


Visual Overview
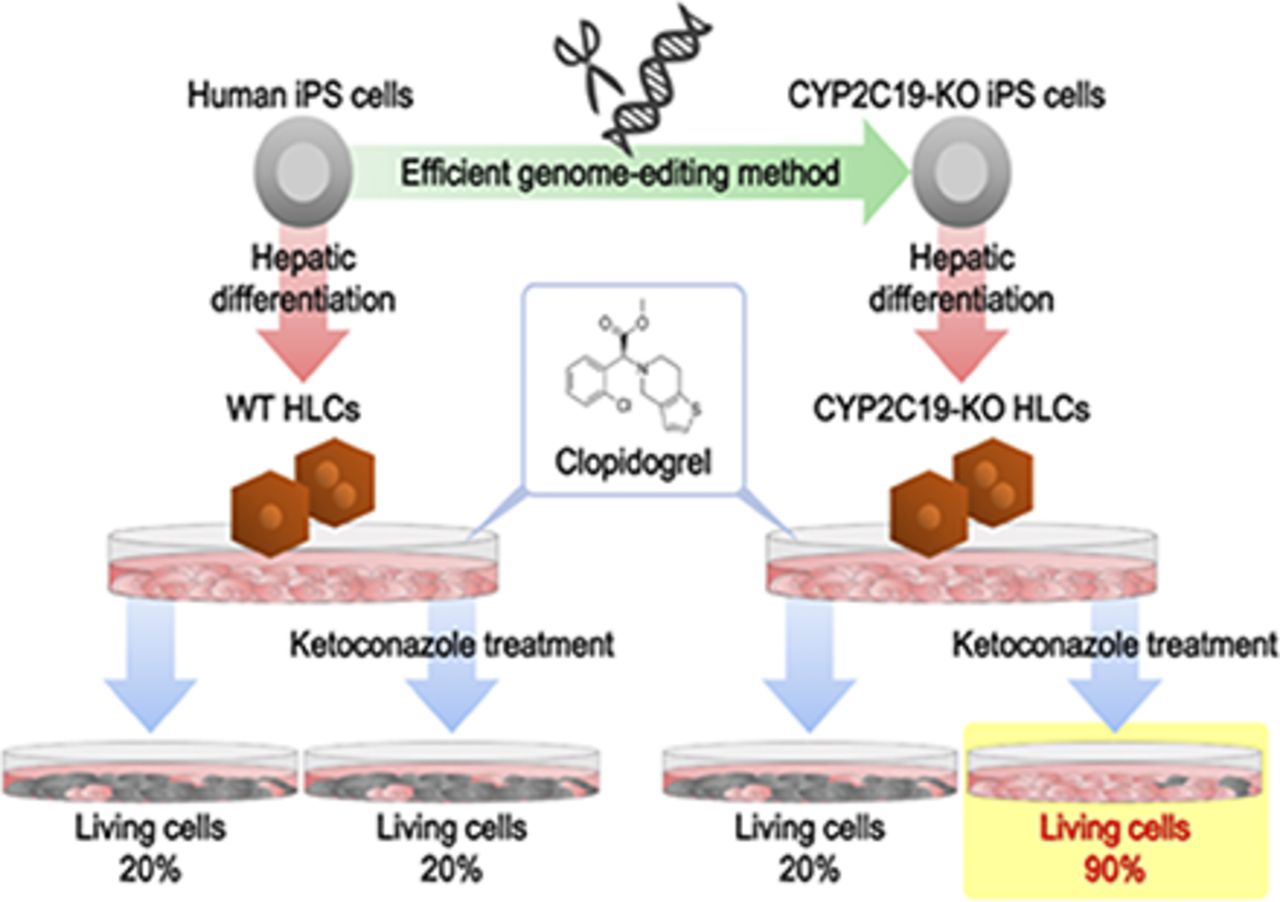
Abstract
Cytochrome P450 family 2 subfamily C member 19 (CYP2C19), in liver, plays important roles in terms of drug metabolism. It is known that CYP2C19 poor metabolizers (PMs) lack CYP2C19 metabolic capacity. Thus, unexpected drug-induced liver injury or decrease of drug efficacy would be caused in CYP2C19 substrate–treated CYP2C19 PMs. However, it is difficult to evaluate the safety and effectiveness of drugs and candidate compounds for CYP2C19 PMs because there is currently no model for this phenotype. Here, using human induced pluripotent stem cells (human iPS cells) and our highly efficient genome-editing and hepatocyte differentiation technologies, we generated CYP2C19-knockout human iPS cell-derived hepatocyte-like cells (CYP2C19-KO HLCs) as a novel CYP2C19 PM model for drug development and research. The gene expression levels of hepatocyte markers were similar between wild-type iPS cell–derived hepatocyte-like cells (WT HLCs) and CYP2C19-KO HLCs, suggesting that CYP2C19 deficiency did not affect the hepatic differentiation potency. We also examined CYP2C19 metabolic activity by measuring S-mephenytoin metabolites using ultra-performance liquid chromatography-tandem mass spectrometry. The CYP2C19 metabolic activity was almost eliminated by CYP2C19 knockout. Additionally, we evaluated whether clopidogrel (CYP2C19 substrate)-induced liver toxicity could be predicted using our model. Unexpectedly, there was no significant difference in cell viability between clopidogrel-treated WT HLCs and CYP2C19-KO HLCs. However, the cell viability in clopidogrel- and ketoconazole (CYP3A4 inhibitor)-treated CYP2C19-KO HLCs was significantly enhanced as compared with that in clopidogrel- and DMSO-treated CYP2C19-KO HLCs. This result suggests that CYP2C19-KO HLCs can predict clopidogrel-induced liver toxicity. We succeeded in generating CYP2C19 PM model cells using human iPS cells and genome-editing technologies for pharmaceutical research.
SIGNIFICANCE STATEMENT Although unexpected drug-induced liver injury or decrease of drug efficacy would be caused in CYP2C19 substrate-treated CYP2C19 poor metabolizers, it is difficult to evaluate the safety and effectiveness of drugs and candidate compounds for CYP2C19 poor metabolizers because there is currently no model for this phenotype. Using human iPS cells and our highly efficient genome editing and hepatocyte differentiation technologies, we generated CYP2C19-knockout human iPS cell-derived hepatocyte-like cells as a novel CYP2C19 poor metabolizer model for drug development and research.
Introduction
