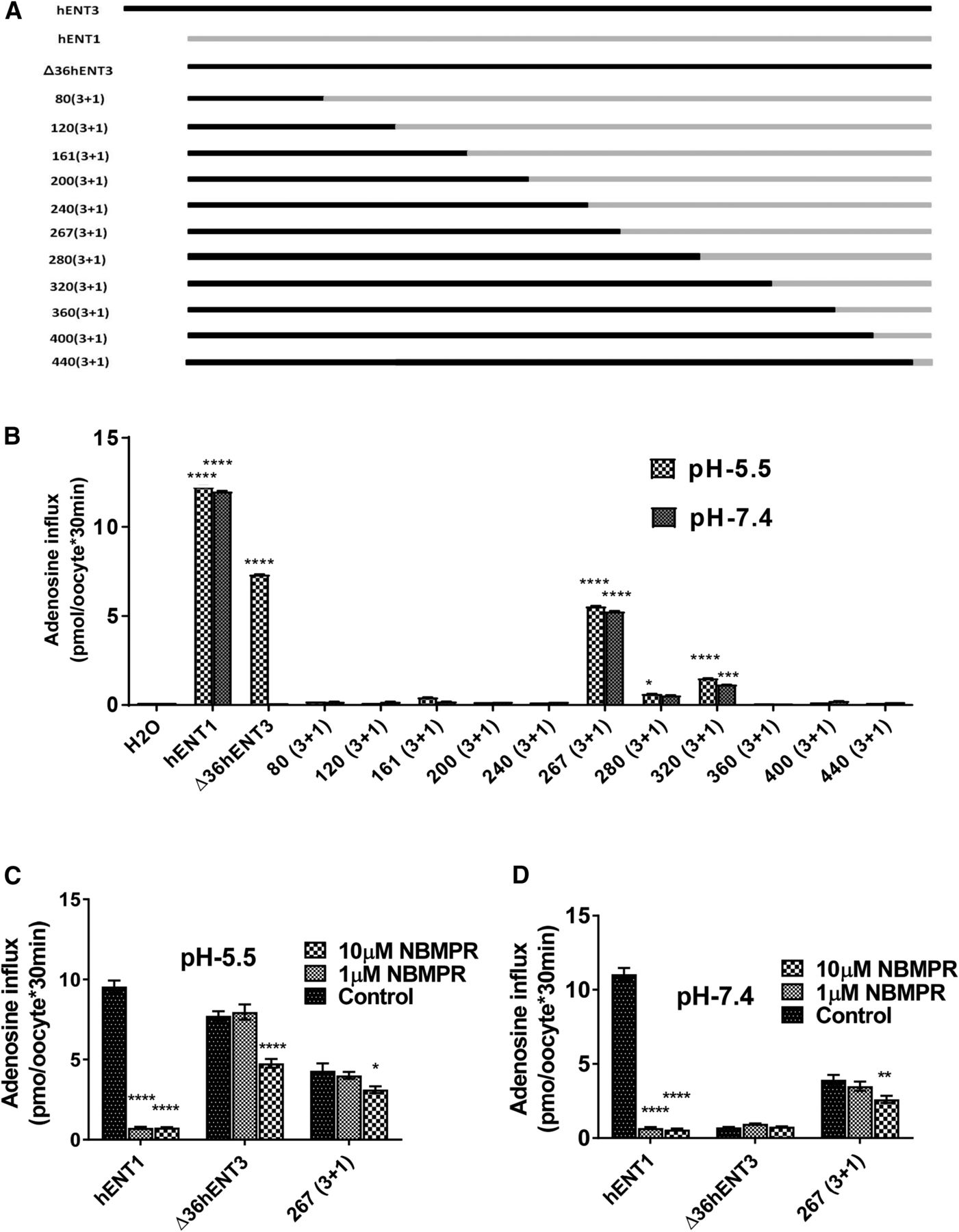
Earlier chimeric studies with ENTs as well as the recently deduced crystal structures of bacterial concentrative nucleoside transporters suggest the possibility of nucleoside transporters functioning in a pseudosymmetric manner to acquire a transport-permissible conformation (Sundaram et al., 1998; Yao et al., 2001, 2002; Baldwin et al., 2005; Zhou et al., 2007, Johnson et al., 2012; Hirschi et al., 2017). Since hENT3 efficiently transported 3′-dideoxy-nucleoside analogs and hENT1 did not show any transport, we decided to try a chimera strategy to determine the putative region(s) of hENT3 dictating the 3′-dideoxy-nucleoside transport. For this purpose, we generated several hENT3-hENT1 chimeras by swapping, at regular intervals, increasing lengths of the N terminus of hENT3 with decreasing lengths of the C terminus of hENT1. These chimeras are designated 80(3+1), 120(3+1), 161(3+1), 200(3+1), 240(3+1), 267(3+1), 280(3+1), 320(3+1), 360(3+1), 400(3+1), and 440(3+1) based on the swap positions in hENT3 (detailed in the Materials and Methods and Fig. 2A; also see Supplemental Fig. 1). To enable cell surface localization, we deleted nucleotides corresponding to the first 36 amino acids of the N terminus of hENT3 in the chimeric constructs and we introduced a start codon to enable transcription of the N-terminal truncated chimeras. The functionality of each of the chimeric proteins was tested by generating cRNAs by in vitro transcription, injecting them into Xenopus oocytes, and measuring 3H-adenosine uptake at both pH 5.5 and pH 7.4. Interestingly, only chimeric proteins 267(3+1), 280 (3+1), and 320(3+1) showed approximately 20%–70% retention of adenosine transport activity at both pH 5.5 and pH 7.4 compared with that seen in wild-type (WT) ∆36hENT3 (Fig. 2B). Intriguingly, these were the only chimeras of hENT3 and hENT1 that were swapped in between transmembranes TM6 and TM7, which divides hENTs roughly into two symmetrical