


Abstract
GDC-0810 (Cheeti et al., 2018) is an orally bioavailable, selective estrogen receptor (ER) degrader developed to treat ER-positive breast cancer. A first-in-human (FIH) dose escalation phase I study (n = 41) was conducted to characterize the pharmacokinetics (PK) of GDC-0810 and its two major metabolites. GDC-0810 demonstrated linear PK from 100 to 600 mg given once daily. The mean terminal half-life following a single 600 mg dose was approximately 8 hours. Since GDC-0810 is a potent in vitro inhibitor of organic anion transporting polypeptide (OATP) 1B1/3, the kinetic profile of coproporphyrin I (CPI), a promising endogenous biomarker for OATP1B1/3, was analyzed retrospectively in a subset of the plasma samples collected in the same FIH study. CPI exhibited a GDC-0810 dose-dependent increase, suggesting in vivo inhibition of OATP1B transporters. To quantitatively predict the magnitude of OATP1B-mediated drug-drug interactions (DDIs) with pravastatin (a known OATP1B substrate), the in vivo unbound inhibition constant was first estimated using a one-compartment model, and then incorporated to a physiologically based pharmacokinetic model. The model showed some underestimation of the magnitude of the DDI when compared with a clinical DDI study result, while prediction had a relatively large uncertainty due to the small effect size, limited sample size, and variability in CPI kinetics. In conclusion, this study characterized the pharmacokinetic profiles of GDC-0810 in breast cancer patients and demonstrated the utility of CPI in detecting OATP1B-mediated DDIs of a new molecular entity as early as FIH study.
SIGNIFICANCE STATEMENT Endogenous biomarkers of transporters have recently been shown to be promising tools in evaluating the risk of clinical transporter-mediated DDIs. This is the first study to report a pharmacokinetic interaction between an investigational molecule and a transporter biomarker in a first-in-human study. The observed interaction and model-based analysis and the prediction provide important insights on the novel approach to quantitatively predict transporter-mediated DDIs as early as FIH studies in the clinical development.
Introduction
Breast cancer (BC) is the most prevalent form of female cancer, of which approximately 70% express estrogen receptors (ERs). In addition to surgery, endocrine therapies that either suppress ER signaling or inhibit aromatase in the biosynthesis of estrogen serve as a major treatment strategy for ER-positive (ER+) BC. With currently available endocrine therapies, many patients relapse or develop resistance. Thus, new ER-targeting therapies are needed to delay disease progression and achieve optimal disease control for ER+ BC patients. GDC-0810 (Cheeti et al., 2018) was a new molecular entity (NME) developed to be an orally bioavailable, selective ER degrader that antagonizes estrogen-mediated ER signaling (Lai et al., 2015; Cheeti et al., 2018). In the first-in-human (FIH) phase 1a study, GDC-0810 was assessed in patients with ER+, human epidermal growth factor receptor 2-negative (HER2-) BC. Based on in vitro studies, GDC-0810 is mainly metabolized via glucuronidation by multiple UDP-glucuronosyltransferase isoforms. It inhibits organic anion transporting polypeptide (OATP) 1B transporters (Liu et al., 2018), which are key players in the hepatic uptake of xenobiotics and have received a significant regulatory spotlight from health authorities as a potential site for clinically relevant drug-drug interactions (DDIs) (European Medicines Agency, 2012; Food and Drug Administration, 2017; Pharmaceuticals and Medical Devices Agency, 2018).
One class of drugs that is a well-known victim of OATP1B-mediated DDI is that which includes the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) inhibitors (or statins), such as pravastatin, rosuvastatin, and atorvastatin, which are lipid-lowering agents prescribed to patients worldwide to reduce risk for cardiovascular events (Kellick et al., 2014). Due to the in vitro evidence, the potential effects of GDC-0810 in perpetrating clinically relevant OATP1B-mediated DDI were warranted. This allowed investigating the in vivo DDI liability of GDC-0810 when given in combination with OATP1B substrates. Recently, novel avenues for measuring the endogenous biomarker kinetics to predict transporter-mediated DDI were explored with growing interest (Chu et al., 2018). Specifically, coproporphyrin I (CPI), the porphyrin metabolite from heme synthesis, was found to be promising as a desirable biomarker of OATP1B transporters.