The liver plays an essential role in drug metabolism. Drugs are metabolized in the liver and transformed into active (or inactive) metabolites. Some drugs and their metabolites can cause hepatocellular toxicity (Macdonald and Robertson, 2009), compelling pharmaceutical companies to discontinue development of these drugs or withdraw them from the market. Therefore, establishment of the highly specific in vitro systems for the evaluation of drug metabolism and toxicity is a pressing issue.
Poor metabolizers (PMs) are individuals who lack the metabolic capacity of a specific drug-metabolizing enzyme. Therefore, the plasma drug concentrations and frequency of side effects differ greatly between PMs and non-PMs (Stingl et al., 2013). Because cytochrome P450 family 2 subfamily C member 19 (CYP2C19) largely contributes to the phase I reaction of drug metabolism (Zanger and Schwab, 2013), there is a concern that the PMs of CYP2C19 (CYP2C19-PMs) would suffer from the unexpected changes of pharmacokinetics and toxicity of CYP2C19 substrate drugs. Additionally, the frequency of CYP2C19-PMs is only 2% in Caucasians but 18%–23% in Asians (Nakamura et al., 1985; Bertilsson, 1995; Gardiner and Begg, 2006; Isomura et al., 2010). For these reasons, to ensure the safety of drugs on the international market, we must develop a drug-metabolism and toxicity evaluation system which considers CYP2C19-PMs and their ethnic differences. However, the available sources of primary human hepatocytes (PHHs), which are widely used in hepatocellular toxicity evaluation tests, are almost entirely limited to Caucasians, making it difficult to perform drug screening for CYP2C19-PMs.
In recent years, genome-editing technologies using human induced pluripotent stem cells (human iPS cells) have progressed rapidly. If we could use these technologies to establish CYP2C19-knockout human iPS cell–derived hepatocyte-like cells (CYP2C19-KO HLCs), these cells could be used to reproduce the drug metabolism in CYP2C19-PMs. However, the genome-editing efficiency depends on the target genes—that is, most of the transcriptionally inactive heterochromatic genes in undifferentiated iPS cells, including CYP2C19, can scarcely be edited (the biallelic-targeting efficiency is no more than 1%). Therefore, we have optimized the conditions of the genome-editing strategy and successfully enhanced the biallelic-targeting efficiency to more than 15% by using RAD51-expressing plasmid and valproic acid (Takayama et al., 2017). Moreover, we have developed an efficient hepatic differentiation protocol from human iPS cells (Takayama and Mizuguchi, 2017). From the aforementioned, we aimed to knockout the CYP2C19 gene using our unique genome-editing technology in human iPS cells and to differentiate the cells into hepatocyte-like cells (HLCs) to develop a cell culture system imitating CYP2C19-PMs.