halves. When we tested the inhibition by a synthetic adenosine uptake inhibitor S-(4-nitrobenzyl)-6-thioinosine (NBMPR), which inhibits hENT1 at nanomolar concentrations (ki of approximately 10 nM) and hENT3 at micromolar concentrations (ki ∼10 µM), the 267(3+1) chimera was found to be partially sensitive to NBMPR, indicating that the chimeric proteins retained the characteristic inhibitory pattern noted with hENTs (Fig. 2, C and D). Although it is possible that a lack of proper folding or improper insertion of transmembrane segments into the membrane may have contributed to the loss of functionality of the other chimeras, the recovery of robust transport activity, particularly with the 267(3+1) chimera, suggested that the major transport domains of hENT3 consist of two similar structures that come together to form a nucleoside translocation pore.

Generation and transport characterization of chimeras of hENT3 and hENT1. (A) Diagrammatic representation of the hENT3 and hENT1 chimeras constructed by replacement of the C-terminal hENT3 region by corresponding regions of hENT1. N-terminal segments of hENT3 and C-terminal segments of hENT1 were joined with increasing increments of approximately 40 amino acids from ∆36hENT3. The constructs were named as in the following example: 80(3+1) is 80 amino acids starting from the N terminus of hENT3 and the remaining length of the protein from hENT1. (B) Uptake of 3H-adenosine (20 µM) into oocytes was measured and plotted at 25°C 24 hours after injection of hENT1, ∆36ENT3, or chimeric transcripts. (C and D) Uptake of 3H-adenosine (20 µM) into oocytes at 25°C 24 hours after injection of hENT1, ∆36ENT3, or chimeric transcripts in the absence and presence of NBMPR at pH 5.5 (C) and pH 7.4 (D), respectively. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (one-way analysis of variance). Data represent the average ± S.E.M. (n = 8–12 oocytes).
The N-Terminal Half of hENT3 Determines 3′-Deoxy-Nucleoside Analog Transport.