Previously, we characterized the kinetic profile of CPI from plasma samples collected in a clinical DDI study that investigated the interaction between GDC-0810 and pravastatin in healthy volunteers (Liu et al., 2018). Furthermore, by incorporating the in vivo unbound inhibition constant (Ki,u), which was estimated from the CPI kinetic profile into physiologically based pharmacokinetic (PBPK) modeling, we showed that the extent of OATP1B-mediated DDI between GDC-0810 and pravastatin could be reasonably predicted (Yoshida et al., 2018). This successful simulation sparked interest in investigating if DDI potential could be captured even earlier in the clinical development of the investigational drug using data from phase I clinical studies that involved patients.
In the current study, we aimed to: 1) determine the pharmacokinetics (PK) of GDC-0810 and its two metabolites in a FIH dose escalation study to inform the recommended phase II dose of GDC-0810; 2) investigate the utility of monitoring the kinetic profile of CPI to predict the magnitude of OATP1B-mediated DDI between GDC-0810 and pravastatin; and 3) determine how this approach would help inform the decision on whether a dedicated DDI study would be warranted by retrospectively measuring the CPI concentrations in plasma samples collected from the same FIH study. One advantage of measuring the endogenous biomarker plasma levels in this type of dose escalation study is that it allows the evaluation of the DDI potential of the NME at a wider range of drug exposure, which can hardly be achieved in typical clinical DDI studies (Chu et al., 2018). Furthermore, assessing the magnitude of DDIs at various drug exposure levels would also provide a quantitative recommendation on potential dose adjustment strategies, if needed. Most importantly, this approach will avoid unnecessary drug exposure in dedicated DDI studies that often enroll healthy subjects.
Materials and Methods
First-in-Patient Study Design.
The FIH study was a multicenter phase Ia open-label, dose escalation study of GDC-0810 in postmenopausal women with locally advanced or metastatic ER+, human epidermal growth factor receptor 2-negative (HER2-) BC (https://clinicaltrials.gov/ct2/show/NCT01823835). The study was conducted in accordance with the principles of the Declaration of Helsinki. The study protocol, informed consent forms, and other supporting information were submitted to and approved by the Institutional Review Board/Ethics Committee prior to study initiation. No modifications were made to the protocol after receipt of the Institutional Review Board/Ethics Committee approval.
GDC-0810 monotherapy was administered orally to the study subjects on a continuous daily dosing regimen with a 7-day lead-in period for single dose PK evaluation prior to the start of daily treatment (Fig. 1). Depending on safety and tolerability, patients were assigned sequentially to escalating doses of GDC-0810 with use of a standard 3+3 design. The starting dose of GDC-0810 was 100 mg once daily, followed by dose escalation to 200 mg (dose level 2), and 200 mg increments thereafter up to 800 mg. The full prescribed dose of GDC-0810 was taken by mouth in the morning, after fasting overnight, and at approximately the same time each day. Food was not to be eaten for at least 1 hour after taking GDC-0810. The dosing regimen at select dose levels in dose escalation was changed based on PK and safety data to a twice daily dosing frequency, with or without fasting requirements. Patients enrolled in this study continued treatment at their assigned dose level for at least two 28-day cycles.

Schematics of study workflow: (A) Study design; (B) Model-based analysis and clinical DDI prediction (NONMEM, nonlinear mixed-effect modeling).
Subjects.
Informed consent was obtained from all individual participants included in the study prior to enrollment. Additional informed consent was obtained from all individual participants for whom identifying information is included in this article.
The demographic analysis is presented in Table 1. A total of 41 female patients were enrolled in the study. PK analyses were conducted for all of these patients who had at least one PK sample collected. Plasma CPI concentrations were retrospectively measured in 21 of 41 subjects for the Day -7 lead-in period samples and in 18 of 41 subjects for the steady-state samples (Fig. 1; Table 2).
Demographics and baseline characteristics
Subgroup of patients with plasma samples available for CPI kinetics analysis
Treatment and Blood Sample Collection.
