


Visual Overview

Abstract
Our previous study suggests that berberine (BBR) lowers lipids by modulating bile acids and activating intestinal farnesoid X receptor (FXR). However, to what extent this pathway contributes to the hypoglycemic effect of BBR has not been determined. In this study, the glucose-lowering effects of BBR and its primary metabolites, berberrubine (M1) and demethyleneberberine, in a high-fat diet–induced obese mouse model were studied, and their modulation of the global metabolic profile of mouse livers and systemic bile acids was determined. The results revealed that BBR (150 mg/kg) and M1 (50 mg/kg) decreased mouse serum glucose levels by 23.15% and 48.14%, respectively. Both BBR and M1 markedly modulated the hepatic expression of genes involved in gluconeogenesis and metabolism of amino acids, fatty acids, and purine. BBR showed a stronger modulatory effect on systemic bile acids than its metabolites. Moreover, molecular docking and gene expression analysis in vivo and in vitro suggest that BBR and M1 are FXR agonists. The mRNA levels of gluconeogenesis genes in the liver, glucose-6-phosphatase and phosphoenolpyruvate carboxykinase, were significantly decreased by BBR and M1. In summary, BBR and M1 modulate systemic bile acids and activate the intestinal FXR signaling pathway, which reduces hepatic gluconeogenesis by inhibiting the gene expression of gluconeogenesis genes, achieving a hypoglycemic effect. BBR and M1 may function as new, natural, and intestinal-specific FXR agonists with a potential clinical application to treat hyperglycemia and obesity.
SIGNIFICANCE STATEMENT This investigation revealed that BBR and its metabolite, berberrubine, significantly lowered blood glucose, mainly through activating intestinal farnesoid X receptor signaling pathway, either directly by themselves or indirectly by modulating the composition of systemic bile acids, thus inhibiting the expression of gluconeogenic genes in the liver and, finally, reducing hepatic gluconeogenesis and lowering blood glucose. The results will help elucidate the mechanism of BBR and provide a reference for mechanism interpretation of other natural products with low bioavailability.
Introduction
Obesity is now prevalent all over the world and causes at least 2.8 million deaths each year. Obesity is also the primary cause of various diseases, including type II diabetes, nonalcoholic fatty liver disease (NAFLD), cardiovascular disease, and hypertension. A common feature of these diseases is a metabolic disorder with abnormal glucose metabolism, lipids, and amino acids (Gaggini et al., 2013; Yki-Järvinen, 2014). Berberine (BBR) is a compound isolated from plants (Coptis chinensis, Hydrastis canadensis, etc.) and has been used for treating diseases including diarrhea, obesity, NAFLD, and various tumors in both animal models and human because of its antimicrobial, antiprotozoal, antioxidant, anti-inflammatory, and immunomodulatory properties (Yin et al., 2008; Zhang et al., 2008; Xing et al., 2011; Yu et al., 2015; Zou et al., 2017). BBR shows good antiobesity and hypoglycemic effects through various mechanisms by stimulating glucose transport and uptake, increasing insulin receptor expression, enhancing gluconeogenesis, and modulating glucose-6-phosphatase and hexokinase to achieve its hypoglycemic effect (Zhou et al., 2007; Zhang et al., 2010; Xia et al., 2011; Li et al., 2014; Pirillo and Catapano, 2015). However, the mechanisms of BBR’s actions are not fully understood. A large amount of BBR is accumulated in the gut after dosing as a result of its low bioavailability. Previous studies reported that the intestine might be the targeting organ for BBR’s hypoglycemic effects, including inhibiting α-glucosidase, decreasing glucose transport through the intestinal epithelium, modulating the production of short-chain fatty acids (primarily butyrate) of the gut microbiota (Pan et al., 2003; Wang et al., 2017), and modulating the composition of gut bacteria and the metabolism of bile acids (Gu et al., 2015; Sun et al., 2017).
