


Abstract
The antiplatelet drug clopidogrel is metabolized to an acyl-β-d-glucuronide, which causes time-dependent inactivation of CYP2C8. Our aim was to characterize the UDP-glucuronosyltransferase (UGT) enzymes that are responsible for the formation of clopidogrel acyl-β-d-glucuronide. Kinetic analyses and targeted inhibition experiments were performed using pooled human liver and intestine microsomes (HLMs and HIMs, respectively) and selected human recombinant UGTs based on preliminary screening. The effects of relevant UGT polymorphisms on the pharmacokinetics of clopidogrel were evaluated in 106 healthy volunteers. UGT2B7 and UGT2B17 exhibited the greatest level of clopidogrel carboxylic acid glucuronidation activities, with a CLint,u of 2.42 and 2.82 µl⋅min−1⋅mg−1, respectively. Of other enzymes displaying activity (UGT1A3, UGT1A9, UGT1A10-H, and UGT2B4), UGT2B4 (CLint,u 0.51 µl⋅min−1⋅mg−1) was estimated to contribute significantly to the hepatic clearance. Nonselective UGT2B inhibitors strongly inhibited clopidogrel acyl-β-d-glucuronide formation in HLMs and HIMs. The UGT2B17 inhibitor imatinib and the UGT2B7 and UGT1A9 inhibitor mefenamic acid inhibited clopidogrel carboxylic acid glucuronidation in HIMs and HLMs, respectively. Incubation of clopidogrel carboxylic acid in HLMs with UDPGA and NADPH resulted in strong inhibition of CYP2C8 activity. In healthy volunteers, the UGT2B17*2 deletion allele was associated with a 10% decrease per copy in the plasma clopidogrel acyl-β-d-glucuronide to clopidogrel carboxylic acid area under the plasma concentration-time curve from 0 to 4 hours (AUC0–4) ratio (P < 0.05). To conclude, clopidogrel carboxylic acid is metabolized mainly by UGT2B7 and UGT2B4 in the liver and by UGT2B17 in the small intestinal wall. The formation of clopidogrel acyl-β-d-glucuronide is impaired in carriers of the UGT2B17 deletion. These findings may have implications regarding the intracellular mechanisms leading to CYP2C8 inactivation by clopidogrel.
Introduction
The platelet P2Y12 adenosine diphosphate receptor inhibitor clopidogrel is widely used to reduce blood clotting in patients at high risk for vascular thrombotic events. Because of its complex pharmacokinetic profile, clopidogrel is susceptible to various drug-drug interactions and genetic polymorphisms resulting in variable drug response (Mega et al., 2009; Floyd et al., 2012; Frelinger et al., 2012; Tornio et al., 2014; Tarkiainen et al., 2015). Clopidogrel is a prodrug that is metabolized via two distinct routes. Approximately 10% of the absorbed dose undergoes a two-step oxidative metabolism pathway, producing the active cis 5-thiol metabolite, whereas most of the dose is metabolized by sequential hydrolysis and conjugation reactions into pharmacodynamically inactive clopidogrel carboxylic acid and clopidogrel acyl-β-d-glucuronide (Savi et al., 2000; Tang et al., 2006; Hagihara et al., 2009; Silvestro et al., 2011) (Fig. 1).

Metabolic pathways of clopidogrel.
The oxidative metabolism of clopidogrel is catalyzed by several cytochrome P450 (P450) isoforms, including CYP2C19, CYP1A2, CYP2B6, and CYP3A4/5 (Clarke and Waskell, 2003; Hagihara et al., 2009; Kazui et al., 2010; Dansette et al., 2012). The CYP2C19 loss-of-function alleles are associated with low plasma concentrations of the active metabolite and poor clopidogrel responsiveness (Hulot et al., 2006; Giusti et al., 2007; Sibbing et al., 2009; Simon et al., 2009; Hochholzer et al., 2010). Interestingly, clopidogrel is also a mechanism-based inhibitor of CYP2C19 and CYP2B6 (Richter et al., 2004; Nishiya et al., 2009). In healthy volunteers, pretreatment with clopidogrel significantly increased the area under the plasma concentration-time curve (AUC) of omeprazole in subjects with homozygous CYP2C19 extensive metabolizer genotype and the AUC of the CYP2B6 substrate bupropion (Turpeinen et al., 2005; Chen et al., 2009). The tendency to form reactive metabolites that irreversibly inactivate P450 enzymes has recently been observed also for the secondary metabolite, clopidogrel acyl-β-d-glucuronide. This glucuronide metabolite was shown to be a strong mechanism-based inhibitor of CYP2C8, leading to a marked increase in the plasma concentrations of repaglinide and pioglitazone (Tornio et al., 2014; Itkonen et al., 2016).
The hydrolysis of clopidogrel into clopidogrel carboxylic acid is catalyzed by carboxylesterase 1 (CES1) (Tang et al., 2006), as demonstrated clinically by the increased antiplatelet effects of clopidogrel in carriers of a loss-of-function single-nucleotide variant of CES1 (Tarkiainen et al., 2015). What is not known, yet, is which UGT enzymes catalyze the glucuronidation of clopidogrel carboxylic acid into clopidogrel acyl-β-d-glucuronide. Identifying the UGT enzymes responsible for this activity is particularly interesting since the metabolic pathway leads to the formation of a bioactive glucuronide metabolite that inactivates CYP2C8. To this end, we carried out an in vitro study to elucidate the role of 13 human UGTs in the formation of clopidogrel acyl-β-d-glucuronide. Moreover, to gain insight into the clinical relevance of the findings, we investigated the effects of common variants in relevant UGT genes on the pharmacokinetics of clopidogrel carboxylic acid glucuronidation in a population comprising healthy volunteers from previous and ongoing pharmacokinetic studies.
Materials and Methods
Materials
Pooled HLMs (UltraPool), pooled HIMs, and microsomes from baculovirus-infected insect cells expressing recombinant human UGT enzymes (Supersomes) were purchased from Corning (Corning, NY), except for His-tagged UGT1A10 (UGT1A10-H), which was prepared in Helsinki as previously described (Kuuranne et al., 2003). The rapid equilibrium dialysis device (RED) was obtained from Thermo Scientific (Waltham, MA). Aprepitant, S-(+)-clopidogrel hydrogen sulfate, (±)-clopidogrel carboxylic acid, (±)-clopidogrel-d4 carboxylic acid, gemfibrozil, N-desethylamodiaquine hydrochloride, and N-desethylamodiaquine-d5 were purchased from Toronto Research Chemicals (North York, ON, Canada). Amodiaquine dihydrochloride dihydrate, (±)-ketamine hydrochloride, β-NADPH and UDPGA were from Sigma-Aldrich (St. Louis, MO). Clopidogrel acyl-β-d-glucuronide was obtained from Santa Cruz Biotechnology (Dallas, TX), fluconazole and mefenamic acid were from Pfizer (Sandwich, UK), and imatinib mesylate was from Sequoia Research Products, (Pangbourne, UK). Other solvents and reagents were of analytical grade.
