


Abstract
Preliminary analysis of ongoing birth surveillance study identified evidence of potential increased risk for neural tube defects (NTDs) in newborns associated with exposure to dolutegravir at the time of conception. Folate deficiency is a common cause of NTDs. Dolutegravir and other HIV integrase inhibitor drugs were evaluated in vitro for inhibition of folate transport pathways: proton-coupled folate transporter (PCFT), reduced folate carrier (RFC), and folate receptor α (FRα)-mediated endocytosis. Inhibition of folate transport was extrapolated to the clinic by using established approaches for transporters in intestine, distribution tissues, and basolateral and apical membranes of renal proximal tubules (2017 FDA Guidance). The positive controls, methotrexate and pemetrexed, demonstrated clinically relevant inhibition of PCFT, RFC, and FRα in folate absorption, distribution, and renal sparing. Valproic acid was used as a negative control that elicits folate-independent NTDs; valproic acid did not inhibit PCFT, RFC, or FRα. At clinical doses and exposures, the observed in vitro inhibition of FRα by dolutegravir and cabotegravir was not flagged as clinically relevant; PCFT and RFC inhibition was not observed in vitro. Bictegravir inhibited both PCFT and FRα, but the observed inhibition did not reach the criteria for clinical relevance. Elvitegravir and raltegravir inhibited PCFT, but only raltegravir inhibition of intestinal PCFT was flagged as potentially clinically relevant at the highest 1.2-g dose (not the 400-mg dose). These studies showed that dolutegravir is not a clinical inhibitor of folate transport pathways, and it is not predicted to elicit clinical decreases in maternal and fetal folate levels. Clinically relevant HIV integrase inhibitor drug class effect on folate transport pathways was not observed.
SIGNIFICANCE STATEMENT Preliminary analysis of ongoing birth surveillance study identified evidence of potential increased risk for neural tube defects (NTDs) in newborns associated with exposure to the HIV integrase inhibitor dolutegravir at the time of conception; folate deficiency is a common cause of NTDs. Dolutegravir and other HIV integrase inhibitor drugs were evaluated in vitro for inhibition of the major folate transport pathways: proton-coupled folate transporter, reduced folate carrier, and folate receptor α−mediated endocytosis. The present studies showed that dolutegravir is not a clinical inhibitor of folate transport pathways, and it is not predicted to elicit clinical decreases in maternal and fetal folate levels. Furthermore, clinically relevant HIV integrase inhibitor drug class effect on folate transport pathways was not observed.
Introduction
Dolutegravir is an integrase inhibitor for the treatment of HIV, a drug class that also includes bictegravir, elvitegravir, raltegravir, and cabotegravir (Han et al., 2017). Preliminary analysis of an ongoing birth surveillance study identified evidence of a potential increased risk for neural tube defects (NTDs) in newborns associated with exposure to dolutegravir at the time of conception (Zash et al., 2018). This observation was unexpected because standard preclinical reproductive toxicity studies in rats and rabbits did not identify fetal abnormalities at exposures up to 27-fold higher than at the maximum indicated human dose (2018).
Folate is critical to proper neural tube embryonic development in the first 4 weeks of human pregnancy (Botto et al., 1999). Women of childbearing potential are therefore encouraged to supplement folic acid (Botto et al., 1999; Kancherla et al., 2018). Since neural tube development occurs before most women know they are pregnant, many countries have implemented folic acid fortification of food via wheat and/or maize flour supplementation (Kancherla et al., 2018). Likewise, rodent chow of laboratory animals is supplemented with folic acid, as these animals have no access to natural vitamin sources. Notably, the food supply is not fortified with folic acid in Botswana, where the dolutegravir birth surveillance study was conducted (Kancherla et al., 2018; Zash et al., 2018).
Drug-induced folate deficiency leading to increased incidence of adverse effects, including neurotoxicity and embryonic NTDs, as well as rescue/prevention with folic acid supplementation, are well established for the antifolate drug methotrexate (DeSesso and Goeringer, 1991; Cohen, 2017). Likewise, the intravenous antifolate pemetrexed is indicated for use only with folic acid premedication and oversupplementation to avoid serious toxicities associated with folate depletion in cancer patients (2019). Notably, folate deficiency is not the sole cause of increased NTD risk associated with drug treatment. For example, the antiepileptic drug valproic acid increases the incidence of NTDs 4-fold relative to other antiepileptic therapies used in pregnant women (2011). As folic acid supplementation does not reduce the incidence of valproic acid NTDs, this drug is considered to elicit NTDs via unknown folate-independent mechanism(s) (Hansen et al., 1995; Craig et al., 1999; Candito et al., 2007).