BBR is extensively metabolized with oxidative demethylation followed by subsequent glucuronidation in vivo to various metabolites, and the concentration of its metabolites in organs is higher than that of BBR in rats (Liu et al., 2010; Tan et al., 2013). Four primary phase I metabolites of BBR have been identified—namely, berberrubine (M1), thalifendine (M2), demethyleneberberine (M3), and jatrorrhizine (M4) in rats and humans (Qiu et al., 2008). The pharmacokinetic properties of exogenous metabolites may be different from the metabolites generated endogenously (Prueksaritanont et al., 2006); thus, we studied the pharmacokinetic property of M1 (Zhao et al., 2017b). Our previous study showed that M1 might have a much greater system exposure than BBR after oral administration in rats, and M1 had a more substantial glucose-lowering effect than BBR, mainly by increasing glucose consumption, enhancing gluconeogenesis, and stimulating the uptake of the glucose (Yang et al., 2016, 2017). M3 can protect against nonalcoholic fatty liver disease by attenuating hepatic steatosis and fibrosis in mice mainly through suppressing nuclear factor kappa-B, activation of adenosine monophosphate–activated protein kinase, and inhibition of oxidative stress (Qiang et al., 2016; Wang et al., 2016). Moreover, the four metabolites of BBR can also upregulate low-density lipoprotein receptor (LDLR) expression in human hepatoma cells (Zhou et al., 2014b). Based on all our previous findings, we hypothesized that the metabolites of BBR might also contribute to its antiobesity and hypoglycemic activity.
Farnesoid X receptor (FXR) in the liver and intestine is critical in bile acid–mediated feedback suppression of bile acid synthesis and cholesterol metabolism. Besides the regulation of bile acid homeostasis, FXR activation also improved hyperglycemia and hyperlipidemia by decreasing hepatic gluconeogenesis and increasing glycogenesis (Schumacher and Guo, 2019), as well as ameliorating inflammation and fibrosis in NAFLD (Claudel et al., 2005; Ma et al., 2006; Adorini et al., 2012). However, nonselective activation of both hepatic and intestinal FXR presented some adverse effects, such as decreased high-density lipoprotein cholesterol and increased low-density lipoprotein cholesterol in the blood. Further studies suggest that intestine-specific FXR activation may be safer than whole-body FXR agonist for the treatment of metabolic syndrome, which leads to an increasing focus on the development of selective FXR modulators to achieve organ-specific FXR functions and reduction of side effects (Massafra et al., 2018). Our previous study showed that BBR could modulate the metabolism of bile acids and activate the intestinal FXR signaling pathway (Sun et al., 2017). However, the direct effect of BBR and its metabolites on the FXR signaling pathway is still unclear. In this study, we determined the effect of BBR and its metabolites on bile acid metabolism, the direct and indirect action on intestinal FXR signaling pathway, and the expression of G6pase and Pepck in the liver and aimed to elucidate the mechanisms of their pharmacological effects.
Materials and Methods
Drug and Reagents.
BBR (≥98%) was purchased from Nanjing Zelang Medical Technology Co., Ltd. (Nanjing, China). M1 (≥95%) and M3 (≥95%) were synthesized by Nanjing Chemzam PharmTech Co., Ltd. (Nanjing, China). Methanol, chloroform, isopropanol, and heptane were purchased from Merck KGaA (Darmstadt, Germany). Ethanol was purchased from Nanjing Chemical Reagent Co., Ltd. Millipore water was produced with a Milli-Q Reagent Water System (Millipore, MA). Trizol kit was obtained from TaKaRa Biotechnology Co., Ltd. (Dalian, China). Myristic-1,2-13C2 acid was purchased from Cambridge Isotope Laboratories (Andover, MA). Cholic-2,2,4,4-d4 acid (d4-CA), taurocholic-2,2,4,4-D4 acid (d4-TCA), and glycocholic-2,2,4,4-D4 acid were purchased from TLC PharmaChem (Concord, ON, Canada). Methoxyamine, pyridine, and N-methyl-N-trimethyl-silyl-trifluoroacetamide were purchased from Sigma-Aldrich (Shanghai, China).
Animal Studies.