In this study, we established CYP2C19-KO iPS cells using a CRISPR-Cas9 system with several modifications. We then differentiated the CYP2C19-KO iPS cells into HLCs (CYP2C19-KO HLCs), which could be used as a hepatocyte model of CYP2C19-PMs. To evaluate whether the generated CYP2C19-KO HLCs would be useful for drug toxicity tests, we examined their response to CYP2C19 substrate drugs.
Materials and Methods
Human iPS Cells.
In this study, the human iPS cell line, FCL-iPS, was used. This iPS cell line was generated from PHHs, and the details are available in our previous report (Takayama et al., 2014). The cells were maintained on iMatrix-511 (Nippi)–coated plates with StemFit AK02N medium (Ajinomoto) according to the manufacturer’s instructions.
Hepatic Differentiation.
Human iPS cells were dissociated into single cells using TrypLE Select Enzyme (Thermo Fisher Scientific) and plated onto a Matrigel Basement Membrane Matrix, Growth Factor Reduced (Corning)–coated plate (MultiWell Plate for Cell/Tissue Culture 12F with lid; Sumitomo Bakelite). The cells were then cultured with StemFit AK02N medium for several days prior to the hepatic differentiation. When they reached approximately 80% confluence, the four-step hepatic differentiation protocol was conducted. The first step is the induction of definitive endoderm cells, which is a 4-day culture of human iPS cells in RPMI 1640 medium (Sigma-Aldrich) containing 1×GlutaMAX (Thermo Fisher Scientific), 1×B27 Supplement Minus Vitamin A (Thermo Fisher Scientific), and 100 ng/ml of Activin A (R&D Systems). The second step is the induction of hepatoblast-like cells, which is a 5-day culture of the definitive endoderm cells in RPMI 1640 medium containing 1×GlutaMAX, 1×B27 Supplement Minus Vitamin A, 20 ng/ml of bone morphogenetic protein 4 (R&D Systems), and 20 ng/ml of FGF4 (R&D Systems). The third step is the induction of HLCs, which is a 5-day culture of the hepatoblast-like cells in RPMI 1640 medium containing 1×GlutaMAX, 1×B27 Supplement Minus Vitamin A, and 20 ng/ml of hepatocyte growth factor (R&D Systems). The fourth step is the maturation of HLCs, which is an 11-day culture of the HLCs in hepatocyte culture medium (HCM; Lonza) without epidermal growth factor but with 20 ng/ml of oncostatin M (R&D Systems).
Real-Time Reverse-Transcription Polymerase Chain Reaction.
By using ISOGEN (NIPPON GENE), total RNA was isolated from the cells. Using 500 ng of the isolated total RNA, cDNA was synthesized with a Superscript VILO cDNA synthesis kit (Thermo Fisher Scientific). Real-time reverse-transcription polymerase chain reaction (PCR) was performed with SYBR Green PCR Master Mix (Applied Biosystems) using a StepOnePlus real-time PCR system (Applied Biosystems). The 2−ΔΔCT method was adopted for the relative quantitation of target mRNA levels. Namely, the values of the target genes were normalized by those of the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase. PCR primer sequences (described in Supplemental Table 1) used in this report were obtained from PrimerBank (https://pga.mgh.harvard.edu/primerbank/).
Albumin Secretion.
To evaluate the albumin (ALB) secretion capacity of the cells, the culture supernatants, which were incubated for 24 hours after the medium was added, were collected. The collected supernatants ware analyzed by ELISA with a Human Albumin ELISA Quantitation Set (Bethyl Laboratories). ELISA was performed according to the manufacturer’s instructions. The amount of ALB secretion was normalized by the protein content per well, which was evaluated with a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific).
Immunohistochemistry.
The human iPS cells and their derivatives were fixed with 4% paraformaldehyde in PBS at 4°C for 15 minutes. After blocking the cells with PBS containing 2% bovine serum albumin and 0.2% Triton X-100 at room temperature for 45 minutes, the cells were incubated with a primary antibody at 4°C overnight and then with a secondary antibody at room temperature for 1 hour. All antibodies used in this report are described in Supplemental Table 2.