Folate’s hydrophilic nature results in negligible passive membrane permeability and transport-mediated disposition. Transport mechanisms governing folate intestinal absorption, distribution, and renal sparing are depicted in Fig. 1 (Kim et al., 2004; Solanky et al., 2010; Zhao et al., 2011). The proton-coupled folate transporter (PCFT), a solute carrier family transporter, is primarily responsible for folic acid intestinal absorption. Distribution of folate into the tissues, including the placenta and brain, involves the solute carriers PCFT and reduced folate carrier (RFC), as well as folate receptor α (FRα)-mediated endocytosis. Like other hydrophilic nutrients and vitamins, folate is not bound to plasma protein in blood and therefore undergoes extensive glomerular filtration in the kidney. Since folate passive tubular reabsorption is negligible owing to low permeability, folate requires efficient tubular reabsorption (a.k.a., renal sparing), so that blood folate is not rapidly excreted in urine (Zhao et al., 2011). All three folate transport processes are involved in folate active tubular reabsorption.

PCFT, RFC, and FRα-mediated endocytosis in folate absorption, distribution, and renal tubular reabsorption (Kim et al., 2004; Solanky et al., 2010; Zhao et al., 2011). CSF, cerebrospinal fluid; GFR, glomerular filtration rate; PT, renal proximal tubule.
Another mechanism of folate deficiency is inhibition of dihydrofolate reductase (DHFR), which converts dietary folic acid into reduced folates used for nucleic and amino acid synthesis. Dolutegravir is not an inhibitor of DHFR (Cabrera et al., 2019), and since this mechanism has already been ruled out, the present study focused on folate transport.
HIV integrase inhibitors were evaluated for inhibition of PCFT folic acid transport, RFC transport of reduced folate, and FRα-mediated folic acid endocytosis. Inhibition of these folate transport pathways has been investigated in vitro and was extrapolated to the clinic by using established approaches based on regulatory recommendations for drug-drug interactions for transport processes in intestine, distribution tissues, and basolateral and apical membranes of the renal proximal tubules (FDA, 2017). The aim of this work was to determine whether dolutegravir is a clinically relevant inhibitor of the three major folate transport pathways, which could increase the risk of maternal and fetal folate deficiency and NTDs, as well as to investigate potential HIV integrase inhibitor drug class effect on folate transport.
Materials and Methods
Materials
Dolutegravir and cabotegravir were provided by GlaxoSmithKline (Zebulon, NC). Bictegravir was purchased from Medkoo Biosciences (Morrisville, NC). Raltegravir and elvitegravir were obtained from Cayman Chemical (Ann Arbor, MI). Methotrexate, pemetrexed, valproic acid, and bromosulfophthalein were purchased from Sigma-Aldrich (St. Louis, MO). [3H]-folic acid and [3H]-methotrexate were purchased from Moravek (Brea, CA). All other chemicals were of reagent grade and are readily available from commercial sources.
Folate Transport Assays
The test system was Madin-Darby canine kidney-II (MDCK-II) cell polarized monolayers grown on permeable supports of 96-well transwell plates. The MDCK-II cells were individually transfected to express PCFT, RFC, FRα, or vector control transiently.
Assay conditions for PCFT, RFC, and FRα are summarized in Fig. 2. PCFT is expressed on the apical membrane in the kidney/MDCK-II renal cell line (Zhao et al., 2011); hence, all incubations in this study were done on the apical side. The test article was preincubated for 30 minutes at pH 7.4 on the apical side to account for potential time-dependent inhibition and to allow the test article to equilibrate with cells, followed by a 5-minute coincubation of 10 nM [3H]-folic acid with the test article at pH 5.5 (PCFT cotransports folic acid with a proton). Folic acid uptake in PCFT versus vector control cells was determined. Samples for test article concentration and lactate dehydrogenase were collected from the apical side.

Diagram of experimental setup for (A) PCFT transport, (B) RFC transport, and (C) FRα-mediated endocytosis studies. LDH, lactate dehydrogenase; TA, test article drug.
RFC is localized on the basolateral membrane of kidney proximal tubules/MDCK-II renal cell line (Zhao et al., 2011); thus, all test article incubations were done on the basolateral side (see Fig. 2B). RFC is the reduced folate carrier and does not transport folic acid, but its reduced form; 0.5 μM [3H]-methotrexate, was used as a reduced form of folate in these studies (Milstein et al., 2001). The 30-minute and 5-minute coincubations were similar to that of PCFT, except the pH of the medium for RFC experiments was 7.4 during coincubation (RFC activity is optimal at neutral pH) (Matherly et al., 2007; Zhao et al., 2011). Methotrexate cellular uptake in RFC versus vector control cells was determined. Samples for test article concentration and lactate dehydrogenase were collected from the basolateral side.