In Vitro Study
UGT Incubation Conditions.
Human recombinant UGTs (hrUGTs), pooled human liver microsomes (HLMs), or pooled human intestinal microsomes (HIMs) were diluted in 0.1 M sodium phosphate buffer (pH 7.4) containing MgCl2 (10 mM). In addition, the pore-forming peptide alamethicin (10 µg⋅ml−1) was added to incubation mixtures containing HLMs or HIMs. The protein concentrations in hrUGT, HLM, and HIM incubations ranged from 0.2 to 0.8 mg⋅ml−1, as described for each assay as follows. Clopidogrel carboxylic acid concentration ranged from 0.5 to 3000 µM, depending on the experiment. HLM and HIM incubation mixtures were kept on ice for 20 minutes before preincubation to allow pore formation by alamethicin. For consistency, the same treatment was applied to hrUGT incubations. The incubation mixtures were then moved to a heated shaker and preincubated for 5 minutes (at 37°C, 300 rpm), followed by the addition of 5 mM uridine-diphosphate glucuronic acid (UDPGA) to initiate the reactions. Incubations were terminated after a designated time by mixing one part of reaction mixture with three parts acetonitrile containing clopidogrel-d4 carboxylic acid as an internal standard. Samples were vortexed and cooled on ice for at least 10 minutes before centrifugation (20,800g for 10 minutes). Finally, the supernatants were transferred to liquid chromatography (LC) microvials for high performance LC-tandem mass spectrometry analysis. The nonspecific binding of 0.5 µM clopidogrel carboxylic acid to HLMs (0.4 and 0.8 mg⋅ml−1) and HIMs (0.2 mg⋅ml−1), 1 and 30 µM clopidogrel carboxylic acid to hrUGT2B17 (0.2 mg⋅ml−1), and 100 µM clopidogrel to HLMs and HIMs (0.8 mg⋅ml−1) was determined by equilibrium analysis using the rapid equilibrium dialysis method. The fraction unbound was at least 0.8 in all incubations. Substrate and inhibitor compounds were dissolved in acetonitrile and methanol, respectively. The final solvent concentrations in incubations were 1% or less. All incubations were performed in at least duplicates.
UGT Screening.
To identify the specific UGTs responsible for clopidogrel carboxylic acid metabolism, glucuronidation was tested in human recombinant UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, His-tagged UGT1A10 (UGT1A10-H), UGT2B4, UGT2B7, UGT2B10, UGT2B15, and UGT2B17 at a final protein concentration of 0.4 mg⋅ml−1. In this experiment, clopidogrel carboxylic acid (10 µM) was incubated with each UGT enzyme for 30 and 90 minutes as described in the preceding section.
Kinetic Analyses.
Based on the results of the preliminary screening, UGT1A3, UGT1A9, UGT1A10-H, UGT2B4, UGT2B7, and UGT2B17 were selected for further kinetic analyses. Incubations were conducted as previously described using a protein concentration of 0.2 mg⋅ml−1 and a minimum of seven different substrate concentrations ranging from 1 to 3000 µM for each hrUGT assay. Reactions were terminated after a 20-minute incubation.
Clopidogrel Carboxylic Acid Metabolism in HLM, HIM, hrUGT2B7, and hrUGT2B17.
To determine the maximal linear range of glucuronidation, depletion of 0.5 µM clopidogrel carboxylic acid and formation of clopidogrel acyl-β-d-glucuronide were measured in HLMs, HIMs, and recombinant UGT2B7 and UGT2B17 (0.4 and 0.8 mg⋅ml−1) for up to 90 minutes. This clopidogrel carboxylic acid concentration is within the clinically relevant range since its average peak plasma concentrations (Cmax) in the clinical data were 35 and 72 µM, corresponding to unbound concentrations (Cmax,u) of 0.7 and 1.4 µM for the 300- and 600-mg dose, respectively.
Inhibition Experiments.
Clopidogrel carboxylic acid glucuronidation in HLMs, HIMs, UGT2B7, and UGT2B17 was investigated in the absence and presence of the potential UGT2B7 inhibitors aprepitant, fluconazole, gemfibrozil, ketamine, and mefenamic acid, as well as the UGT2B17 inhibitor imatinib (Uchaipichat et al., 2006, 2011; Knights et al., 2009; House et al., 2015; Zhang et al., 2015). In addition, the selectivity of inhibition by gemfibrozil, mefenamic acid, and imatinib was further investigated in UGT1A3, UGT1A9, UGT1A10-H, and UGT2B4. Inhibition experiments were performed using clopidogrel carboxylic acid concentration of 0.5 µM for HLM and HIM incubations (0.4 mg⋅ml−1) and 5 µM for hrUGT incubations (0.2 mg⋅ml−1). Concentrations of aprepitant, fluconazole, gemfibrozil, ketamine, mefenamic acid, and imatinib in the incubations were 10, 2500, 100, 500, 5, and 5 µM, respectively.
Metabolism of Parent Clopidogrel in HLMs and HIMs.
Clopidogrel carboxylic acid and clopidogrel acyl-β-d-glucuronide formation in HLMs and HIMs (0.8 mg⋅ml−1) was investigated by incubating 10 or 100 µM clopidogrel for 90 minutes. Incubation mixtures were prepared as described for UGT incubation conditions, except that UDPGA was added to the mixture before preincubation and the reaction was initiated by the addition of the substrate.
Clopidogrel Carboxylic Acid Metabolism and CYP2C8 Inhibition in HLMs.
The formation of clopidogrel acyl-β-d-glucuronide from clopidogrel carboxylic acid and the subsequent inactivation of CYP2C8 were studied in HLMs using amodiaquine N-desethylation as a marker reaction for CYP2C8 activity. Briefly, incubation mixtures containing clopidogrel carboxylic acid (50 µM) in HLMs (0.5 mg⋅ml−1) were prepared as described previously; however, after preincubation for 5 minutes, a combined cofactor solution containing both 5 mM UDPGA (to initiate glucuronidation) and 1 mM NADPH (to initiate metabolism-dependent inactivation of CYP2C8 by clopidogrel acyl-β-d-glucuronide) was added to the mixtures. At determined time points, aliquots of 7.5 µl of the reaction mixture were transferred to other tubes containing amodiaquine (10 µM) and NADPH (1 mM) in buffer in a final incubation volume of 150 µl to measure CYP2C8 activity. Protein and inhibitor concentrations were thus diluted 20-fold, and direct inhibition by clopidogrel acyl-β-d-glucuronide was minimized by using an amodiaquine concentration greater than its Km for CYP2C8. The reaction mixture was incubated for 2 minutes (37°C, 300 rpm), and the reaction was stopped by mixing a 50-µl sample with 150 µl of stop solution (0.3% formic acid in acetonitrile:methanol:water 90:5:5 v/v/v) containing deuterium-labeled N-desethylamodiaquine as an internal standard. Samples were vortexed, kept on ice for at least 10 minutes, and centrifuged (20,800g for 10 minutes). The supernatant was transferred to silanized glass tubes and evaporated to dryness under nitrogen. The residue was reconstituted with 50 µl of mobile phase A/mobile phase B (95:5, v/v) (see Quantification of Drug Concentrations). To confirm inhibition by clopidogrel acyl-β-d-glucuronide, controls without clopidogrel carboxylic acid and controls without either NADPH or UDPGA in the predilution incubation mixture were prepared as described herein. Separate samples for clopidogrel carboxylic acid and clopidogrel acyl-β-d-glucuronide quantification were also collected directly after N-desethylamodiaquine sampling. The samples were treated and prepared as described for UGT incubation conditions.