Thirty-six male C57BL/6J mice (6 weeks old; obtained from Comparison Medical Center of Yangzhou University, Yangzhou, China) were fed with a standard chow diet [AIN-93M; containing 10% fat, 75.9% carbohydrate, and 14.1% protein in total calories (3601 kcal/kg); Trophic Animal Feed High-Tech Co., Ltd, Nantong, China] and tap water ad libitum. They were housed at a temperature- and light-controlled facility [temperature 25 ± 2°C, relatively humidity (50% ± 5%), and 12-hour light/dark cycle (06:00 AM to 6:00 PM)] for 1 week to acclimate to the environment before the study.
To determine the effects of BBR, M1, and M3 on hyperglycemia and their antiobesity activities, we weighed the mice and randomly divided them into six groups (n = 6), as shown in Supplemental Fig. 1. The vehicle group continued feeding with the standard diet, and the other groups were fed with a high-fat diet (HFD; Trophic Animal Feed High-Tech Co., Ltd) containing 60% fat, 25.9% carbohydrate, and 14.1% protein in total calories (5018 kcal/kg) and 1% cholesterol to induce obesity and hyperglycemia. For intragastric administration, BBR, M1, and M3 were ground and suspended in 0.5% sodium carboxymethyl cellulose aqueous solution to achieve concentrations of 15 mg/ml (150 mg/kg of BBR), 2.5 mg/ml (25 mg/kg of M1), 5 mg/ml (50 mg/kg of M1), and 18 mg/ml (180 mg/kg of M3), respectively, and the dose was 10 ml/kg. All groups were weighed weekly, and the food intake was recorded daily. After feeding for 7 weeks, the mice were placed in metabolic cages to collect feces. After administration with BBR and its metabolites for another week, all mice were fasted overnight for 12 hours, and serum samples were collected and serum glucose levels were measured. Liver, ileum, and cecal content were snap-frozen in liquid nitrogen until molecular analyses were conducted. All animal experiments were performed with the approval of the Animal Ethics Committee of China Pharmaceutical University.
Metabolomics Study.
Metabolite profiling of mouse liver by gas chromatography-mass spectrometry (GC-MS)–based metabolomics method was described previously (Aa et al., 2005). Briefly, metabolites from 20 mg of liver tissue were extracted by 600 μl of 80% methanol (containing 5 μg/ml of myristic-1,2-13C2 acid as internal standard). An aliquot of 100 μl supernatant was transferred to a gas chromatograph vial and dried by Savant SPD2010 Speedvac concentrator (Thermo Scientific, MA). The extract was then oximated with 30 μl pyridine containing 10 mg/ml methoxyamine for 16 hours at room temperature, followed by derivatization with 30 μl N-methyl-N-trimethylsilyl-trifluoroacetamide+1% trimethylchlorosilane (MSTFA+1%TMCS) for 1 hour. Subsequently, 30 μl of heptane was added to the sample, and 0.5 μl of sample was used to inject the Shimadzu QP2010Ultra/SE GC-MS system (Shimadzu Co., Kyoto, Japan) with an RTx-5MS fused silica capillary column (30 m × 0.25 mm inner diameter; J&W Scientific, Bellefonte, PA) at split mode.
Compound Identification.
The raw data acquired by GC-MS were processed by GCMS solution version 4.11 (Shimadzu Co.), and the metabolites were identified by comparing the mass spectra and retention time of the detected peaks with National Institute of Standards and Technology/Environmental Protection Agency/National Institutes of Health Mass Spectral Library (NIST14; National Institute of Standards and Technology, Gaithersburg, MD) and Wiley Registry of Mass Spectral Data, 9th Edition (Wiley 9; John Wiley and Sons, Inc., Hoboken, NJ) and an in-house mass spectra library data base. The peak area of identified compounds in each sample was normalized to that of the internal standard.
Multivariate and Univariate Data Analysis.