FRα is expressed on both the apical and basolateral membranes of the MDCK-II renal cell line (Kim et al., 2004). As such, test articles were incubated on both apical and basolateral sides (see Fig. 2C). Preincubation with the test article (30 minutes) was followed by a 2-hour coincubation of 50 nM [3H]-folic acid with the test article at pH 7.4. Folic acid cellular uptake in FRα cells versus the vector control cells was determined. Samples for test article concentration were collected from both basolateral and apical sides; lactate dehydrogenase leakage was measured only on the apical side.
Specific transport activity was determined by the subtraction of the mean cellular uptake in vector control cells (incubated at the same time under same conditions) from that observed in PCFT, RFC, or FRα overexpressing cells (n = 4 wells/cell type per condition). Nominal test drug concentration ranges started at the highest soluble concentration based on visual inspection and were decreased in half-log increments for a total of eight concentrations: 0.03–100 µM for dolutegravir, cabotegravir, elvitegravir and 0.3–1000 µM for bictegravir, raltegravir, methotrexate, pemetrexed, and valproic acid. Actual test drug concentrations at the end of the experiment were measured and used for determination of inhibitory concentrations (see Sample Analysis and Data Analysis sections).
The following criteria were used to deem transport or endocytosis activity acceptable in any assay run: 1) PCFT-mediated folic acid uptake, ≥31.6 fmol/min/cm2; RFC-mediated methotrexate uptake, ≥46.3 fmol/min/cm2; FRα-mediated folic acid endocytosis, ≥4.51 fmol/min/cm2; and 2) ≥70% inhibition of transporter-mediated uptake or receptor-mediated endocytosis by prototypical inhibitors: 300 µM bromosulfophthalein for PCFT-mediated folic acid uptake, 1000 µM pemetrexed for RFC-mediated methotrexate uptake, and 300 µM pemetrexed for FRα-mediated folic acid endocytosis.
Sample Analysis
The transport or endocytosis of each assay substrate was determined by radiometric detection. In the transport and endocytosis studies, the concentrations of test articles from the donor sides at the end of coincubation were measured by liquid chromatography-tandem mass spectrometry (see Supplemental Material, Method 1 for summary of liquid chromatography-tandem mass spectrometry methods). These measured concentrations were used in the calculation of all inhibitory concentrations (except second positive control, pemetrexed, for which nominal concentrations were used). Potential cytotoxicity was evaluated by the release of lactate dehydrogenase, whose activity was quantified by spectrophotometric quantification (λ = 590 nm) of formazan formation using a commercial kit (G-Biosciences, St. Louis, MO).
Data Analysis (eq. 1–4)
In Vitro Data Analysis.
 (1)
(1) (2)Inhibitory concentration 50 (IC50) values were estimated by nonlinear least-squares fitting of the following equation to the uptake data (Prism v.5; GraphPad, San Diego, CA):
(2)Inhibitory concentration 50 (IC50) values were estimated by nonlinear least-squares fitting of the following equation to the uptake data (Prism v.5; GraphPad, San Diego, CA): (3)where n is the Hill coefficient. Wherever ≥50% inhibition was achieved, the associated inhibitory concentration (IC50) values are reported. Otherwise, the greatest extent of inhibition observed ≥25% is reported and discussed. This ≥25% inhibition level was selected on the basis that ≥25% cumulative inhibition of folate absorption, distribution, and renal tubular reabsorption (sparing) would decrease fetal folate levels >2-fold (Zamek-Gliszczynski et al., 2009, 2013).
(3)where n is the Hill coefficient. Wherever ≥50% inhibition was achieved, the associated inhibitory concentration (IC50) values are reported. Otherwise, the greatest extent of inhibition observed ≥25% is reported and discussed. This ≥25% inhibition level was selected on the basis that ≥25% cumulative inhibition of folate absorption, distribution, and renal tubular reabsorption (sparing) would decrease fetal folate levels >2-fold (Zamek-Gliszczynski et al., 2009, 2013).
 (4)where LDH = lactate dehydrogenase. Lactate dehydrogenase activity at any given inhibitor concentration had to be statistically significant (t test with Bonferroni correction) versus vehicle control, and percent cytotoxicity had to be >12.5% (>half of minimal inhibition level considered in this study) to disqualify uptake inhibition data due to confounding cytotoxicity.
(4)where LDH = lactate dehydrogenase. Lactate dehydrogenase activity at any given inhibitor concentration had to be statistically significant (t test with Bonferroni correction) versus vehicle control, and percent cytotoxicity had to be >12.5% (>half of minimal inhibition level considered in this study) to disqualify uptake inhibition data due to confounding cytotoxicity.
Clinical Extrapolation (eq. 5–8)
Since clinical extrapolation of in vitro nutrient-drug transport inhibition is not well established, clinical extrapolation of observed in vitro inhibitory potency values was based on established thresholds (FDA, 2017). This extrapolation framework is considered the best available and considers the worst-case scenario, although it was validated for drug-drug interactions, and not drug-folate interactions with the transport pathways studied herein specifically (eq. 5–8).