Quantification of Drug Concentrations.
The drug concentrations were measured using a Nexera X2 (Shimadzu, Kyoto, Japan) or an Agilent 1100 series liquid chromatograph (Agilent Technologies, Waldbronn, Germany) coupled to a 5500 Qtrap, API3000 or API2000 tandem mass spectrometer (AB Sciex, Toronto, ON, Canada). Clopidogrel carboxylic and clopidogrel acyl-β-d-glucuronide were separated on a Sun Fire C18 analytical column (150 × 2.1 mm, 3 µm particle size; Waters, Milford, MA) using isocratic mobile phase conditions (2 mM ammonium acetate; pH 5.5:acetonitrile 58:44 v/v). The flow rate and the column oven temperature were set at 0.26 ml⋅min−1 and 30°C. The mass spectrometer was operated in positive turbo ion spray mode with the target ion transitions (m/z) of 308–198 and 484–308 for clopidogrel carboxylic acid and clopidogrel acyl-β-d-glucuronide, respectively. Deuterium-labeled clopidogrel carboxylic acid served as an internal standard for both analytes. The lower limits of quantitation of clopidogrel carboxylic acid and clopidogrel acyl-β-d-glucuronide were 0.0038 and 0.0023 µM. The chromatographic separation for N-desethylamodiaquine was performed on an Atlantis T3 column (100 × 2.1 mm, 3 µm particle size; Waters, Milford, MA) using gradient elution as previously described (Filppula et al., 2012). The mobile phases consisted of 0.3% formic acid in 5 mM ammonium formate (mobile phase A) and 0.3% formic acid in 90/5/5 acetonitrile/methanol/water (mobile phase B), and the limit of quantification for N-desethylamodiaquine was 0.001 µM. The day-to-day coefficients of variation were less than 15% at relevant concentrations for all analytes, except for the lowest quality control (3 nM) of N-desethylamodiaquine, whose coefficient of variation was 18%. None of the other drugs or drug metabolites interfered with the assays.
Data Analysis.
The kinetics of clopidogrel acyl-β-d-glucuronide formation were analyzed using SigmaPlot, version 13.0 (Systat Software, San Jose, CA). Selection of the best-fit enzyme model was based on the Akaike information criterion, the r2 values, 95% confidence intervals (CI), and examination of Michaelis-Menten and Eadie-Hofstee plots. The data were analyzed with the following models:
Michaelis-Menten
 (1)
(1)

where v is the initial velocity of the enzyme reaction, Vmax is the maximal reaction velocity, [S] is the substrate concentration, and Km is the Michaelis-Menten constant.
Substrate inhibition
(2)
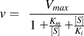
where Ki represents the dissociation constant for the inhibitory substrate-enzyme-substrate ternary complex.
Sigmoidal (Hill)
(3)
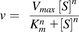
where n is the Hill coefficient.
Two binding sites
(4)
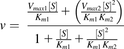
where Km1 and Km2 represent the Michaelis-Menten constants for the binding of the substrate molecules to the first and second binding site, respectively (Korzekwa et al., 1998).
Intrinsic clearance for clopidogrel acyl-β-d-glucuronide formation (CLint) was calculated according to CLint = Vmax/Km. This equation can be used for determination of CLint and reaction phenotyping when the substrate concentration is less than 10% of the Km. In our clinical study, the average Cmax,u was less than 10% of the lowest Km of the recombinant UGTs. Likewise, the substrate concentrations used for CLint,u,dep determination and the inhibition experiments in HLMs and HIMs were only 3% of the lowest Km, corresponding to the clinically relevant concentration range. Pseudo-first-order depletion rate constants (kdep) were determined for the depletion of 0.5 µM clopidogrel carboxylic acid in HLMs and HIMs using nonlinear regression analysis (SigmaPlot). The intrinsic clearance in depletion experiments was calculated as CLint,dep = kdep/[M], where [M] is the microsomal protein concentration (Venkatakrishnan et al., 2003). The relative contributions of UGT1A3, UGT1A9, UGT2B4, UGT2B7, and UGT2B17 in the hepatic metabolism of clopidogrel carboxylic acid were estimated from the determined CLint values using a conversion factor, which was determined from previously published peptide-based quantification studies by dividing the molar abundancy of each UGT isoform in human liver microsomes with the molar abundancy of the respective hrUGT isoform in baculovirus-infected insect cells (Supersomes) (Fallon et al., 2013a,b; Sato et al., 2014). The relative contribution of each UGT isoform in clopidogrel carboxylic acid glucuronidation was calculated by multiplying recombinant CLint values with the respective conversion factor. The CLint, in vivo value for the hepatic glucuronidation of clopidogrel carboxylic acid was calculated using the standard in vitro-in vivo scaling paramaters of 40 mg of microsomal protein per gram liver (Houston and Galetin, 2008) and 25.7 g of liver per kilogram body weight (Davies and Morris, 1993).
Clopidogrel Pharmacokinetics and Pharmacogenetic Analysis in Healthy Volunteers
Subjects and Study Design.
A total of 106 healthy Finnish volunteers from eight of our previous or ongoing pharmacokinetic studies (49 women and 57 men; mean ± S.D.: age 24 ± 4 years, height 175 ± 9 cm, weight 71 ± 13 kg, and body mass index 23 ± 3 kg/m2), including six drug-drug interaction studies and two genotype panel studies, were available for pharmacogenetic analysis (Supplemental Table 1) (Holmberg et al., 2014; Tornio et al., 2014; Itkonen et al., 2015, 2016; Tarkiainen et al., 2015 Holmberg et al., unpublished data; Itkonen et al., unpublished data). No individuals with a non-Caucasian background or excess relatedness were included in the study. During these studies, each subject was given a single 300 mg (n = 49) or 600 mg (n = 57) dose of clopidogrel in the morning after an overnight fast. Timed EDTA blood samples, including a sample for DNA extraction, were drawn before and at designated times after clopidogrel ingestion. Plasma concentrations of clopidogrel and its metabolites were quantified using LC-mass spectrometry, as described in the respective studies. The health of the subjects was confirmed by their medical history, physical examination, and routine laboratory tests before entering the study. All subjects had normal blood platelet counts and hemoglobin values. None was a tobacco smoker, and none used any continuous medication (e.g., oral contraceptives). A written informed consent was obtained from all study participants for participation in the pharmacogenetic studiy, and each study was approved by the Coordinating Ethics Committee of the Helsinki and Uusimaa Hospital District and the Finnish Medicines Agency Fimea.