On day -7, for the lead-in period, patients enrolled in the dose escalation cohorts received a single oral dose of GDC-0810 in clinic and returned to the clinic for the next two consecutive days for PK sample collection. During cycle 1 of the dose escalation, patients returned to the clinic on a weekly basis for safety evaluation and collection of predose PK samples. Similarly, on day 1 of every subsequent new cycle of dosing, beginning with cycle 2, GDC-0810 was administered in the clinic following all assessments. All other doses of GDC-0810 were administered on an outpatient basis.
Bioanalysis of Plasma Samples.
Predose PK samples were collected on all days when patients returned to the clinic on a weekly basis (days 1, 8, 15, and 22). On day -7 and cycle 2 day 1 (steady state), blood samples were collected at the following predefined time points: predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 24 (only lead-in) hours to investigate the kinetics of GDC-0810 and CPI in patients following single dose and multiple dose treatments. Plasma concentrations of GDC-0810 and its two glucuronide metabolites (GDC-0810-N-glucuronide and GDC-0810-acyl-glucuronide) were determined by Q2 Solutions (Ithaca, NY) using a validated liquid chromatography/tandem mass spectrometry method. Full plasma PK profiles were obtained for GDC-0810 and analyzed using noncompartmental methods for all patients in the dose escalation portion of the study. As previously described, CPI plasma concentrations were determined by a high-performance liquid chromatography–tandem mass spectrometry bioanalytical method with multiple reactions monitoring and electrospray ionization in the positive mode (Liu et al., 2018).
Model-Based Analysis of CPI Interactions with Inhibitor Kinetics.
Fig. 1B depicts the work flow for the model-based analysis. The plasma concentration-time profiles of GDC-0810 and CPI were used for model-based analysis as previously described (Yoshida et al., 2018). The model diagram for this analysis has also been reported in the same study. Briefly, portal vein unbound GDC-0810 concentrations at the various studied dosages were first predicted using SimCYP (version 17 a Certara Company, Sheffield, UK). PK analysis revealed that the PK profiles for 600 and 800 mg GDC-0810 were similar. The predicted drug concentration in portal vein for 800 mg was based on that predicted for 600 mg when the data were input to nonlinear mixed-effect modeling to reflect this clinical observation instead of the predicted value in SimCYP, which assumed linear kinetics at all doses. The observed CPI concentrations at various time points where blood samples were collected together with the predicted portal vein concentrations of GDC-0810 were used as data input for the one-compartment model of CPI in nonlinear mixed-effect modeling that has been previously reported: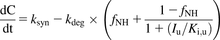 where ksyn, kdeg, fNH, and Iu represent the synthesis rate, degradation rate constant, fraction eliminated via the nonhepatic pathway, and unbound inhibitor (GDC-0810) concentration, respectively. This model was used to estimate the CPI disposition parameters and Ki,u (Yoshida et al., 2018). Nonparametric bootstrap with dose-stratified sampling and 500 iterations was conducted to assess uncertainty of the parameter estimates. Additional statistical analysis, goodness-of-fit plots, and graphical analyses were conducted in (R Core Team, 2019) R and GraphPad Prism version 7.04 for Windows (GraphPad Software, La Jolla, California).
where ksyn, kdeg, fNH, and Iu represent the synthesis rate, degradation rate constant, fraction eliminated via the nonhepatic pathway, and unbound inhibitor (GDC-0810) concentration, respectively. This model was used to estimate the CPI disposition parameters and Ki,u (Yoshida et al., 2018). Nonparametric bootstrap with dose-stratified sampling and 500 iterations was conducted to assess uncertainty of the parameter estimates. Additional statistical analysis, goodness-of-fit plots, and graphical analyses were conducted in (R Core Team, 2019) R and GraphPad Prism version 7.04 for Windows (GraphPad Software, La Jolla, California).
Prediction of Clinical DDI with the Physiologically Based Pharmacokinetic Model.
The estimated in vivo Ki,u values of GDC-0810 were incorporated into the previously established PBPK model, and the clinical DDIs between GDC-0810 and pravastatin were predicted using the SimCYP (version 17) default model file for pravastatin (Chen et al., 2018; Yoshida et al., 2018). Sensitivity analyses were conducted to evaluate the effect of varying the in vivo Ki,u value based on parameter uncertainty on the predicted DDI magnitude. The simulated area under the curve ratios (AUCRs) were compared with that reported in the clinical DDI study in healthy volunteers (Liu et al., 2018).