Intestinal absorption: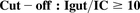 (5)where Igut = highest clinical dose/250 ml or maximal aqueous solubility and IC = inhibitory concentration eliciting noted percentage of inhibition (50% or ≥25% for the purpose of this study).
(5)where Igut = highest clinical dose/250 ml or maximal aqueous solubility and IC = inhibitory concentration eliciting noted percentage of inhibition (50% or ≥25% for the purpose of this study).
Distribution: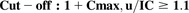 (6)Proximal tubule reabsoprtion (apical proximal tubule transport pathways: PCFT and FRα):
(6)Proximal tubule reabsoprtion (apical proximal tubule transport pathways: PCFT and FRα):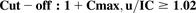 (7)Proximal tubule reabsoprtion (basolateral proximal tubule transporter: RFC):
(7)Proximal tubule reabsoprtion (basolateral proximal tubule transporter: RFC):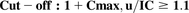 (8)where Cmax, u = unbound plasma Cmax at highest clinical dose; IC = inhibitory concentration eliciting noted percentage of inhibition (50% or ≥25% for the purpose of this study).
(8)where Cmax, u = unbound plasma Cmax at highest clinical dose; IC = inhibitory concentration eliciting noted percentage of inhibition (50% or ≥25% for the purpose of this study).
Data Presentation
All data are reported as mean ± S.D. (n = 4 replicates), unless otherwise noted. Statistical significance was assessed by student’s t test with Bonferroni correction for multiple comparisons. All reported inhibitory concentrations refer to experimentally determined concentrations measured at the end of the coincubation period in the MDCK-II assays (except assay positive controls, bromosulfophthalein or pemetrexed, for which nominal concentrations were used).
Results
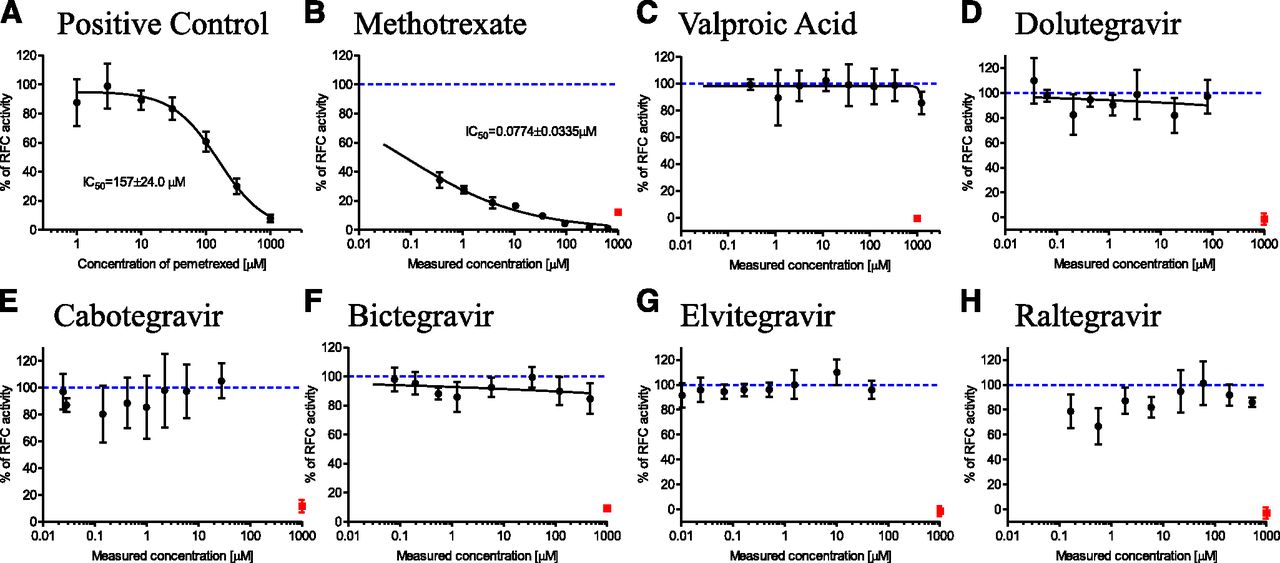
Functional verification of optimized folate in vitro transport assays (Fig. 2) is summarized in Supplemental Fig. 2. Briefly, PCFT functional activity was confirmed with folic acid uptake that was, on average, 31.7-fold (CV = 18.3%, n = 3 independent experiments) enhanced in PCFT versus vector control cells. RFC did not transport unreduced folic acid in this experimental system (data not shown), consistent with the literature (Milstein et al., 2001). Instead, methotrexate was selected as a reduced form of folic acid and demonstrated 2.1-fold (CV = 21.7%, n = 3 independent experiments) enhanced cellular uptake in RFC versus vector control cells. This relatively lower magnitude of RFC functional enhancement in overexpressing cells has been reported previously (Kneuer et al., 2005) and emphasizes the importance of vector-control cell activity correction to determine overexpressed RFC activity. FRα-mediated folic acid endocytosis was 10.7-fold (CV = 16.9%, n = 3 independent experiments) enhanced in FRα overexpressing versus vector control cells (1.2–1.3 × 105 FRα mRNA overexpression was confirmed by polymerase chain reaction; data not shown). Prototypical inhibitors, bromosulfophthalein for PCFT (Nakai et al., 2007) and pemetrexed for RFC and FRα (Chattopadhyay et al., 2007), inhibited these respective folate transport pathways in a concentration-dependent manner (IC50 = 78.1 ± 7.5, 157 ± 24, and 17.5 ± 1.4 µM for PCFT, RFC, and FRα, respectively), with nearly complete inhibition at the highest 1 mM inhibitor concentration (Fig. 3-5).