Pharmacokinetics.
For the present analysis, Cmax, time to Cmax (tmax), the elimination half-life (t1/2), and the area under the plasma concentration-time curve from 0 to 4 hours (AUC0–4) of clopidogrel carboxylic acid and clopidogrel acyl-β-d-glucuronide were calculated for each of the 106 subjects using uniform noncompartmental methods with Phoenix WinNonlin, version 6.3 (Certara, St Louis, MO). The clopidogrel acyl-β-d-glucuronide to clopidogrel carboxylic acid plasma AUC ratio was used as an index of the rate of clopidogrel carboxylic acid glucuronidation. The concentration-time points that were available from all the eight studies (0, 1, 1.5, 2, 3, and 4 hours postdose) were used in the pharmacokinetic calculations.
DNA Extraction, Genotyping, and Copy Number Determination.
Genomic DNA was extracted using the Maxwell 16 LEV Blood DNA kit on a Maxwell 16 Research automated nucleic acid extraction system (Promega, Madison, WI). The samples were genotyped for the UGT2B7*2 allele by genotyping the rs7668258 (c.-161T>C) single nucleotide variation, which is in a complete linkage disequilibrium (r2 = 1.00) (1000 Genomes Project, http://www.internationalgenome.org) with the rs7439366 (c.802T>C, p.Y268H) single nucleotide variation defining *2, with a TaqMan genotyping assay (C__27827970_40) on a QuantStudio 12K Flex Real-Time PCR system (Thermo Fisher Scientific Waltham, MA). The UGT2B17*2 deletion allele was genotyped with the TaqMan copy number assay Hs03185327_cn, targeting the UGT2B17 exon 1 and intron 1 region, with RPPH1 (Ribonuclease P RNA component H1) as the reference gene, on the QuantStudio 12K Flex Real-Time PCR System.
Statistical Analysis.
The in vitro results are expressed as arithmetic means with S.D. The pharmacokinetic variables were logarithmically transformed before analysis and presented as geometric means with 95% CI. The comparisons between genotypes were carried out using forward stepwise linear regression with UGT2B7 and UGT2B17 genotypes, weight, body mass index, age, sex, and dose as independent variables. Additive coding was used for the genotypes. Differences were considered statistically significant when P was below 0.05. The data were analyzed using IBM SPSS Statistics, version 24 (Chicago, IL).
Results
UGT Screening and Kinetic Analyses.
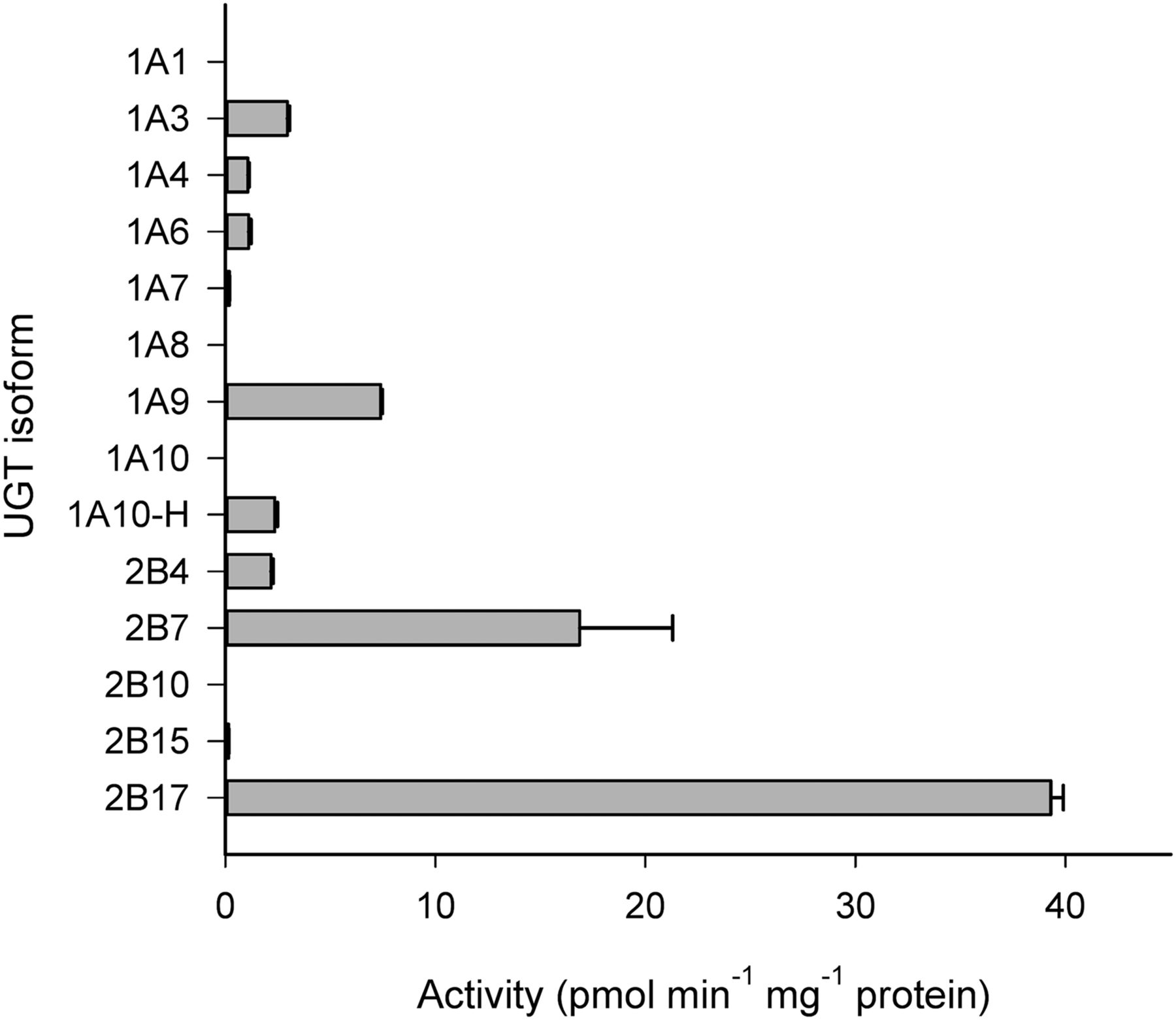
Among the 13 UGT enzymes screened, UGT2B17 and UGT2B7 displayed the highest level of activities at 10 µM clopidogrel carboxylic acid concentration (i.e., 39.3 and 16.9 pmol⋅min−1⋅mg−1 protein at 90 minutes, respectively) (Fig. 2). UGT1A3, UGT1A9, UGT1A10-H, and UGT2B4 also showed significant activities, between 5% and 20% of the UGT2B17 rates. The glucuronidation activities of UGT1A4, UGT1A6, UGT1A7, and UGT2B15 were very low or negligible, and these enzymes were thus excluded from detailed kinetic analyses. UGT1A1, UGT1A8, UGT1A10, and UGT2B10 did not form any detectable clopidogrel acyl-β-d-glucuronide.