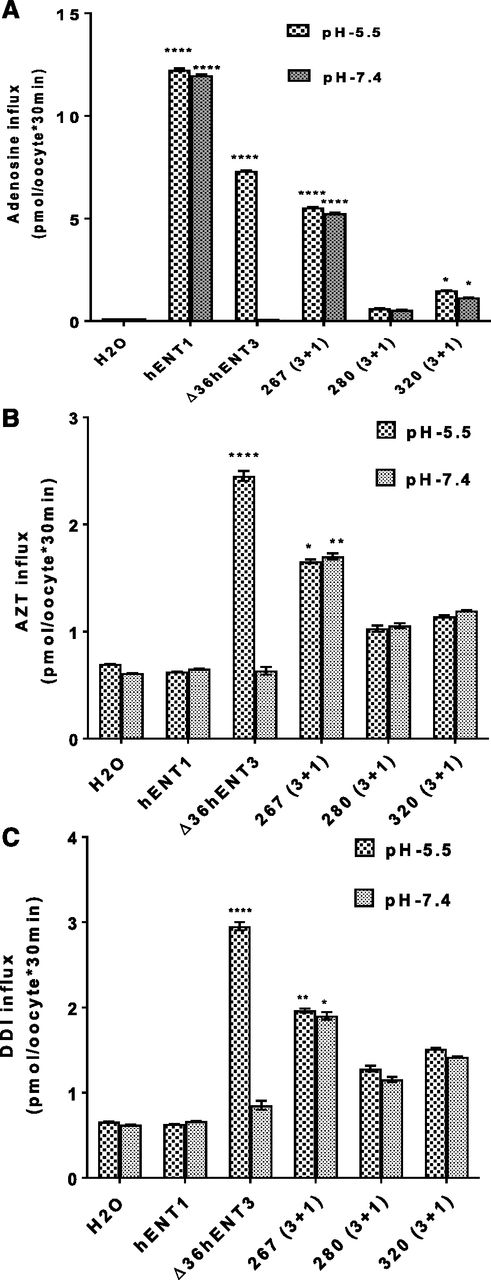
To test whether the N-terminal or C-terminal half of hENT3 is involved in 3′-deoxy-nucleoside analog transport, we investigated whether the 267(3+1) chimera, which retained maximal adenosine transport activity, is capable of transporting 3′-dideoxy-nucleoside analogs AZT and DDI. We hypothesized that if this chimera transports 3′-dideoxy-nucleoside analogs, then the N-terminal half of hENT3 is the major site of interaction with 3′-deoxy-nucleoside analogs, and if not, the C-terminal half of hENT3 is involved in the interaction. Interestingly, in addition to adenosine (Fig. 3A), our findings showed that 267(3+1) robustly transported both AZT and DDI at both pH 5.5 and pH 7.4 (Fig. 3, B and C). Furthermore, whereas the 267(3+1) chimera transported only 45% and 76% of adenosine as WT hENT1 and ∆36hENT3, respectively (Fig. 3A), it retained more than 65% transport activity of both AZT and DDI at both pH 5.5 and pH 7.4 compared with the transport capabilities of WT ∆36hENT3 at pH 5.5 in the same batch of oocytes (Fig. 3, B and C). Similar results were obtained with the 280(3+1) and 320(3+1) chimeras (Fig. 3, B and C). Taken together, these results identified the N-terminal 37–267 residues of hENT3 and not the C-terminal half of hENT3 as primarily interacting with 3′-dideoxy-nucleoside analogs.

Identification of the region(s) of hENT3 responsible for 3′-deoxy-nucleoside analog transport by hENT1 and hENT3 chimeras. (A) Uptake of 3H-adenosine (20 µM) into oocytes was measured and plotted at 25°C 24 hours after injection of hENT1, ∆36ENT3, or chimeric transcripts. (B) Uptake of 3H-AZT (20 µM) into oocytes at 25°C 24 hours after injection of hENT1, ∆36ENT3, or chimeric transcripts. (C) Uptake of 3H-DDI (20 µM) into oocytes at 25°C 24 hours after injection of hENT1, ∆36ENT3, and chimeric transcripts. *P < 0.05; **P < 0.01; ****P < 0.0001 (one-way analysis of variance). Data represent the average ± S.E.M. (n = 8–12 oocytes).