PCFT transport of 10 nM [3H]-folic acid in the presence of (A) assay positive control inhibitor, bromosulfophthalein (nominal concentrations and IC50); (B) clinical positive control, methotrexate; (C) clinical negative control, valproic acid; (D) dolutegravir; (E) cabotegravir; (F) bictegravir; (G) elvitegravir; and (H) raltegravir. Black symbols represent PCFT [3H]-folic acid uptake activity at various test article concentrations, solid black line is IC50 curve fit with IC50 estimate shown wherever >50% inhibition was observed, red square symbols (B–H) denote PCFT activity in the presence of assay positive control (bromosulfophthalein at 300 μM nominal concentration), dashed blue line (B–H) represents 100% PCFT activity (i.e., no inhibition). All test article concentrations were determined experimentally at the end of the experiment, except bromosulfophthalein concentrations are nominal. Mean ± S.D., n = 4.

RFC transport of 0.5 μM [3H]-methotrexate in the presence of (A) assay positive control inhibitor, pemetrexed (nominal concentrations and IC50); (B) clinical positive control, methotrexate; (C) clinical negative control, valproic acid; (D) dolutegravir; (E) cabotegravir; (F) bictegravir; (G) elvitegravir; and (H) raltegravir. Black symbols represent RFC [3H]-methotrexate uptake activity at various test article concentrations, solid black line is IC50 curve fit with IC50 estimate shown wherever >50% inhibition was observed, red square symbols (B–H) denote RFC activity in the presence of assay positive control (pemetrexed at 1 mM nominal concentration), dashed blue line (B–H) represents 100% RFC activity (i.e., no inhibition). All test article concentrations were determined experimentally at the end of the experiment, except pemetrexed concentrations are nominal. Mean ± S.D., n = 4.

FRα-mediated endocytosis of 50 nM [3H]-folic acid in the presence of (A) assay positive control inhibitor, pemetrexed (nominal concentrations and IC50); (B) clinical positive control, methotrexate; (C) clinical negative control, valproic acid; (D) dolutegravir; (E) cabotegravir; (F) bictegravir; (G) elvitegravir; and (H) raltegravir. Black symbols represent FRα-mediated [3H]-folic acid uptake activity at various test article concentrations, solid black line is IC50 curve fit with IC50 estimate shown wherever >50% inhibition was observed, red square symbols (B–H) denote FRα activity in the presence of assay positive control (pemetrexed at 300 μM nominal concentration), and dashed blue line (B–H) represents 100% FRα activity (i.e., no inhibition). All test article concentrations were determined experimentally at the end of the experiment, except pemetrexed concentrations are nominal. Although elvitegravir elicited an apparent 14%–38% decrease in FRα-mediated folic acid cellular accumulation at the top two concentrations tested, this decrease was comparable in magnitude to 14%–27% cytotoxicity observed at these elvitegravir concentrations in this assay (Supplemental Table 3); therefore, no apparent inhibition of FRα by elvitegravir was concluded. Mean ± S.D., n = 4.
PCFT inhibition results are summarized in Fig. 3. The clinical positive control, methotrexate, inhibited PCFT (IC50 = 2.9 ± 0.3 µM); in contrast, no inhibition was observed for the clinical negative control, valproic acid (Fig. 3, B and C). Dolutegravir and cabotegravir at all test concentrations (up to maximum soluble in assay buffer) did not inhibit PCFT (Fig. 3, D and E). Bictegravir inhibited PCFT (IC50 = 370 ± 23 µM; Fig. 3F). Elvitegravir elicited 28.1% ± 3.3% PCFT inhibition at 30.0 ± 2.0 µM, and raltegravir inhibited PCFT 32.4% ± 2.4% at 564 ± 38 µM (Fig. 3, G and H).