Glucuronidation of 10 µM clopidogrel carboxylic acid in human recombinant UGTs. Each column represents the mean ± S.D. of duplicate incubations.
The kinetics of clopidogrel carboxylic acid glucuronidation by UGT1A3, UGT1A9, UGT2B7, and UGT2B17 followed the Michaelis-Menten model (Fig. 3). For UGT1A10-H, several kinetic models gave a reasonable fit, and the Michaelis-Menten model was chosen based on confidence in the parameter estimates. Clopidrogel carboxylic acid glucuronidation by UGT2B4 displayed nonhyperbolic kinetics, and the best fit was obtained with the two binding sites model. The calculated kinetic parameters, including parameters that are not shown in Fig. 3, are summarized in Table 1.

Enzyme kinetics of clopidogrel carboxylic acid glucuronidation by human recombinant UGT1A3 (A), UGT1A9 (B), UGT1A10-H (C), UGT2B4 (D), UGT2B7 (E), and UGT2B17 (F). Eadie-Hofstee plots are shown as insets. Each point represents the mean ± S.D. of at least duplicate incubations.
Kinetic parameters (mean ± S.D.) of clopidogrel carboxylic acid glucuronidation by UGT1A3, UGT1A9, UGT1A10-H, UGT2B4, UGT2B7, and UGT2B17 and scaling of their contributions to the total microsomal intrinsic clearance in the liver and intestine
To allow estimation of the clinical relevance of each UGT in the metabolism of clopidogrel, we scaled the in vitro CLint,u data to the average hepatic and intestinal microsomal intrinsic clearance on the basis of published protein expression data. Despite its weaknesses, this scaling method could be used to approximate the contributions of most UGTs identified in this study. UGT2B17 and UGT2B7 exhibited the highest CLint,u values of clopidogrel carboxylic acid glucuronidation (Table 1). After abundancy-based scaling, UGT2B7 was estimated to have the highest unbound intrinsic liver microsomal clearance (CLint,u,HLM), accounting for about 55%–60% of the total (combined) intrinsic clearance (CLint,u,HLM,tot). UGT2B7 was followed by UGT1A9, UGT2B4, and UGT2B17, whose estimated combined contribution to the metabolism of clopidogrel carboxylic acid was approximately 40% of the CLint,u,HLM,tot. However, the estimated contribution of UGT1A3 was only minor. In the intestine, the scaled unbound intrinsic clearance (CLint,u,HIM) of UGT2B17 accounted for 87% of the total unbound intrinsic clearance (CLint,u,HIM,tot), whereas the contribution of UGT2B7 to CLint,u,HIM,tot was only about 10%, and those of UGT1A10-H and UGT1A3 were <1%.
Clopidogrel Carboxylic Acid Depletion in HLM, HIM, UGT2B7, and UGT2B17.
In a substrate depletion experiment, approximately 15%–30% of the initial clopidogrel carboxylic acid concentration (0.5 µM) was consumed during a 90-minute incubation with UDPGA in HLMs or HIMs. In recombinant UGT2B7 and UGT2B17, 5%–10% of the initial substrate concentration was consumed during similar incubations. In all experiments, depletion was log-linear until at least 30 minutes, allowing for estimation of the intrinsic clearance from the results (Table 2).
Kinetic parameters of clopidogrel carboxylic acid (0.5 µM) depletion in HLMs, HIMs, and human recombinant UGT2B7 and UGT2B17 at protein concentration of 0.4 mg⋅ml−1
Inhibition of Clopidogrel Carboxylic Acid Glucuronidation by Chemical Inhibitors of UGTs.
In preliminary experiments in hrUGT2B7 and hrUGT2B17, aprepitant (10 µM), fluconazole (2.5 mM), and ketamine (500 µM) inhibited both UGT2B7- and UGT2B17-mediated clopidogrel carboxylic acid glucuronidation by >90% (Fig. 4A). Gemfibrozil (100 µM) showed very strong UGT2B7 inhibition (almost 100%), but it also showed moderate inhibition (30%–70%) of other UGTs, including UGT1A9, UGT1A10-H, UGT2B4, and UGT2B17 (Fig. 4B). Mefenamic acid (5 µM) had a strong inhibitory effect on both UGT2B7 (95%) and UGT1A9 (98%), but it had no inhibitory effect on UGT1A3, UGT2B4, and UGT2B17. In contrast, 5 µM imatinib strongly and selectively inhibited UGT2B17 activity (almost 100% inhibition), with only a small inhibitory effect on UGT2B7 (16% inhibition) and a moderate effect on UGT1A10-H (59%).

Effects of chemical inhibitors on clopidogrel carboxylic acid glucuronidation in vitro. Aprepitant (10 µM), fluconazole (2.5 mM), and ketamine (500 µM) (A) and gemfibrozil (100 µM), mefenamic acid (5 µM), and imatinib (5 µM) (B) were incubated with 5 µM clopidogrel carboxylic acid in human recombinant UGTs (0.2 mg⋅ml−1) or 0.5 µM clopidogrel carboxylic acid in HLMs and HIMs (0.4 mg⋅ml−1) (C) for 15 minutes. Complete inhibition of UGT2B7 was caused by gemfibrozil (100 µM) and of UGT2B17 by imatinib (5 µM). Each column represents the mean ± S.D. of duplicate incubations.
The UGT2B7 and UGT1A9 inhibitor mefenamic acid inhibited clopidogrel acyl-β-d-glucuronide formation in HLMs by 38% but only slightly in HIM (12%), whereas gemfibrozil inhibited clopidogrel glucuronidation in both HLMs and HIMs (34%–43%) (Fig. 4C). The preferential UGT2B17 inhibitor imatinib inhibited clopidogrel carboxylic acid glucuronidation only in HIMs (by 85%). The nonselective UGT2B inhibitors aprepitant, fluconazole, and ketamine substantially inhibited clopidogrel acyl-β-d-glucuronide formation in both HLMs and HIMs (by 60%–90%).
Metabolism of Parent Clopidogrel in HLMs and HIMs.