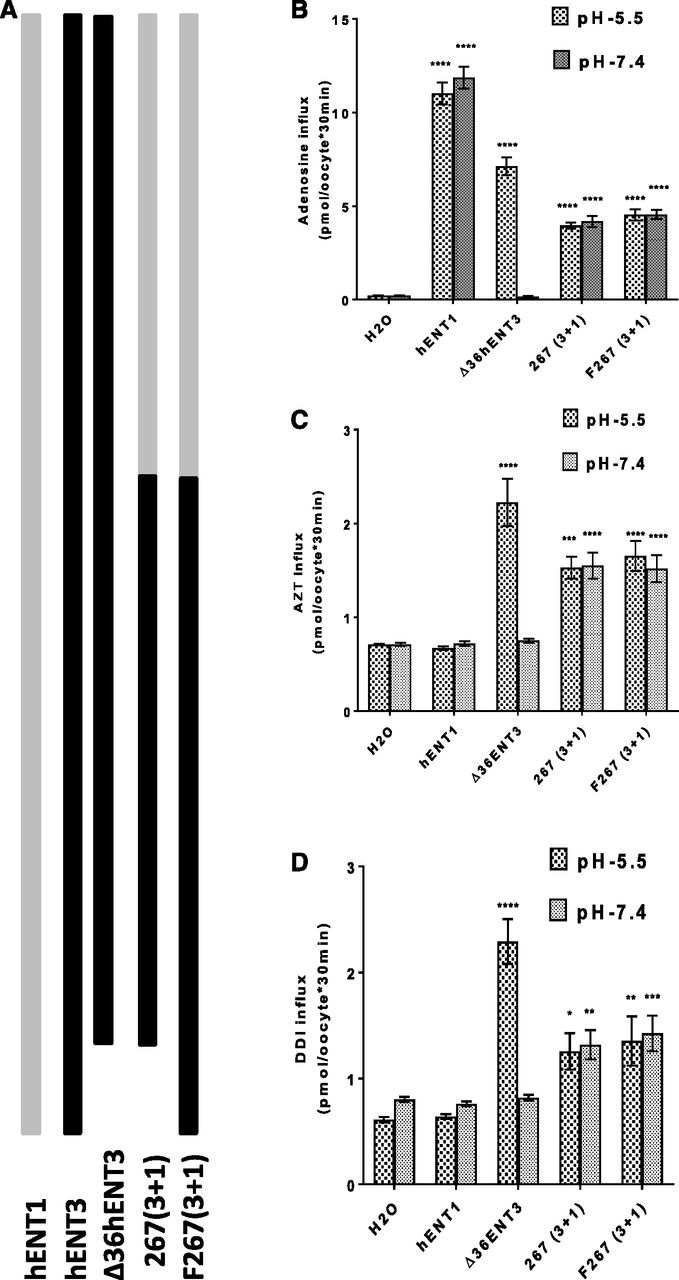
Full-length hENT3 does not show cell surface transport activity in oocytes without the removal of the N-terminal 36 amino acid residues because of the presence of intracellular targeting signals within this region of hENT3 that causes its localization to organelles (Baldwin et al., 2005). These findings were also confirmed in our earlier studies (Govindarajan et al., 2009). Intriguingly, a recent study showed that removal of the C-terminal 32 amino acid residues resulted in no plasma membrane localization of hENT1, suggesting that cell surface–targeting signals of hENT1 may be present in the C terminus of hENT1 (Aseervatham et al., 2015). Therefore, we wondered whether the transport activity of the 267(3+1) chimera observed at the oocyte cell surface was due to the loss of the 36 amino acid residues in the N terminus of hENT3 or the presence of the C-terminal half of hENT1. To test this further, we restored the intracellular targeting signal of hENT3 in the chimeric construct by including the N-terminal 36 amino acid residues in the 267(3+1) chimeric construct and tested the influx of various permeants in the same batch of oocytes (Fig. 4A). Despite the presence of intracellular targeting signals in the N terminus of hENT3, intriguingly, the full-length 267(3+1) chimera [F267(3+1)] retained permeant transport activity for both natural nucleoside (adenosine) and nucleoside analogs (AZT and DDI) (Fig. 4, B–D). Since hENT1 does not transport 3′-deoxy-nucleoside analogs, these results further suggested that the N-terminal half of hENT3 facilitated the transport of 3′-deoxy-nucleoside analogs by the chimera, whereas the C-terminal half of hENT1 facilitated the cell surface localization of the chimera.