RFC inhibition results are summarized in Fig. 4. The clinical positive control, methotrexate, inhibited RFC (IC50 = 0.08 ± 0.03 µM); no inhibition was noted for the clinical negative control, valproic acid (Fig. 4, B and C). The five integrase inhibitors drugs did not inhibit RFC transport (Fig. 4, D–H) at any concentration tested, up to the maximum soluble in assay buffer.
Inhibition of FRα-mediated folic acid endocytosis is summarized in Fig. 5. The clinical positive control, methotrexate, inhibited FRα (IC50 = 50.4 ± 5.3 µM); inhibition exceeding the 25% predefined threshold for this study was not observed for the clinical negative control, valproic acid (Fig. 5, B and C). Dolutegravir inhibited FRα 36.0% ± 5.7% at 37.3 ± 4.2 µM, and cabotegravir inhibited FRα 36.7% ± 5.8% at 25.8 ± 5.0 µM (Fig. 5, D and E). Bictegravir inhibited folic acid FRα-mediated endocytosis with an IC50 of 268 ± 66 µM (Fig. 5F). Although elvitegravir elicited an apparent 14%–38% decrease in FRα-mediated folic acid cellular accumulation at the top two concentrations tested, this decrease was comparable in magnitude to 14%–27% cytotoxicity observed at these elvitegravir concentrations in this assay (Supplemental Table 3); therefore, no apparent inhibition of FRα by elvitegravir was concluded. Raltegravir did not inhibit FRα (Fig. 4H).
Drug parameters necessary for clinical extrapolation of in vitro folate transport inhibition results are presented in Table 1. Clinical extrapolations of PCFT, RFC, and FRα inhibition in folate intestinal absorption, distribution, and renal tubular reabsorption are summarized in Fig. 6. Extrapolations for folate intestinal absorption were made only for PCFT because the other two pathways are not involved in this process, whereas all three pathways are involved in folate distribution and renal sparing (Fig. 1).
Drug parameters relevant for clinical extrapolation of in vitro transport inhibition

Clinical extrapolation summary for PCFT, RFC, and FRα inhibition in folate absorption, distribution, and renal sparing.
The antifolate drugs, methotrexate and pemetrexed, were flagged as clinically relevant inhibitors of PCFT, RFC, and FRα in folate absorption, distribution, and renal tubular reabsorption (Fig. 6; FDA, 2017). Inhibition of PCFT transport of folic acid by bictegravir, raltegravir, and elvitegravir was flagged only as potentially clinically relevant for 1200-mg oral raltegravir in intestinal folate absorption (Igut/IC32 > 10). Otherwise, 400 mg oral raltegravir did not meet the criteria for clinically relevant intestinal PCFT inhibition; bictegravir, raltegravir, and elvitegravir were not clinically relevant systemic PCFT inhibitors in folate distribution and renal tubular reabsortion (Fig. 6; FDA, 2017). None of the five HIV integrase inhibitor drugs were considered clinical inhibitors of RFC because in vitro RFC inhibition was not observed up to their respective maximal soluble concentrations, which were sufficiently high to rule out intestinal and systemic inhibition at clinical doses and exposures (Table 1). Inhibition of FRα-mediated folic endocytosis by dolutegravir, cabotegravir, and bictegravir observed in vitro was well below the thresholds for potential clinical relevance of transport pathways at the level of tissue distribution (1 + Cmax,u/IC < < 1.1) and renal tubular reabsorption on apical membrane of renal proximal tubules (1 + Cmax,u/IC < < 1.02) (Fig. 6; FDA, 2017).
Discussion
Preliminary analysis of an ongoing clinical birth surveillance study suggested increased risk of NTDs in newborns from mothers taking dolutegravir at the time of conception (Zash et al., 2018). Dolutegravir is an integrase inhibitor for the treatment of HIV, a drug class that also includes cabotegravir, bictegravir, raltegravir, and elvitegravir (Han et al., 2017). Folate deficiency increases the incidence rate of NTDs (Daly et al., 1995; Botto et al., 1999; Crider et al., 2014; Kancherla et al., 2018). Folate relies on transport via PCFT for intestinal absorption, FRα-mediated endocytosis along with transport by PCFT and RFC for tissue distribution, including to the fetus, and renal sparing via active tubular reabsorption after extensive glomerular filtration (Fig. 1, Solanky et al., 2010; Zhao et al., 2011).
The aim of the present work was to determine whether dolutegravir and other integrase inhibitor drugs may be clinically relevant inhibitors of the three major folate transport pathways, which would support an increased likelihood of folate deficiency leading to increased NTD risk. Of the five HIV integrase inhibitor drugs evaluated for inhibition of PCFT, RFC, and FRα in folate absorption, distribution, and renal tubular reabsorption, only raltegravir at the highest (i.e., 1.2 g) clinical dose was flagged as a potential clinical inhibitor of PCFT intestinal folate absorption. Otherwise, these five HIV integrase inhibitor drugs are not predicted to inhibit the three major folate transport pathways at clinical therapeutic doses and concentrations (Fig. 6; FDA, 2017).