Results
Pharmacokinetic Profiles of GDC-0810 and Its Metabolites.
Of the 41 patients, 36 [87.8%] were white and the mean age at baseline was 59.8 years (33–78 years). Baseline Eastern Cooperative Oncology Group (ECOG) values were either 1 [22 patients (53.7%)] or 0 [19 patients (46.3%)], suggesting that patients were either fully active without restriction or only restricted in physically strenuous activity, but work of light or sedentary nature was not affected (Oken et al., 1982). The mean number of sites of disease was 2.20 (1.0–9.0). Approximately two-thirds of the patients had visceral disease at baseline [26 patients (63.4%)].
Samples from all 41 patients were analyzed to study the PK of GDC-0810 and its two metabolites (Fig. 2; Table 3). A total of nine cohorts were studied with doses ranging from 100 to 800 mg, given either once or twice daily, under fasted or fed state (Table 3). GDC-0810 was rapidly absorbed with peak concentrations achieved at 1–3 hours [median time to reach the maximum concentration (Tmax)] after dosing. GDC-0810 exhibited linear PK with the mean exposure (the area under the concentration-time curve from time zero to infinity), which increased proportionally to doses up to 600 mg. The PK profile for 800 mg was similar to that of 600 mg. Food effect was explored at 600 and 800 mg. In general, the Cmax and area under the concentration-time curve at 24 hours observed in the fed state were numerically higher than that in the fasted state, but statistical significance was not evaluated due to the small sample size. The mean terminal half-life following a 600-mg single dose of GDC-0810 under the nonfasting condition was approximately 8 hours. Consistent with its half-life, minimal drug accumulation was observed following multiple dosings for both once and twice daily regimens.

Mean GDC-0810 plasma concentration-time profiles following oral administration of a single dose (A) and multiple doses (at steady state) (B) of GDC-0810 in postmenopausal women with ER+ breast cancer (BID, twice a day).
Mean (± S.D.) PK parameters of GDC-0810 following oral administration of single and multiple doses (at steady state) of GDC-0810 in postmenopausal women with ER+ breast cancer
Unless otherwise specified, all doses were given once a day (fasted state). The half-life, CL/F, and AUC0–inf values were reported only for patients on the once daily regimen with Tlast ≥ 24 h.
The PK parameters of the two metabolites of GDC-0810 (GDC-0810-N-glucuronide and GDC-0810 acyl-glucuronide) following single and multiple doses of the parent drug are reported in Table 4. In general, the mean exposures of the N-glucuronide and acyl-glucuronide metabolites ranged from 1% to 5% and 10% to 25%, respectively, of the parent.
Mean (± S.D.) PK parameters of GDC-0810-N-glucuronide and GDC-0810-acyl-glucuronide following oral administration of single and multiple doses (at steady state) of GDC-0810 in postmenopausal women with ER+ breast cancer
Unless otherwise specified, all doses were given once a day (fasted state).
Kinetic Profile of CPI.
For both single dose and steady-state analyses, the exposure of CPI changed with the plasma concentration of GDC-0810 in a dose-dependent manner over the studied time course (Supplemental Figs. 1 and 2). Figure 3 illustrates the trend of increasing CPI concentration from baseline at 3 hours (approximately the median Tmax of GDC-0810) with increasing doses of GDC-0810 [Spearman r = 0.48 (P = 0.026) and 0.51 (P = 0.038), respectively, for single dose and steady-state analyses].

Positive correlation between dose and fold change of CPI plasma concentration from baseline at 3 hours postdose for a single dose (lead in) (A) and multiple doses (steady state) (B), respectively. A linear model (black solid lines) has been fitted to illustrate the positive correlation. Spearman’s coefficient (rs) and the P value are also reported. Black dots represent the observed ratio from patients; gray horizontal bars represent the median of the fold change for each dose cohort.
The kinetic profiles of CPI when 600 mg of GDC-0810 was given as a single dose and at steady state were compared with that previously reported (Liu et al., 2018), where healthy female subjects were enrolled (Fig. 4). At the same administered dose, the CPI kinetics in cancer patients and healthy subjects appeared to be comparable.