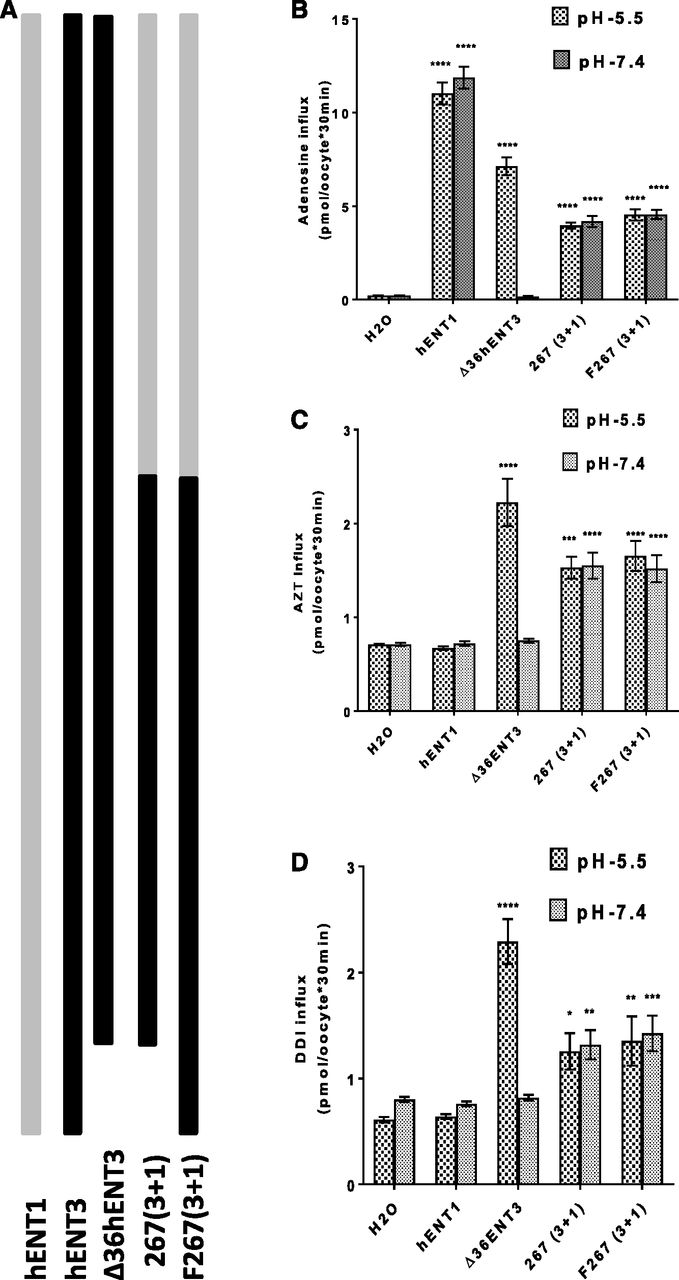
Determination of relative dominance of the intracellular targeting signal(s) of N-terminal 36 amino acids of hENT3 and the C-terminal half of hENT1 in deciding cell surface transport activity. (A) Diagrammatic representation of the 267(3+1) chimera with or without the N-terminal 36 amino acids from hENT3. (B) Uptake of 3H-adenosine (20 µM) into oocytes was measured and plotted at 25°C 24 hours after injection of hENT1, ∆36ENT3, or chimeric transcripts. (C) Uptake of 3H-AZT (20 µM) into oocytes at 25°C 24 hours after injection of hENT1, ∆36ENT3, or chimeric transcripts. (D) Uptake of 3H-DDI (20 µM) into oocytes at 25°C 24 hours after injection of hENT1, ∆36ENT3, or chimeric transcripts. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (one-way analysis of variance). Data represent the average ± S.E.M. (n = 8–12 oocytes).
Residues 225D and 231L in hENT3 Partially Contribute to 3′-Deoxy-Nucleoside Analog Transport.