Based on the current results, dolutegravir is not predicted to elicit clinical decreases in maternal and fetal folate levels. Furthermore, dolutegravir is not an inhibitor of DHFR, the first step in folic acid metabolism in the synthesis of nucleic and amino acids (Cabrera et al., 2019). Taken together, these results do not support dolutegravir-induced folate deficiency as a mechanistic explanation for the reported preliminary observations of increased NTD risk (Zash et al., 2018). As such, these studies do not lend mechanistic support for folic acid supplementation to overcome this proposed increased NTD risk (Zash et al., 2018); however, there is no harm to folic acid supplementation, and it is an established good practice known to reduce the risk of NTDs in the general population who may not otherwise consume adequate amounts of dietary folic acid (Botto et al., 1999; Kancherla et al., 2018). Although studies have repeatedly failed to demonstrate folic acid supplementation to reduce the 4-fold increased NTD rate in epileptic women on valproic acid treatment, folate supplementation is still recommended for women of childbearing potential taking valproic acid as a safe precaution supported by evidence in the general population ((Depakene Prescribing Information, 2011; Hansen et al., 1995; Craig et al., 1999; Candito et al., 2007; 2011)).
HIV integrase inhibitor drug class effect on folate transport pathways was not observed in the present study (Fig. 6), which is conceptually consistent with the lack of clinical evidence for increased NTD risk by this class of drugs ((Viteka Prescribing Information, 2015, Isentress (raltegravir), 2019, Stribild Prescribing Information (2019))). Even raltegravir, which was flagged as a potentially clinically relevant intestinal PCFT inhibitor at the highest 1.2 g clinical dose is not known to increase the clinical risk of NTDs or cause birth defects in preclinical embryofetal toxicology studies (Isentress (raltegravir), 2019).
After the report of potentially increased prevalence of NTDs in the ongoing dolutegravir birth surveillance study (Zash et al., 2018), in vitro FRα receptor binding experiments were used to demonstrate that dolutegravir is a noncompetitive FRα antagonist (Cabrera et al., 2019). Dolutegravir inhibited folic acid binding to FRα up to 44% at concentrations between 16 and 64 μM, with an apparent IC50 of 4.4 μM, where IC50 was defined as dolutegravir concentration eliciting 22% inhibition of folic acid binding to FRα (i.e., half the maximal inhibitory effect) (Cabrera et al., 2019). Considering that Cabrera et al. examined receptor binding as opposed to functional receptor-mediated endocytosis reported in this study, the dolutegravir FRα antagonism results (44% inhibition at dolutegravir concentrations 16–64 μM) are in good agreement with 36% inhibition of FRα-mediated folic acid at 37 μM dolutegravir (Fig. 5D; Fig. 6); however, Cabrera et al. (2019) concluded that dolutegravir’s apparent FRα inhibitory potency of 4.4 μM occurred at therapeutic concentrations based on direct comparison with clinical total plasma concentrations (Table 1; Tivicay, Prescribing Information, 2018). Dolutegravir is highly plasma protein bound (≥98.9%; Table 1; Tivicay, Prescribing Information, 2018), and because only unbound drug is available for pharmacologic activity (i.e., on- and off-target binding, interactions with transporters and drug-metabolizing enzymes, tissue distribution, etc.), direct comparison of in vitro potencies to total plasma concentrations is not clinically pertinent (Smith et al., 2010). Such a direct in vitro to total plasma comparison may have been possible if 4% albumin were added to the buffer in the FRα-binding study to mimic plasma protein binding, but the comparison was made directly between in vitro potency in a protein-free buffer and clinical total plasma concentrations for a highly bound drug (Cabrera et al., 2019), where clinical unbound plasma concentrations are two orders of magnitude lower than total concentrations (Table 1; Tivicay, Prescribing Information, 2018).
Using the accepted clinical extrapolation approach for the dolutegravir apparent FRα inhibitory potency of 4.4 μM in the receptor binding assay, 1 + Cmax,u/IC50 = 1.017 (Table 1; FDA, 2017; Cabrera et al., 2019), which is not flagged as clinically relevant at the level of tissue distribution (1 + Cmax,u/IC < 1.1) and renal sparing on apical membrane of renal proximal tubules (1 + Cmax,u/IC < 1.02) (FDA, 2017). Since 50% FRα antagonism was not achieved, the apparent IC50 is really an IC22 (Cabrera et al., 2019), and it is not accepted practice to use this apparent IC50 parameter in clinical extrapolations (FDA, 2017). Applying the present extrapolation approach to inhibition of folate transport pathways, where the greatest inhibitory potency ≥25% is used wherever 50% inhibition was not achieved, FRα antagonism IC44 = 16 μM (Cabrera et al., 2019), such that 1 + Cmax,u/IC50 = 1.005 and well below thresholds for potential clinical significance (Table 1; FDA, 2017; Cabrera et al., 2019).