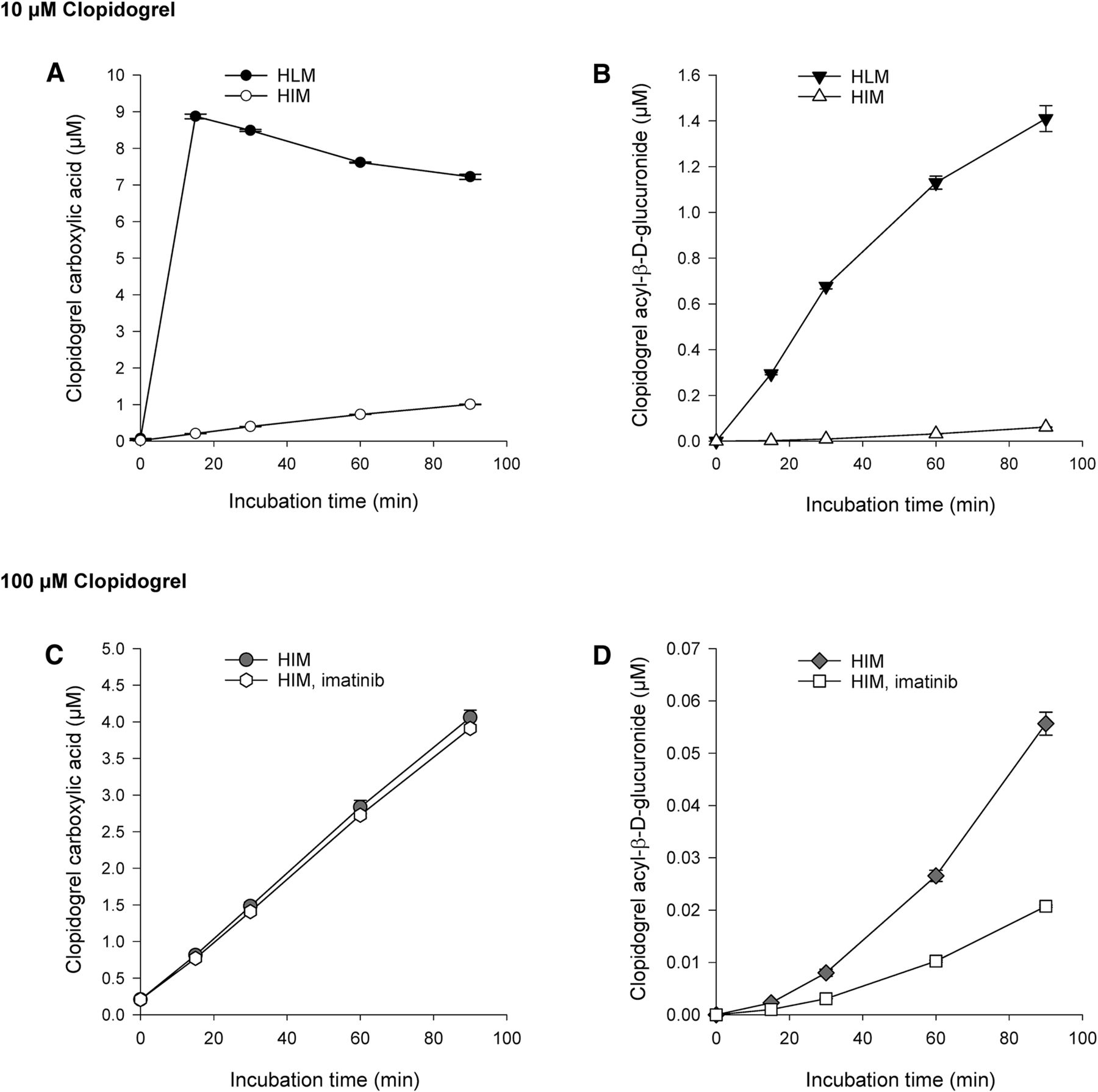
During incubation of 10 µM parent clopidogrel in HLMs, clopidogrel carboxylic acid concentration rose to 8.9 µM rapidly, in 15 minutes, followed by a gradual decrease (Fig. 5A). At the same time, clopidogrel carboxylic acid was further metabolized to clopidogrel acyl-β-d-glucuronide, whose concentration reached 19% of that of clopidogrel carboxylic acid at 90 minutes (Fig. 5B). In HIMs, the formation of clopidogrel carboxylic acid and clopidogrel acyl-β-d-glucuronide from 10 µM clopidogrel was substantially slower than in HLMs. At 90 minutes, clopidogrel carboxylic acid and clopidogrel acyl-β-d-glucuronide concentrations were 1.0 and 0.06 µM, respectively (Fig. 5, A and B). After incubating 100 µM clopidogrel in HIMs for 90 minutes, clopidogrel carboxylic acid concentration rose to 4.1 µM, whereas the concentration of clopidogrel acyl-β-d-glucuronide rose to the same level (0.06 µM) as in the experiment with 10 µM clopidogrel (Fig. 5, C and D). Coincubation with 5 µM imatinib in HIMs did not affect clopidogrel carboxylic acid formation but markedly reduced clopidogrel acyl-β-d-glucuronide formation (by 62% at 90 minutes) (Fig. 5D).

Metabolism of 10 µM clopidogrel in HLMs and HIMs (A and B) and 100 µM clopidogrel in HIMs (C and D) (0.8 mg⋅ml−1). Each point represents the mean ± S.D. of duplicate incubations.
UGT-Mediated Bioactivation of Clopidogrel Carboxylic Acid and Subsequent Inactivation of CYP2C8.
Preincubation of 50 µM clopidogrel carboxylic acid for 90 minutes in HLMs in the presence of UDPGA or a combined cofactor solution containing both UDPGA and NADPH produced approximately 19 µM clopidogrel acyl-β-d-glucuronide. After preincubation of clopidogrel carboxylic acid with both UDPGA and NADPH for 90 minutes, amodiaquine N-desethylation (CYP2C8 marker reaction) was inhibited by 53% (Fig. 6). No inhibition of amodiaquine N-desethylation could be observed when amodiaquine was incubated with a corresponding sample from preincubation mixtures containing only either one of the two cofactors.

The effect of a preincubation containing UDPGA, NADPH, and clopidogrel carboxylic acid (50 µM) on the metabolism of amodiaquine (10 µM) by CYP2C8. Each column represents the mean ± S.D. of duplicate incubations.
Clopidogrel Pharmacokinetic and Pharmacogenetic Analysis in Healthy Volunteers.
Linear regression analysis of the pharmacokinetic data obtained in 106 healthy volunteers revealed that of the tested genetic covariates, the UGT2B17*2 deletion allele was independently associated with pharmacokinetic variables of clopidogrel carboxylic acid and its acyl-β-d-glucuronide. In addition, the clopidogrel dose was independently associated with the AUC and AUC ratio values of these clopidogrel metabolites (P < 0.05). Each UGT2B17*2 deletion allele reduced the clopidogrel acyl-β-d-glucuronide to clopidogrel carboxylic acid plasma AUC0–4 ratio by 10.1% (95% CI, 1.7%–17.8%; P = 0.021) (Fig. 7A). This AUC ratio was 47% higher with the 600-mg dose than with the 300-mg dose of clopidogrel (95% CI, 28%–68%; P < 0.001). Moreover, UGT2B17*2 had a significant association with the t1/2 of clopidogrel carboxylic acid and clopidogrel acyl-β-d-glucuronide and the AUC0–4 of clopidogrel acyl-β-d-glucuronide (Fig. 7B). Each UGT2B17*2 allele prolonged the t1/2 of clopidogrel carboxylic acid by 8.7% (95% CI, 2.2%–15.7%; P = 0.008) and the t1/2 of clopidogrel acyl-β-d-glucuronide by 6.5% (95% CI, 0.34%–13.0%; P = 0.039) and decreased the AUC0–4 of clopidogrel acyl-β-d-glucuronide by 11.9% (95% CI, 1.1%–21.5%; P = 0.032). On the other hand, the *2 polymorphism in the UGT2B7 gene did not have a significant association with the AUC0–4 or t1/2 values of clopidogrel, the active metabolite, clopidogrel carboxylic acid, and clopidogrel acyl-β-D-glucuronide or with the clopidogrel acyl-β-d-glucuronide to clopidogrel carboxylic acid plasma AUC ratio.