Next, we aimed to determine the amino acid residues within the N-terminal half of hENT3 responsible for interaction with 3′-deoxy-nucleoside analogs. Since hENT3 and hENT2 were both capable of transporting 3′-deoxy-nucleoside analogs and hENT1 was not, we hypothesized that the amino acid determinants for 3′-deoxy-nucleoside transport should be commonly present in hENT2 and hENT3 but not hENT1. When the sequences of the N-terminal halves of all three hENTs were examined, we found 12 amino acid residues in hENT3, including six branched chain amino acids (55I, 72I, 131V, 181V, 231L, and 239V), three serine residues (158S, 179S, and 198S), two acidic residues (156D and 225D), and one alanine (213A) common between hENT2 and hENT3 but not in hENT1 (Fig. 5A). To test their putative involvements in interaction with 3′-deoxy-nucleoside analogs, we employed site-directed mutagenesis and substituted each of these 12 residues in hENT3 to corresponding residues present in hENT1 (e.g., 55I>L, 72I>M, 131V>I, 156D>Q, 158S>D, 179S>G, 181V>I, 198S>T, 213A>S, 225D>E, 231L>F, and 239V>A). Again, the functionalities of each of the chimeric transporters were tested by generating and injecting mutated cRNAs into oocytes and measuring mediated transport of 3H-permeants (adenosine, AZT, DDI) in the same batch of oocytes at pH 5.5. We anticipated the amino acid(s) in mutant positions that demonstrated decreased activity in transporting AZT and DDI but maintained adenosine transport were likely to be involved in 3′-deoxy-nucleoside analog transport by hENT3, although the residues that are commonly involved in transport of both adenosine and 3′-deoxy-nucleoside analogs could not be distinguished by this approach. Of 12 mutants examined, nine mutants showed transport functionality between 60% and 100% in transporting adenosine compared with WT hENT3, except the 72I>M, 131V>I, and 213A>S mutants, which showed drastic reductions in adenosine transport activity (but not 3′-deoxy-nucleosides transport), suggesting their essential role in adenosine transport functionality (Fig. 5B). When AZT transport was conducted, of 12 mutants, 225D>E and 231L>F showed decreased transport activity for AZT while maintaining adenosine transport (Fig. 5C). When DDI transport was carried out, of the 12 mutants, 225D>E and 231L>F showed decreased activity in transporting DDI while maintaining adenosine transport (Fig. 5D). These results suggested that residues 225D and 231L are responsible, at least partially, for the 3′-deoxy-nucleoside analog transport by hENT3 with residues 225D and 231L acting as common determinants for both AZT and DDI transport.

Identification of residues dictating the selectivity of hENT3 in transporting 3′-deoxy-nucleoside analogs. (A) Sequence alignment of hENT1, hENT2, and hENT3: the residues that are common in hENT2 and hENT3 but not in hENT1 are indicated in the boxes. (B–D) Transport activities of 3H-adenosine (20 µM; B), 3H-AZT (20 µM; C), and 3H-DDI (20 µM; D) of H2O, ∆36hENT3, and mutant transcript-injected oocytes. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (one-way analysis of variance of mutants compared with ∆36hENT3). For (C) and (D), statistical analysis was performed for mutants retaining adenosine transport activity. Data represent the average ± S.E.M. (n = 8–12 oocytes).
Discussion
Antiviral 3′-deoxy-nucleoside analogs (e.g., AZT and DDI) generally require carrier-mediated transport to exert their efficacy and toxicity (Yao et al., 2001; Damaraju et al., 2011; Moss et al., 2012) although some drugs within the class have higher membrane diffusion capabilities (e.g., AZT) than others (e.g., DDI) (Zimmerman et al., 1987; Chan et al., 1992; Yao et al., 2001). Earlier studies showed that the absence of the 3′-hydroxyl group in 3′-deoxy-nucleoside analogs makes them poor substrates of hENT1 but not of hENT2 (Yao et al., 2001). This is consistent with the fact that ribose recognition is less important for hENT2 but not for hENT1, which primarily recognizes the 3′ oxygen of ribose in the nucleoside for transport (Lum et al., 2000; Yao et al., 2001). This study recognizes 3′-deoxy-nucleoside analogs as one of the primary drug substrates for intracellularly localized hENT3, which exhibits transport capabilities similar to that seen with cell surface hENT2 (Yao et al., 2001). In addition, our study provides new insights into the structural and molecular features of hENT3 transport of 3′-deoxy-nucleosides. We demonstrated that the N-terminal half of the hENT3 transporter is the region of interaction with 3′-deoxy-nucleoside analogs and amino acid residues 225D and 231L partly contribute to that process.