DNA Microarray.
By using the RNeasy Mini Kit (QIAGEN), total RNA was isolated from human iPS cell–derived HLCs. Then, all operations related to the microarray, cDNA amplification, labeling, hybridization, and analysis were performed at Takara Bio Inc. The platform of the microarray was SurePrint G3 Human Gene Expression 8×60K v3 (Agilent Technologies). The data are available at Gene Expression Omnibus, and the accession number is GSE123937. Gene ontology (GO) enrichment clustering analysis was performed by using DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/).
CYP2C19 and CYP3A4 Activity.
To examine the CYP2C19 and CYP3A4 activity, an ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS) analysis was performed. Human iPS cell–derived HLCs were cultured with medium containing 50 μM S-mephenytoin (a substrate whose metabolite is 4′-hydroxymephenytoin; Toronto Research Chemicals) or 5 μM midazolam (a substrate whose metabolite is 1′-hydroxymidazolam; FUJIFILM Wako). The supernatant was collected at 24 hours after the treatment with the substrate. The collected supernatant was immediately mixed with two volumes of acetonitrile (FUJIFILM Wako). Samples were filtrated with AcroPrep Advance 96-Well Filter Plates (Pall Corporation) for 5 minutes at 1750g, and then the supernatant was analyzed by UPLC-MS/MS to measure the concentration of the metabolite according to the standard curve. UPLC analysis was performed using an Acquity UPLC (Waters), and MS/MS was performed on a Q-Premier XE (Waters). The mass spectrometer was set to the multiple-reaction monitoring (MRM) mode and was operated with the electrospray ionization source in positive ion mode. The MRM transitions (m/z of precursor ion/m/z of product ion) for 4′-hydroxymephenytoin and 1′-hydroxymidazolam were 342.2/203.1 and 342.2/203.1, respectively. For transition, the cone voltage and collision energy were set at 40 V and 26 eV, respectively. The dwell time for each MRM transition was set at 100 ms. LC separations were carried out at 40°C with an Acquity UPLC BEH C18 column, 1.7 μm, 2.1 × 50 mm (Waters). The mobile phase was delivered at a flow rate of 0.5 ml/min using a gradient elution profile consisting of solvent A (0.1% formic acid/distilled water) and solvent B (0.1% formic acid/acetonitrile). The initial composition of the binary solvent was 10% B from 0 to 0.5 minute. Solvent B was increased from 10% to 100% over 2.0 minutes. The composition of solvent remained for 1.0 minute at 100% B. Ten microliters of sample solution was injected into the column. The concentrations of the metabolite were calculated according to the standard followed by normalization to the protein content per well.
Cell Viability.
The cell viability of wild-type (WT) and CYP2C19-KO HLCs was assessed by WST-8 assay after 48-hour exposure to different concentrations of clopidogrel (0, 100, and 300 μM) in the presence or absence of the CYP3A4 inhibitor ketoconazole (10 μM) and CYP2C19 inhibitor N-3-benzyl-nirvanol (1 μM). WST-8 assay was performed using Cell Counting Kit-8 was purchased from Dojindo Laboratories. WST-8 assay was performed according to the manufacturer’s instructions. The cell viability was calculated as a percentage of that in the cells treated with vehicle (DMSO) only.
Primary Human Hepatocytes.
Three lots of cryopreserved human hepatocytes (lots YOW, OHO, and FCL; Veritas) were used. The average values of the three lots are shown as “PHH” in this report. The vials of hepatocytes were rapidly thawed in a shaking water bath at 37°C; the contents of each vial were emptied into prewarmed Cryopreserved Hepatocyte Recovery Medium (Thermo Fisher Scientific), and the suspension was centrifuged at 900 rpm for 10 minutes at room temperature. The hepatocytes were seeded at 1.25 × 105 cells/cm2 in HCM containing 10% fetal calf serum (GIBCO) onto type I collagen-coated 12-well plates. The medium was replaced with HCM 6 hours after seeding. The hepatocytes, which were cultured 48 hours after plating the cells, were used in the experiments.
Results
Generation of CYP2C19-KO Human iPS Cells.