In the present study, clinical extrapolation of observed in vitro inhibitory potency values were based on established thresholds for clinically relevant transporter inhibition in the intestine (Igut/IC50 ≥ 10), systemically (1 + Cmax,u/IC50 ≥ 1.1), and on the apical membrane of renal proximal tubules (1 + Cmax,u/IC50 ≥ 1.02) (FDA, 2017). Although it was validated for transporter-based drug-drug interactions, the principles should apply to drug-nutrient interactions. This extrapolation framework is considered the best available and a conservative approach. As theoretically expected, intensive study of clinical translation of in vitro transport pathway inhibition demonstrated that the same translational pharmacokinetic principles apply to different transporter processes located at the same site in the body (i.e., the same principles and thresholds apply to different transporters localized in the intestine, liver, and basolateral or apical membrane of renal proximal tubules) (Giacomini et al., 2010), Hillgren et al., 2013; FDA, 2017; Zamek-Gliszczynski et al., 2018). Furthermore, these translational approaches have precedent in the extrapolation of in vitro intestinal and systemic thiamine transport by drugs as an alert to drug-induced Wernicke's disease (Giacomini et al., 2017). Notably, these approaches are based on IC50 values where >50% transport function impairment was observed in vitro, and in the present study any inhibitor concentration associated with the greatest extent of inhibition observed ≥25% was reported and extrapolated (Fig. 6). This ≥25% inhibition level was selected on the basis that ≥25% of the cumulative inhibition of folate absorption, distribution, and renal tubular reabsorption (sparing) would decrease fetal folate levels >2-fold (Zamek-Gliszczynski et al., 2009, 2013). The present approach of extrapolating any noted in vitro inhibition ≥25% of folate transport pathways was deliberately conservative and intended to flag any potential clinical inhibition by the five integrase inhibitor drugs. Even with this conservative ≥25% inhibition approach, the present studies do not support 1) dolutegravir as a clinical inhibitor of the three major folate transport pathways or 2) clinically relevant effect of the integrase inhibitor drug class on folate transport.
The present studies are limited to acute inhibition effects by parent drugs. Parent dolutegravir is the predominant circulating moiety with negligible systemic metabolite exposure (Moss et al., 2015). As such, total radioactivity can be assumed to approximate parent drug concentrations, and relevant tissue-to-blood ratios can be estimated from whole-body autoradiography studies in pregnant rats (unpublished data GlaxoSmithKline study 2012N137348_00): placenta-to-blood ratio of 0.59 ± 0.10, fetal-to-maternal blood ratio of 0.13 ± 0.02, and kidney-to-blood ratio of 0.42 ± 0.03. The clinical translational framework based on unbound systemic drug levels is supported by these tissue-to-blood ratios, which do not indicate preferential fetal or renal drug partitioning, as well as systemic exposure primarily to parent dolutegravir and not metabolites; however, potential effects of dolutegravir-glucuronide, the major form of dolutegravir in urine (Moss et al., 2015), on folate tubular reabsorption have not been ruled out by the present studies. Furthermore, potential regulation effects on folate transport and metabolism that may occur upon chronic dolutegravir administration remain to be investigated. These would be most relevant to study clinically in terms of blood folate, folate renal clearance, and maternal-to-neonate blood ratio, because clinical translation of in vitro folate transport and metabolism regulation is not established.
Acknowledgments
We thank Drs. Kim Adkison, Joseph Polli, Dinesh Stanislaus, and Nassrin Payvandi for their scientific input.
Authorship Contributions
Participated in research design: Zamek-Gliszczynski, Zhang, Mudunuru, Taskar, J. Huang, Y. Huang, Romach.
Conducted experiments: Zhang, Du, Chen.
Contributed new reagents and analytic tools: Zhang, Du, Chen, J. Huang, Y. Huang.
Performed data analysis: Zamek-Gliszczynski, Zhang, Mudunuru, Chen, Taskar, Romach.
Wrote or contributed to the writing of the manuscript: Zamek-Gliszczynski, Zhang, Mudunuru, Taskar, Romach.
Footnotes
- Received April 18, 2019.
- Accepted June 4, 2019.
↵1 M.J.Z.-G. and X.Z. contributed equally to this work.
This study was funded by ViiV Healthcare.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- DHFR
- dihydrofolate reductase
- FRα
- folate receptor α
- IC
- inhibitory concentration
- MDCK-II
- Madin-Darby canine kidney-II
- NTDs
- neural tube defects
- PCFT
- proton-coupled folate transporter
- RFC
- reduced folate carrier
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}