The hENT3 protein contains 11 predicted transmembrane domains, a large intracellular loop between TM5 and TM6, and an N-glycosylation site (Baldwin et al., 2005). The overall topology of hENT3 is very similar to all ENTs, except that hENT3 has an extended N terminus with putative endosomal/lysosomal and mitochondrial targeting motifs (Baldwin et al., 2005; Govindarajan et al., 2009). Consistently, hENT3 is partially localized to late endosomes, lysosomes, and mitochondria in human cell lines. Truncation of the N terminus (Δ36hENT3) redirects it from the intracellular domains to the cell surface, which allows functional characterization of the transporter in Xenopus oocytes (Baldwin et al., 2005; Govindarajan et al., 2009; Kang et al., 2010). Unlike hENT2, which shows transport of 3′-deoxy-nucleoside at pH 7.4, Δ36ENT3 shows maximum transport only at an acidic pH of 5.5 (Baldwin et al., 2005; Rahman et al., 2017). In the mammalian cell context, the acidic pH-activated transport activity of hENT3 facilitates transport of permeants from lower pH to higher pH such as from the lysosome to the cytosol and from intermembrane mitochondrial spaces to the mitochondrial matrix (Govindarajan et al., 2009; Kang et al., 2010; Singh and Govindarajan, 2017). Thus, despite hENT3 being an equilibrative nucleoside transporter, the pH dependence characteristic of this transporter may allow it to function in a unidirectional manner in lysosomal or mitochondrial contexts. These unique transport characteristics of hENT3 could cause increased expulsion of 3′-deoxy-nucleoside analogs from late endosomes and lysosomes and/or increased accumulation of 3′-deoxy-nucleoside analogs inside mitochondria.
Several successful attempts have been made to elucidate the structure-function relation of ENTs by generating chimeric constructs of hENTs (ENT1 and ENT2) or human and rodent ENTs (hENT1 and rENT1) (Sundaram et al., 1998; Yao et al., 2001, 2002; Baldwin et al., 2005; Zhou et al., 2007). This approach has led to the identification of precise structural elements and/or regions involved in the binding of endogenous substrates, drugs, and nucleoside transport inhibitors such as dipyramidole and NBMPR (Sundaram et al., 1998; Endres and Unadkat, 2005). The same approach has also facilitated the identification of the N-terminal half of hENT2 as a major site of interaction of 3′-deoxy-nucleoside analogs (Yao et al., 2001). Furthermore, recent elucidation of the crystal structures of two concentrative nucleoside transporters from bacterial species Vibrio cholerae and Neisseria wadsworthii demonstrates a pseudosymmetrical conformation of two major structural domains within the transporter that form a transport pore for nucleoside permeation (Johnson et al., 2012; Hirschi et al., 2017). Using a chimeric approach, here we found that replacement of TM1–6 of hENT1 with TM1–6 of hENT3 was able to robustly retain nucleoside transport capability in a pH-independent manner. Although residue 219D is one of the primary determinants of pH sensitivity in hENT3 (not present in the chimera), the loss of the pH-sensing residue 447E from the hENT3 segment could be responsible for the pH-independent activities of the chimeras (Rahman et al., 2017). Alternatively, the pH-independent characteristics could have also arisen from the C-terminal half of the hENT1 segment of the chimeras. In this regard, we reported earlier that hENT1 has the capability to function in a pH-independent manner within the physiologic pH range (pH 5.0–8.0) (Kang et al., 2010). Importantly, the findings from transport analyses of chimeras demonstrated that the first half (267 amino acid residues) of hENT3 is the major domain that interacts with the 3′-deoxy-nucleoside analogs, which is similar to that identified for hENT2. The chimera also showed less sensitivity to NBMPR inhibition, which mimicked the characteristics of hENT3 and not hENT1. Consistently, the NBMPR binding region in hENT1 was shown to be present in the N terminus of hENT1 (Sundaram et al., 1998; Endres and Unadkat, 2005) and was not present in the chimera. Although it appears that the C-terminal half of the hENT3 region is involved in NBMPR binding, our attempts to generate a functional N-terminal half of hENT1 and a C-terminal half of hENT3 chimeras were futile and precluded further understanding of NBMPR’s interaction with hENT3. Therefore, further studies are warranted to determine the precise NBMPR binding sites of hENT3 (Kwong et al., 1993).