Kinetic profile of CPI in cancer patients in the 600 mg cohort [lead in (single dose) and at steady state (multiple doses)] compared with that in healthy subjects who were also given 600 mg GDC-0810 in the DDI study.
Model-Based Analysis of CPI Kinetics and Estimation of In Vivo Unbound Inhibition Constant.
The estimated parameters for the CPI kinetics and Ki,u values of GDC-0810 for OATP1B are summarized in Table 5. The parameters previously estimated from CPI analysis using samples from healthy female volunteers in the clinical DDI study are also presented in Table 5 for comparison (Yoshida et al., 2018). The baseline CPI levels in cancer patients were approximately 0.5, which is slightly lower than that in healthy subjects (0.873). The degradation rate constant values in cancer patients ranged from 0.6 to 2.4 hour−1. The Ki,u value in the present study was higher than that previously reported in healthy volunteers (0.00437–0.00469 vs. 0.00174). In addition, the interindividual variability of the Ki,u value in this study was also significantly greater than that in the previous study. The changes in CPI plasma concentrations from baseline at 3 hours at the tested doses were simulated based on the parameter estimates and are presented in Fig. 5.
Summary of parameter estimates for CPI kinetics and inhibition constants of GDC-0810 for OATP1B

The simulated fold change of CPI plasma concentration from baseline at 3 hours postdose during the lead-in (single dose) phase (A) and at steady state (B) of the GDC-0810 FIH study. The simulated CPI concentrations showed a positive trend with the administered GDC-0810 dose despite the large empirical bootstrap confidence region (shaded gray areas). Black dots represent observed fold change in BC patients.
Prediction of Clinical DDI with Pravastatin.
Figure 6 describes the magnitude of the DDIs observed in the clinical study, and that predicted using the in vivo Ki,u values estimated from nonlinear mixed-effect modeling. The predicted AUCR was lower than that for both the single dose (lead in) and multiple doses at steady state observed (geometric mean ∼1.2 vs. 1.41). The bootstrapped Ki,u values at the fifth and 95th percentiles were also used to predict the AUCR to capture the uncertainty of the prediction based on the empirical distribution.

Predicted and observed AUCRs of pravastatin in the presence of GDC-0810. The magnitude of the DDI is described by the AUCR. For the observed AUCR, the geometric mean (1.41) with 90% confidence interval is shown here. The DDI prediction was based on the single dose (lead in) and multiple dose (steady state) analyses. As a comparison, the DDI prediction using CPI kinetics from the clinical DDI study is also presented. The upper and lower bounds shown here correspond to the AUCR predicted based on the bootstrapped Ki,u value at the fifth and 95th percentiles, respectively.
Discussion
To the best of our knowledge, this is the first study that measured CPI kinetics in a FIH dose escalation study to evaluate the transporter-mediated DDI potential of an NME.
Most PK parameters for GDC-0810 administered after a 600 mg dose to BC patients in this study were similar to those in healthy subjects reported in the DDI study with pravastatin (Liu et al., 2018). Food intake numerically increased the Cmax and area under the concentration-time curve at 24 hours, but statistical significance was not assessed given the limited sample size and exhibited large variability. The PK of 800 mg once daily was similar to that of 600 mg once daily, yet 800 mg was deemed intolerable due to gastrointestinal side effects (safety data on file). Collectively, the recommended phase II dose was determined to be 600 mg once daily administered under fed conditions.
The in vitro inhibition study suggested that the two major glucuronide metabolites of GDC-0810 were potent inhibitors of OATP1B with IC50 comparable to that of the parent drug (data on file). However, considering the much lower plasma concentrations (3% and 10% of parent drug at 600 mg for N-glucuronide and acyl-glucuronide, respectively), we concluded that these two metabolites would not contribute significantly to the OATP1B-mediated DDI. Therefore, only the impact of GDC-0810 on OATP1B-mediated DDI was considered in subsequent analyses.