To establish CYP2C19-KO iPS cells, we designed donor plasmids targeting CYP2C19 (Fig. 1A). To efficiently perform the homologous recombination-mediated gene editing at the CYP2C19 locus in human iPS cells, we used single guide RNA/Cas9-coexpressing plasmids [pX330 (Le et al., 2013)] and RAD51-expressing plasmids with donor plasmids. Human iPS cells were electroporated with these three plasmids. After the positive selection with puromycin, we isolated 24 single cell-derived colonies. To examine whether single cell-derived human iPS cells carry the transgene cassette at the targeted locus, genomic DNA was extracted and genotyping analysis was performed using the primers indicated by arrows in Fig. 1A. PCR primers were designed to distinguish WT and mutant alleles; WT cells show a 2750-bp band, biallelically targeted cells show a 5000-bp band, and monoallelically targeted cells show two bands (a 2750- and a 5000-bp band). As shown in Fig. 1B, “clone 3” shows integration of the transgene into both alleles of CYP2C19. From this result, we could successfully establish the human iPS cells with a defect in exon 1 of the CYP2C19 gene, which includes its start codon (CYP2C19-KO iPS cells).

Generation of CYP2C19-knockout human iPS cells. (A) A schematic overview of the targeting strategy for CYP2C19 is shown. PCR primers that can distinguish the wild-type and mutant alleles are shown with arrows. The following donor plasmids were used to target the CYP2C19 locus: EF1α, elongation factor 1 α promoter; PuroR, puromycin-resistant protein; and pA, polyadenylation sequence. The CRISPR-Cas9 system was applied to produce CYP2C19 sequence-specific double-strand breaks. (B) Genotyping was performed to examine whether the human iPS cell clones were correctly targeted.
Effect of CYP2C19 Deficiency on the Undifferentiated State.
To characterize the effect of CYP2C19 deficiency on the undifferentiated state, we performed several assays (Fig. 2). Phase images showed that there was no morphologic difference between WT iPS cells and CYP2C19-KO iPS cells (Fig. 2A). Real-time reverse-transcription PCR analysis showed that there was no significant difference in gene expression levels of pluripotent markers (NANOG, OCT3/4, and SOX2) between the two groups (Fig. 2B). Immunostaining analysis also showed that there was no significant difference in the protein expression levels of OCT3/4 and SOX2 between the two groups (Fig. 2C). Taken together, these results suggested that CYP2C19 deficiency does not affect the undifferentiated state of human iPS cells.

Effect of CYP2C19 deficiency on the undifferentiated state. (A) Phase contrast images of WT iPS cells (iPSCs) and CYP2C19-KO iPSCs are shown. Scale bar, 500 μm. (B) The gene expression levels of pluripotent markers (NANOG, OCT3/4, and SOX2) were measured by real-time reverse-transcription PCR analysis in WT iPS cells and CYP2C19-KO iPS cells. The gene expression levels in undifferentiated WT iPS cells were taken as 1.0. (C) WT and CYP2C19-KO iPS cells were subjected to immunostaining with anti-OCT3/4 (green) and anti-SOX2 (red) antibodies. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 100 μm. All data are represented as means ± S.D. (n = 3). *P < 0.05; **P < 0.01.
Hepatic Differentiation of CYP2C19-KO Human iPS Cells.
To examine whether the hepatic differentiation capacity of CYP2C19-KO iPS cells is similar to that of WT iPS cells, both WT iPS cells and CYP2C19-KO iPS cells were differentiated into HLCs as described in Fig. 3A, and then the gene expression levels of hepatic markers in wild-type iPS cell–derived hepatocyte-like cells (WT HLCs) and CYP2C19-KO HLCs were analyzed (Fig. 3B). The gene expression levels of ALB, α-fetoprotein, α-1 antitrypsin, transthyretin, hepatocyte nuclear factor 4 α, and CYP3A4 in CYP2C19-KO HLCs were similar to those in the WT HLCs. ELISA analysis showed that there were no significant differences in the ALB secretion capacity between WT HLCs and CYP2C19-KO HLCs (Fig. 3C). Immunostaining analysis also showed that there was no significant difference in the protein expression levels of ALB and α-1 antitrypsin between WT HLCs and CYP2C19-KO HLCs (Fig. 3D). These results suggest that CYP2C19 deficiency does not affect the hepatic differentiation capacity of human iPS cells.