Intriguingly, chimeric analyses of hENT3 and hENT1 swapped at various regions showed that maximal transport activity only occurred when the splice region was present in the middle region of the transporter (i.e., in the large intracellular loop connecting TM1–6 and TM7–11 or at the beginning of TM7). These observations suggest the conservation of each of these structures in the 267(3+1) chimera (conserved the TM2–6 from hENT3 and the TM7–11 from hENT1) may be crucial for maximal recovery of nucleoside transport function. However, the activity of the chimera was not hindered if each of these structures came from different hENTs. Since this feature was also observed in several previously conducted ENT chimeric studies (hENT1-rENT1, hENT1-hENT2, and hENT1-hENT4) (Sundaram et al., 1998; Yao et al., 2002; Zhou et al., 2007), it is tempting to speculate that a 2-fold pseudosymmetric conformation is operational for all ENTs involved in nucleoside influx with TM2–6 forming one structure and TM7–11 forming the other structure. Future studies and crystal structures of mammalian ENTs are expected to shed more light on these predictions. The chimeric studies also provided other useful structural insights, particularly on the localization signals in hENT1 and hENT3. Although a putative dileucine motif and a putative mitochondrial targeting signal of hENT3 were identified in the N terminus of the protein (Baldwin et al., 2005; Govindarajan et al., 2009), the localization signals responsible for basolateral cell surface targeting of hENT1 is uncertain. Since the 267(3+1) chimera, in the presence or absence of intracellular targeting signals of hENT3, was both functional in transporting nucleosides at the oocyte cell surface, this suggests that the C-terminal half of hENT1 facilitated the localization of the chimera at the cell surface. Although these observations provide the beginning insight into understanding the cell surface localization signals in hENT1, further studies are required to map the cell surface–targeting motif(s) in hENT1. Finally, site-directed mutagenesis analyses allowed characterization of individual amino acid residues in the hENT3 transporter possibly interacting with nucleosides and 3′-deoxy-nucleosides analogs. Indeed, these results suggested that several residues including 225D and 231L in the N-terminal half of hENT3 may have individual contributing roles in dictating 3′-deoxy-nucleoside analog transport of hENT3. Thus, our findings revealed the region of interaction of hENT3 with the antiviral 3′-deoxy-nucleoside analogs AZT and DDI. The identification of molecular and structural elements of hENT3 transport of 3′-deoxy-nucleosides provides useful information for rational development of novel nucleoside analogs that may avoid mitochondrial accumulation and drug toxicity.
Authorship Contributions
Participated in research design: Rahman, Govindarajan.
Conducted experiments: Rahman, Raj.
Contributed new reagents or analytic tools: Rahman.
Performed data analysis: Rahman.
Wrote or contributed to the writing of the manuscript: Rahman, Govindarajan.
Footnotes
- Received November 9, 2017.
- Accepted March 7, 2018.
The work was supported by the National Institutes of Health National Institute of Arthritis and Musculoskeletal and Skin Diseases [Grant R03-AR063326] and the National Institutes of Health National Cancer Institute [Grants R15-CA176528 and R01-CA188464].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AZT
- 3′-azido-3′-deoxythymidine
- DDI
- 2′3′-dideoxyinosine
- hCNT
- human concentrative nucleoside transporter
- hENT
- human equilibrative nucleoside transporter
- NBMPR
- S-(4-nitrobenzyl)-6-thioinosine
- PCR
- polymerase chain reaction
- TM
- transmembrane
- WT
- wild type
- Copyright © 2018 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}