A positive trend between the plasma concentrations of CPI and GDC-0810 exposure was observed (Fig. 3). As expected, the extent of the change in CPI plasma concentrations was comparable to that observed in the dedicated DDI study, illustrating that CPI is a sensitive biomarker for the OATP1B-mediated DDI pathway (Liu et al., 2018). There are a few reported studies evaluating the interaction between endogenous biomarkers and multiple dose levels of transporter inhibitors (Tsuruya et al., 2016; Takehara et al., 2018). This is the first time that the extent of DDI potential of a perpetrator was studied for a wide range of drug exposures in patients using an endogenous compound. For NMEs that cause potent in vivo inhibition of transporters, strategies to mitigate the risk of DDI at various doses of the perpetrator drug can be formulated based on the change in CPI plasma concentrations from baseline and predicted magnitude of DDI at different exposures observed in the FIH study.
The kinetic profiles and the estimated parameters for CPI, such as the rate of degradation, Cmax, and Tmax in this study, were compared with those from the previously reported dedicated DDI study in healthy volunteers (Liu et al., 2018; Yoshida et al., 2018), and were found to be comparable in the presence of GDC-0810. When the BC patients were given 600 mg GDC-0810, they had similar CPI baseline values as healthy subjects in the clinical DDI study (Fig. 4); however, in general, when all patients were considered in the population kinetic analysis as reported in Table 5, the CPI baseline levels were slightly lower in patients than that in healthy female volunteers (Yoshida et al., 2018). This difference may be due to interindividual variability instead of the disease state. Among healthy volunteers, previous studies reported that the CPI baseline levels could range from approximately 0.6 nM (Kunze et al., 2018; Shen et al., 2018) to 0.97 nM (Lai et al., 2016). The estimated Ki,u value in the present study was more than 2-fold higher than the previous study, accompanied by much larger interindividual variability, indicating that the point estimate is less reliable. However, the estimated Ki,u value in this study was still approximately 100-fold smaller than the in vitro IC50 values [<0.3 µM (OATP1B1) and 0.9 µM (OATP1B3)], which is consistent with the previous observation (Liu et al., 2018; Yoshida et al., 2018). Although part of the discrepancy might be explained by the reduced effective concentration in the in vitro study for this highly bound molecule (the plasma unbound fraction is 0.008%) (Chen et al., 2018), quantitative extrapolation of in vitro IC50 to in vivo Ki remained challenging. This once again highlights an advantage of using biomarker kinetic profiles to estimate the Ki,u value, which results in a more accurate prediction of clinical DDI. Notably, the Tmax value of CPI in the single dose analysis (Fig. 4) was slightly delayed to 6 hours from the median of 3 hours. This difference can be explained by the PK of GDC-0810, which changed the plasma concentration of CPI. A delayed peak after one dose of 600 mg GDC-0810 was seen in two patients in this cohort, which consequently led to the delayed CPI peak. It is uncertain if other disease states would affect the CPI levels and its kinetic parameters; for instance, reduced OATP1B1/3 expression has been reported in patients with hepatocellular carcinoma (Obaidat et al., 2012). Therefore, measuring CPI levels in patients in a FIH study instead of a DDI study, which typically enrolls healthy volunteers, may provide a better reflection of the extent of an OATP1B-mediated DDI in prospective patients.
The simulated fold change in CPI plasma concentrations from baseline at 3 hours postdose using a developed model (Fig. 5) showed a positive trend despite the large empirical bootstrap confidence intervals. Furthermore, our PBPK DDI simulation based on the estimated Ki,u value resulted in underestimation of the point estimate, and again with a large extent of uncertainty in the predicted AUCR of the victim probe drug, which ranged from no DDI to mild DDI (AUCR = 1.02–1.35) (Fig. 6). This uncertainty in the simulation was likely attributed to the relatively small sample size per cohort in this substudy. Therefore, and more importantly, since the magnitude of interindividual variability is also wide, it is challenging to quantitatively characterize the interaction between GDC-0810 and CPI from the current data. Interestingly, Barnett et al. (2018) evaluated sample size requirement for CPI to detect different levels of inhibition and found that 15 subjects are needed to achieve 80% power at α = 0.05 or 0.01 for weak (AUCR > 1.25) inhibition. Indeed, we had <5 patients in the 600 or 800 mg groups with CPI measurement. As a comparison, in the clinical DDI study where 15 healthy subjects were enrolled and administered 600 mg GDC-0810, the uncertainty in the predicted AUCR of pravastatin was much narrower (Fig. 6). These observations support the need for adequate sample size to accurately quantify the magnitude of DDI, especially when the effect of the NME on OATP1B is small. In contrast, for strong DDIs, which are more likely to be clinically important, a standard FIH cohort can be sufficient to capture the interaction. Collectively, our study illustrates the utility of model-based analysis of an endogenous biomarker in a FIH study to provide a quantitative estimation of the extent of DDIs. The proposed approach, accompanied by sufficient sample size, may pave the way to justifying that GDC-0810 has low clinically relevant potential of driving OATP1B-mediated DDI.