Hepatic differentiation capacity of CYP2C19-knockout human iPS cells. Human iPS cells (FCL-iPS) were differentiated into the HLCs as described in the Materials and Methods section. (A) The schematic overview shows the protocol for hepatic differentiation. (B) The gene expression levels of hepatocyte markers [ALB, α-fetoprotein (AFP), α-1 antitrypsin (AAT), transthyretin (TTR), hepatocyte nuclear factor 4 α (HNF4A), and CYP3A4] were examined in undifferentiated WT iPS cells, undifferentiated CYP2C19-KO iPS cells, and WT iPS cell- and CYP2C19-KO iPS cell–derived HLCs by real-time reverse-transcription PCR. The gene expression levels in WT iPS cell–derived HLCs were taken as 1.0. (C) The amounts of ALB secretion were examined in WT iPS cell- and CYP2C19-KO iPS cell–derived HLCs by ELISA. (D) The expression of the hepatocyte markers (ALB and AAT) in human iPS cell–derived HLCs was examined by immunohistochemistry. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 50 μm. All data are represented as means ± S.D. (n = 3). *P < 0.05; **P < 0.01. BMP4, bone morphogenetic protein 4; HGF, hepatocyte growth factor; OsM, oncostatin M.
To characterize the global gene expression profiles in WT HLCs and CYP2C19-KO HLCs, microarray analysis was performed. This result showed that the gene expression of CYP2C19 was almost entirely eliminated in CYP2C19-KO HLCs (Fig. 4, shown in red letters). There was little difference in the gene expression levels of most genes (>99%) between WT HLCs and CYP2C19-KO HLCs (Fig. 4, shown in black dots). In addition, we extracted the 134 genes whose expression levels were up- or downregulated more than 3-fold and analyzed them by the GO enrichment clustering analysis. As shown in Supplemental Table 3, the top GO cluster—i.e., the cluster with the highest enrichment score—indicated that the 134 genes may not affect either hepatic differentiation or metabolic capacity. Furthermore, the gene expression levels of other CYP2 forms were not changed by knockout of CYP2C19 (Supplemental Fig. 1). We also confirmed that the expression levels of 231 genes that had the highest potential for off-target cleavage were not largely changed (Supplemental Fig. 2). These results suggest that the specific genome editing of the CYP2C19 locus was successfully accomplished by our strategy.

Transcriptomic analysis of CYP2C19-knockout human iPS cell–derived hepatocyte-like cells. Human iPS cells (FCL-iPS) were differentiated into the HLCs as described in the Materials and Methods section, and then the cells were analyzed by DNA microarray. A scatter plot of the gene expression signals in WT HLCs and CYP2C19-KO HLCs is shown. Red dots and blue dots indicate the genes whose expression levels were up- and downregulated more than 3-fold, respectively.
Drug-Metabolism Capacity of CYP2C19-KO Human iPS Cell–Derived Hepatocyte-Like Cells.
To determine whether CYP2C19-KO HLCs have a defect in the drug-metabolism capacity of CYP2C19, UPLC-MS/MS was performed (Fig. 5). The WT HLCs and CYP2C19-KO HLCs were treated with S-mephenytoin, which is known to be a substrate of CYP2C19, and the amount of 4′-hydroxymephenytoin, which is the metabolite of S-mephenytoin, was measured by UPLC-MS/MS analysis. The metabolic activity in CYP2C19-KO HLCs was significantly lower than that in WT HLCs (WT HLCs = 4408.4 pg/ml per minute per milligram protein, CYP2C19-KO HLCs = below the limit of detection). This result indicates that CYP2C19-KO HLCs defect the drug-metabolism capacity of CYP2C19.

Drug-metabolism capacity of CYP2C19-knockout human iPS cell–derived hepatocyte-like cells. Human iPS cells (FCL-iPS) were differentiated into the HLCs as described in the Materials and Methods section. The cytochrome P450–mediated drug-metabolizing capacities in WT iPS cell- and CYP2C19-KO iPS cell–derived HLCs were evaluated by quantifying the metabolites of cytochrome P450 substrates (S-mephenytoin; substrate for CYP2C19). The quantity of metabolites (4′-hydroxymephenytoin) was measured by UPLC-MS/MS. The results are shown as means ± S.D. (n = 6). Statistical significance was evaluated by unpaired two-tailed Student’s t tests (**P < 0.01).
Drug Response of CYP2C19-KO Human iPS Cell–Derived Hepatocyte-Like Cells.