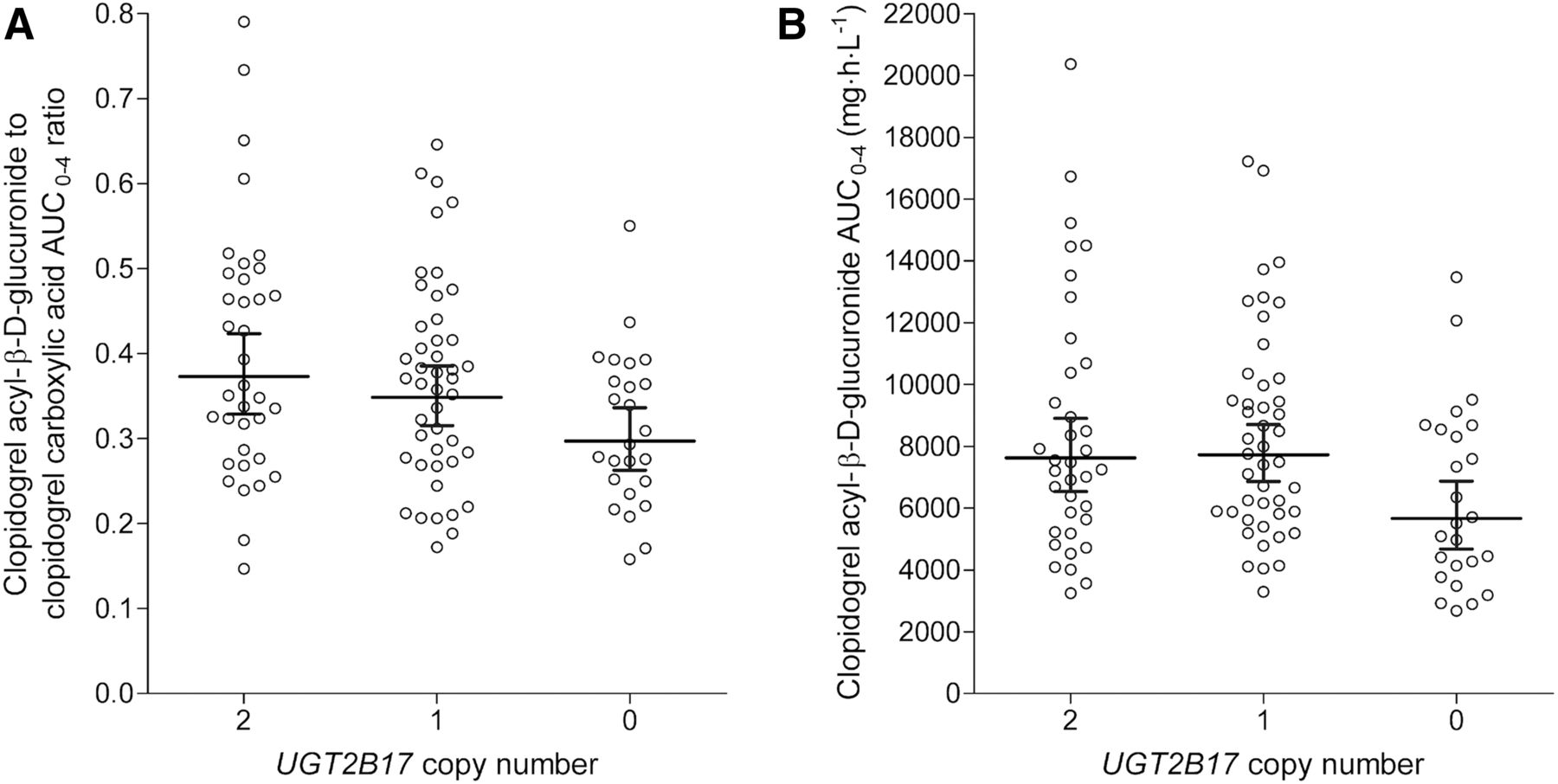
Effect of UGT2B17 copy number variation on the dose-adjusted clopidogrel acyl-β-d-glucuronide to clopidogrel carboxylic acid plasma AUC0–4 ratio (A) and the dose-adjusted clopidogrel acyl-β-d-glucuronide AUC0–4 (B). Central line shows the geometric mean, and bars show the 95% CI. Dose adjustments to the 300-mg dose were carried out on the basis of the corresponding regression coefficients.
Discussion
In the present study, we investigated the glucuronidation of clopidogrel carboxylic acid in vitro and assessed the relative contributions of the different UGT enzymes to clopidogrel acyl-β-d-glucuronide formation by using two in vitro approaches. Both an approach with chemical UGT inhibitors in HLMs and a scaling approach of recombinant UGT-mediated clearances using peptide-based quantification data from previous publications (Fallon et al., 2013a,b; Sato et al., 2014) were consistent with an important role of UGT2B7 and UGT2B17 in the glucuronidation of clopidogrel carboxylic acid. In addition, UGT1A3, UGT1A9, UGT1A10-H, and particularly UGT2B4 were estimated to participate in clopidogrel carboxylic acid glucuronidation. The role of UGT2B17 could also be verified in a clinical pharmacogenetic study.
Fluconazole and ketamine strongly inhibit UGT2B4, UGT2B7, and UGT2B17, with only minor effects on other relevant UGTs (Uchaipichat et al., 2006, 2011; Raungrut et al., 2010; House et al., 2015). In HLMs, they reduced clopidogrel carboxylic acid glucuronidation by up to 90%, suggesting that, collectively, UGT2B4, UGT2B7, and UGT2B17 are responsible for most of the clopidogrel acyl-β-d-glucuronide formation in the liver. Furthermore, the nonselective UGT2B7 inhibitors mefenamic acid and gemfibrozil inhibited clopidogrel carboxylic acid glucuronidation in HLMs by about 40%. Accordingly, UGT2B7 is likely to play a large role in the formation of clopidogrel acyl-β-d-glucuronide in the liver. On the other hand, the UGT2B17 inhibitor imatinib had practically no effect on clopidogrel carboxylic acid glucuronidation, suggesting a very small role for UGT2B17 in the liver.
In the liver, UGT2B7, UGT2B4, and UGT1A9 are relatively abundant, whereas the expression of UGT2B17 is low (Oda et al., 2012; Achour et al., 2014; Sato et al., 2014). Consequently, the abundancy-based scaling suggested that UGT2B7 accounts for almost 60% of the total unbound hepatic microsomal intrinsic clearance of clopidogrel carboxylic acid, whereas particularly UGT2B4, but also UGT2B17 and UGT1A9, account for the remaining CLint,u,HLM,tot. This result is in agreement with the inhibition results. Yet, considering the effects of the various UGT2B inhibitors, this scaling probably slightly overestimated the contributions of UGT2B7 and UGT1A9 and underestimated the contribution of UGT2B4.