Recently, a PBPK model for CPI has been established that predicts the DDI caused by rifampicin via the OATP1B and multidrug resistance-associated protein pathway (Mori et al., 2019). This model enables the mechanistic understanding of the complex DDI mediated by the studied perpetrator based on in vitro information; however, it also requires in-depth understanding of the substrate-dependent, and potentially patient-dependent, biomarker kinetic processes for model development. Our current approach complements this PBPK model; it allows the evaluation of interindividual variability in the estimated parameters by using a population PK approach, and hence the magnitude of the DDI can be better characterized and would be more reflective of the expected DDI potential of the perpetrator in a real patient population. Finally, it is noteworthy that in addition to CPI, there are other endogenous biomarkers for OATP1B that are being studied, which may be measured concurrently with CPI in the future to predict transporter-mediated DDIs with greater confidence (Takehara et al., 2018; Barnett et al., 2019).
Here, we mention two limitations of this study. First, the effect of OATP1B polymorphism on the CPI baseline and kinetics in the presence of GDC-0810 was not investigated. OATP1B polymorphism has been associated with many clinically relevant impacts on the disposition of drug substrates (Niemi et al., 2005; Ho et al., 2007; Deng et al., 2008), and recently with CPI and other biomarkers (Mori et al., 2019). Therefore, it is of great interest to study how the interactions between CPI and inhibitors for OATP1B would be affected by the transporter pharmacogenomics. The second limitation is the inevitably confounding effect of concomitant medications; all patients enrolled in this study reported taking at least one medication before or after study initiation. While known strong OATP1B inhibitors, such as cyclosporine, were restricted in the study, some medications might weakly inhibit the transporters.
In conclusion, the PK results of GDC-0810 and its two glucuronide metabolites in ER+ human epidermal growth factor receptor 2-negative (HER2-) BC patients were characterized in this FIH dose escalation study. Furthermore, the dose-dependent increase in CPI concentration suggested the presence of weak in vivo inhibition of OATP1B. The prediction of DDI potential based on population PK and PBPK modeling approaches resulted in some underestimation with relatively large uncertainty, likely due to limited sample size. This study provides important insights into the novel avenue to quantitatively predict transporter-mediated DDI as early as in FIH studies of the NME using endogenous biomarkers.
Acknowledgments
The authors thank Drs. Scott Lam and Yuan Chen for the contributions to this study.
Authorship Contributions
Participated in Research design: Cheung, Yoshida, Cheeti, Morley, Chan, Sahasranaman, Liu.
Conducted experiments: Cheung, Yoshida.
Contributed new reagents or analytic tools: Chen.
Performed data analysis: Cheung, Yoshida, Liu.
Wrote or contributed to the writing of the manuscript: Cheung, Yoshida, Cheeti, Morley, Chan, Liu.
Footnotes
- Received May 9, 2019.
- Accepted June 26, 2019.
K.Y., S.C., B.C., R.M., I.T.C., S.S., and L.L. were employees of Genentech, a member of the Roche Group, when the work was performed. The employees are also holders of Roche Holding Ltd. stock. K.W.K.C. was a postdoctoral fellow at Genentech and the University of California, San Francisco, when the work was performed.
↵1 Current affiliation: BeiGene, San Mateo, California.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUCR
- area under the curve ratio
- BC
- breast cancer
- CPI
- coproporphyrin I
- DDI
- drug-drug interaction
- ER
- estrogen receptor
- ER+
- estrogen receptor positive
- FIH
- first-in-human
- HER2-
- Human epidermal growth factor receptor 2-negative
- Ki,u
- in vivo unbound inhibition constant
- NME
- new molecular entity
- OATP
- organic anion transporting polypeptide
- PBPK
- physiologically based pharmacokinetic
- PK
- pharmacokinetics
- Tmax
- time to reach the maximum concentration
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}