Clopidogrel, a platelet aggregation inhibitor, is a prodrug that is metabolized into an active metabolite mainly by CYP2C19 (Wallentin et al., 2010; Gillette et al., 2016; Kiss et al., 2018). In a study using primary rat hepatocytes and HepG2 cells, Zhai et al. (2016) revealed that the hepatocellular toxicity of clopidogrel is associated with CYP2C19-mediated metabolism. Therefore, we expected that CYP2C19-KO HLCs would exhibit a different response to clopidogrel as compared with WT HLCs. To examine this, we treated WT HLCs and CYP2C19-KO HLCs with clopidogrel and measured the cell viability 48 hours later (Fig. 6). Contrary to our expectation, the cell viabilities of CYP2C19-KO HLCs as well as WT HLCs decreased in a dose-dependent manner. This result suggests that not only CYP2C19 but also other drug-metabolizing enzymes might contribute to the metabolism and toxicity of clopidogrel.

Drug response of CYP2C19-knockout human iPS cell–derived hepatocyte-like cells. Human iPS cells (FCL-iPS) were differentiated into the HLCs as described in the Materials and Methods section. The cell viability of WT iPS cell- and CYP2C19-KO iPS cell–derived HLCs was assessed by WST-8 assay after 48-hour exposure to different concentrations of clopidogrel. The cell viability was calculated as a percentage of cells treated with solvent only. The results are shown as means ± S.D. (n = 3).
Drug Response of Ketoconazole-Treated CYP2C19-KO Human iPS Cell–Derived Hepatocyte-Like Cells.
The results in Fig. 6 suggest the existence of other drug-metabolizing enzymes that contribute to the metabolism of clopidogrel, in addition to CYP2C19. Using the hepatoma cell line HepG2, Zahno et al. (2013) previously demonstrated that high CYP3A4 activity may be a risk factor for clopidogrel-associated hepatocellular toxicity. From these facts, we expected that CYP3A4 metabolizes clopidogrel into a hepatotoxic metabolite. To inhibit the CYP3A4 activity, we treated WT HLCs and CYP2C19-KO HLCs with ketoconazole, a CYP3A4 inhibitor, and assessed the cell viability after 48 hours of clopidogrel treatment (Fig. 7). As shown in Fig. 7A, the cell viabilities of both ketoconazole-treated WT HLCs and nontreated WT HLCs similarly decreased in a dose-dependent manner. However, the cell viability of ketoconazole-treated CYP2C19-KO HLCs was significantly higher than that of nontreated CYP2C19-KO HLCs (Fig. 7B). Taken together, these results suggest that the hepatocellular toxicity of clopidogrel can be predicted by using CYP2C19-KO HLCs in the presence of ketoconazole.

Drug response of ketoconazole-treated CYP2C19-knockout human iPS cell–derived hepatocyte-like cells. Human iPS cells (FCL-iPS) were differentiated into the HLCs as described in the Materials and Methods section. The HLCs were treated with ketoconazole (a CYP3A4 inhibitor) for 48 hours. (A, B) The cell viability of WT iPS cell- and CYP2C19-KO iPS cell–derived HLCs was assessed by WST-8 assay after 48-hour exposure to different concentrations of clopidogrel (A: WT HLC, B: CYP2C19-KO HLC). The cell viability was calculated as a percentage of cells treated with solvent only. The results are shown as means ± S.D. (n = 3). Statistical significance was evaluated by one-way ANOVA followed by Bonferroni’s post hoc tests (**P < 0.01: compared with “vehicle”).
We also examined whether similar results could be obtained using selective inhibitors of CYP3A4 and CYP2C19. WT HLCs were treated with both ketoconazole (CYP3A4 inhibitor) and N-3-benzyl-nirvanol (CYP2C19 inhibitor) (Suzuki et al., 2002) followed by clopidogrel treatment, and then the cell viability was examined (Supplemental Fig. 3). However, the cell viability was not enhanced by using these inhibitors. This result might be because the N-3-benzyl-nirvanol–mediated inhibition level of CYP2C19 activity was quite low (approximately 50%) (Supplemental Fig. 4). As shown here, it is difficult to synthesize a highly selective cytochrome P450 inhibitor, thus our CYP2C19-KO HLCs might be a useful model for PM.