UGT2B17, UGT2B7, and UGT1A10 are expressed in the intestinal wall, but most other relevant UGTs are virtually absent (Sato et al., 2014). The strong (85%) inhibition of clopidogrel acyl-β-d-glucuronide formation by the UGT2B17 inhibitor imatinib in HIMs indicates that UGT2B17 is mostly responsible for clopidogrel carboxylic acid metabolism in the intestinal wall. Since UGT1A9 is only minimally expressed in enterocytes (Oda et al., 2012), the minor (12%) inhibition by the UGT2B7 and UGT1A9 inhibitor mefenamic acid in HIMs suggests that UGT2B7 also has a small role. In excellent agreement, the scaling approach estimated a major role for UGT2B17 in the unbound intestinal intrinsic clearance, UGT2B7 having a less significant role. In addition to these UGT2B enzymes, UGT1A10 may contribute slightly to the metabolism of clopidogrel carboxylic acid in the intestinal wall since imatinib weakly inhibits UGT1A10 and the scaling suggested a minimal role for UGT1A10 in the CLint,u,HIM,tot.
Analysis of clinical data provided the final approach to investigate the contributions of UGT2B7 and UGT2B17 to the glucuronidation of clopidogrel carboxylic acid. No statistically significant associations between UGT2B7 variants and clopidogrel pharmacokinetics were observed. However, the common UGT2B7*2 allele has rarely had any effect on the glucuronidation kinetics of UGT2B7 substrates and is thought to have little impact on enzyme activity (Bhasker et al., 2000; Peterkin et al., 2007; Parmar et al., 2011; Rae et al., 2012). Therefore, the clinical data do not allow direct estimation of the contribution of UGT2B7 to clopidogrel metabolism.
In contrast to UGT2B7, there is a common deletion in the UGT2B17 gene, which leads to complete UGT2B17 deficiency (Murata et al., 2003). The absence of UGT2B17 that results from homozygous deletion of the UGT2B17 gene is common in Caucasian and African populations (∼10%–40%) and occurs frequently in East Asia (up to ∼90%) (Wilson et al., 2004; Jakobsson et al., 2006; Xue et al., 2008; Chen et al., 2010; Giroux et al., 2012). As expected, the UGT2B17 deletion was common in our Finnish population, with an allele frequency of 45%. Moreover, clopidogrel acyl-β-d-glucuronide formation was impaired in carriers of the deletion, leading to a 10% or 19% lower clopidogrel acyl-β-d-glucuronide to the clopidogrel carboxylic acid plasma AUC0–4 ratio in heterozygous and homozygous carriers, respectively, than in individuals carrying two functional copies of UGT2B17. Given that homozygous carriers of the deletion lack any metabolic activity by UGT2B17, the results suggest that the combined contribution of intestinal and hepatic UGT2B17 to clopidogrel carboxylic acid glucuronidation is about 10% in heterozygous carriers and 20% in noncarriers, leaving 90% or 80% to other UGTs, respectively. As the in vitro findings indicate that UGT2B17 is largely responsible for clopidogrel carboxylic acid glucuronidation in the intestinal wall, with about 10% contribution in the liver, it could be estimated that up to about 10% of clopidogrel acyl-β-d-glucuronide is formed in the intestine, depending on UGT2B17 genotype. Moreover, as both clopidogrel carboxylic acid and acyl-β-d-glucuronide were formed from clopidogrel in HIM, there seems to be an interplay of CES1 and UGT2B17 in the intestinal wall.
The P450s and UGTs are located intracellularly on opposite sides of the endoplasmic membranes, and there is evidence for a protein-protein interplay between certain UGT and P450 enzymes (Takeda et al., 2005; Ishii et al., 2010; Miyauchi et al., 2015). This interplay and the close proximity of the enzymes could theoretically facilitate interaction of glucuronide metabolites with P450 enzymes. In the present study, we demonstrated UDPGA-dependent formation of clopidogrel acyl-β-d-glucuronide (19 µM in 90 minutes) and simultaneous NADPH-dependent 60% inhibition of CYP2C8 activity in HLMs. This CYP2C8 inhibitory effect was slightly stronger than what we expected based on the obtained clopidogrel acyl-β-d-glucuronide concentrations, suggesting that the proximity of UGTs and CYP2C8 may facilitate access of the acyl-β-d-glucuronide to CYP2C8. Such a mechanism might also partially explain why the clopidogrel-repaglinide interaction was best predicted when the unbound concentration of clopidogrel acyl-β-d-glucuronide was assumed to be almost 20 times higher in hepatocytes than in plasma (Tornio et al., 2014).
In conclusion, the present study indicates that UGT2B7 is the main enzyme involved in clopidogrel carboxylic acid glucuronidation in the liver, whereas at least UGT2B4, and possibly also UGT1A9 and UGT2B17, play a smaller role. On the other hand, UGT2B17 has a major role in the intestinal metabolism of clopidogrel. Consequently, the formation of clopidogrel acyl-β-d-glucuronide is impaired in carriers of the UGT2B17 gene deletion. The findings may have implications for drug-drug interactions caused by inactivation of CYP2C8 by clopidogrel acyl-β-d-glucuronide.
Acknowledgments
The authors thank Jouko Laitila, Eija Mäkinen-Pulli, and Lisbet Partanen for skillful technical assistance.
Authorship Contributions
Participated in research design: Kahma, Filppula, Backman.
Conducted experiments: Kahma, Filppula, M. Neuvonen, Tarkiainen, Tornio, Holmberg, Itkonen, P. J. Neuvonen, Niemi, Backman.
Contributed new reagents or analytic tools: Finel.
Performed data analysis: Kahma, Filppula, Tarkiainen, Finel, Niemi, Backman.
Wrote or contributed to the writing of the manuscript: all authors.
Footnotes
- Received August 21, 2017.
- Accepted November 11, 2017.
This study was financially supported by grants from the Academy of Finland [Grant decision 278123, 2014] and the Sigrid Juselius Foundation [Grant 4704583] (Helsinki, Finland) and by State funding for university-level health research.
This work was previously presented as posters at the 11th International Meeting of the International Society for the Study of Xenobiotics (ISSX), June 12–16, 2016, Busan, South Korea, and at the 13th Congress of the European Association for Clinical Pharmacology and Therapeutics (EACPT), June 24–27, 2017, Prague, Czech Republic.
↵
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC0–4
- area under the plasma concentration-time curve from 0 to 4 hours
- CES
- carboxylesterase
- CI
- confidence interval
- CLint
- intrinsic clearance
- Cmax
- peak plasma concentration
- HIM
- human intestine microsome
- HLM
- human liver microsome
- kdep,
- pseudo-first-order depletion rate constant
- Km
- Michaelis–Menten constant
- P450
- cytochrome P450
- t1/2
- elimination half-life
- tmax
- time at which peak plasma concentration occurs
- UDPGA
- uridine diphosphate glucuronic acid
- UGT
- UDP-glucuronosyltransferase
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}