Discussion
In this study, we established CYP2C19-KO iPS cells using our unique and highly efficient genome-editing technology. By differentiating the human iPS cells into HLCs, we obtained CYP2C19-KO HLCs. The CYP2C19 metabolic capacity decreased to below the limit of detection by deleting CYP2C19. The clopidogrel-induced hepatotoxicity test revealed a significant difference between the cell viability of WT HLCs and that of CYP2C19-KO HLCs in the presence or absence of ketoconazole. These results indicated that our CYP2C19-KO HLCs could be useful model cells for predicting the CYP2C19-mediated hepatocellular toxicity.
In general, clopidogrel is administered orally. After oral administration, 85% of clopidogrel is hydrolyzed at the intestine into inactive metabolites by carboxylesterase 1 and excreted, and the residual 15% reaches the liver and is metabolized there into active metabolites by cytochrome P450s (Simon et al., 2009; Kahma et al., 2018). Therefore, to perform a more specific pharmacokinetics study of orally administered drugs such as clopidogrel, a combined model consisting of hepatocytes and intestinal enterocytes will be needed. One such cell model has already been reported: a microphysiological system using PHHs as hepatocytes and Caco-2 cells as intestinal enterocytes (Tsamandouras et al., 2017). By applying this technology, it is anticipated that a microphysiological system consisting of enterocyte-like cells and HLCs, both of which are differentiated from the same individual-derived human iPS cells (e.g., CYP2C19-PM iPS cells), will be established in the new future. In other words, we can create an isogenic intestine and liver systems by using our CYP2C19-KO iPS cells, which would help us to predict the bioavailability of orally administered drugs in CYP2C19-PM individuals. Such a system would make a substantial contribution to the field of personalized medicine.
In addition to CYP2C19-PMs, CYP2C9-PMs or CYP2D6-PMs have also been reported (Gardiner and Begg, 2006). By adopting the technology that we showed in this report, it would be possible to obtain these PM model HLCs. Use of these cells is expected to establish the diverse hepatocyte panel, enabling the in vitro early-stage prediction of drug-induced hepatocellular toxicity with consideration for a multigenetic or multiethnic background. However, to mimic the pharmacokinetics in PM precisely, the model cells which do not have a deletion but have single nucleotide polymorphism (SNP) at the CYP2C19 locus, such as CYP2C19*2 or *3, will be needed (Gardiner and Begg, 2006; Isomura et al., 2010; Kiss et al., 2018). Recently, genome-editing technology that can introduce an SNP to human iPS cells has been developed (Kim et al., 2018). Applying this technology, we would like to introduce (or correct) an SNP at the CYP2C19 locus in human iPS cells and then generate HLCs from the cells. In addition, new SNPs that affect the symptoms of PM could be discovered from the candidate SNPs identified by genome-wide association studies.
In this study, we established a hepatocyte model of CYP2C19-PMs and demonstrated its usefulness as a tool for drug discovery. We hope our technology contributes to the advance of CYP-PM studies, and to the safe and appropriate use of drugs for CYP-PMs.
Acknowledgments
We thank Yasuko Hagihara, Natsumi Mimura, and Ayaka Sakamoto for their excellent technical support.
Authorship Contributions
Participated in research design: Takayama.
Conducted experiments: Deguchi, Igai, Harada, Takayama.
Performed data analysis: Deguchi, Yamashita, Harada, Toba, Takayama.
Wrote or contributed to the writing of the manuscript: Deguchi, Yamashita, Harada, Hirata, Takayama, Mizuguchi.
Footnotes
- Received January 9, 2019.
- Accepted April 2, 2019.
This research is supported by the Japan Agency for Medical Research and Development [Grants 19mk0101125h0002 and 19be0304320h0003].
The authors declare no competing financial interests.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ALB
- albumin
- CYP2C19-KO HLC
- CYP2C19-knockout human iPS cell-derived hepatocyte-like cell
- GO
- gene ontology
- HCM
- hepatocyte culture medium
- HLC
- hepatocyte-like cell
- human iPS cells
- human induced pluripotent stem cells
- MRM
- multiple-reaction monitoring
- PCR
- polymerase chain reaction
- PHH
- primary human hepatocyte
- PM
- poor metabolizer
- SNP
- single nucleotide polymorphism
- UPLC-MS/MS
- ultra-performance liquid chromatography–tandem mass spectrometry
- WT
- wild type
- WT HLC
- wild-type iPS cell–derived hepatocyte-like cell
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}