SIMCA 13.0 software (Umetrices, Umeå, Sweden) was used to perform principal components analysis (PCA) and partial least-squares discriminant analysis (PLS-DA). The PCA model was used to check the overall distribution of all the samples, and the PLS-DA model was used to confirm the general separation among each group. The variable of importance in the projection was used to identify the endogenous metabolites contributing to the classification. The strength of a pattern recognition model was evaluated by the parameters R2X or R2Y and Q2Y parameters. Orthogonal partial least-squares discriminant analysis and shared and unique structure plot (SUS-plot) were used to analyze the differences between two or three groups. Metabolomics pathway analysis of the metabolic biomarkers was carried out using MetaboAnalyst (www.metaboanalyst.ca) (Xia and Wishart, 2016).
Measurement of Bile Acids.
The abundance of bile acids in serum, liver, and feces was analyzed by liquid chromatography-tandem mass spectrometry (Shimadzu High Performance Liquid Chromatography system coupled to an AB SCIEX 4000 mass spectrometer) as previously described (Zhou et al., 2014a; Sun et al., 2017). Briefly, bile acids were extracted with 70% ethanol containing glycocholic-2,2,4,4-D4 acid as an internal standard at 55°C for 4 hours, and the extract was dried by Savant SPD2010 Speedvac concentrator (Thermo Scientific) and redissolved in 100 μl of 50% methanol:water. After centrifugation, 80 μl of supernatant was transferred to a liquid chromatography vial, and 10 μl was injected into a Waters Atlantis T3 column (2.1 × 100 mm, 3 μm) for chromatographic separation. The mobile phase consisted of solvent A (0.1% formic acid in water) and solvent B (methanol).
Bile Salt Hydrolase Activity Measurement.
Bile salt hydrolase (BSH) activity of the gut flora was measured as previously described (Zhao et al., 2014; Sun et al., 2017). The conversion of d4-CA from d4-TCA by BSH was analyzed by LC-MS/MS.
Quantitative Real-Time Polymerase Chain Reaction.
Total RNA from mouse liver, ileum, and LS174T cells was extracted by Trizol reagent according to the manufacture’s protocol and they were reverse-transcribed to cDNA by PrimeScript 1st Strand cDNA Synthesis kit were obtained from Takara Biotechnology. The real-time PCR assay was performed using a SYBR Green–based method (iTaq universal SYBR Green supermix; Bio-Rad, Hercules, CA). The relative mRNA levels of all detected genes were normalized to that of β-actin in each sample (using the Δ-Δ cycle threshold method). Results are expressed as fold change relative to the vehicle group, and primers used for the quantitative real-time PCR study were listed in Table 1.
Primers used in qRT-PCR analysis
Molecular Docking.
A computer-based modeling program (Autodock vina) was employed to evaluate the binding of BBR and its metabolites with FXR. The chemical structures of BBR and its metabolites [generated from PubChem (https://pubchem.ncbi.nlm.nih.gov)] and the crystal structure of human FXR [from Protein Data Bank (4WVD) (https://www.rcsb.org)] were used for docking.
The Effects of BBR and Its Metabolites on FXR Signaling Pathway In Vitro.
To investigate the effects of BBR, M1, and M3 on the activity of FXR and expression of Fgf19, LS174T cells (purchased from American Type Culture Collection, Manassas, VA) were plated in a 12-well plate at 1 × 105 cells per well. After 24 hours, cells were treated with 0.1% DMSO (negative control), various concentrations of BBR (0.1, 1, 10, and 50 μM), M1 (0.1, 1, 10, and 100 μM), and M3 (0.1, 1, 10, and 100 μM) and 100 μM of chenodeoxycholic acid (CDCA) (positive control). Total RNA was isolated by Trizol reagent (Takara). At 24 hours after treatment, gene expression levels of Fxr and Fgf19 were measured by real-time PCR. To test the effects of FXR antagonism by BBR and its metabolites, cells were treated for 24 hours with 100 μM of CDCA in the absence or presence of BBR and its metabolites or 0.1% DMSO as a negative control. Furthermore, the expression of Fgf19 was determined after FXR was silenced with small interfering RNA (siRNA) (5′-GAAUUCGAAAUAGUGGUAUCUCUGA-3′; Invitrogen, Thermo Fisher Scientific) for 24 hours and treated with BBR (25 μM), M1 (100 μM), and M3 (100 μM) for another 24 hours.
Statistical Analysis.
All data are expressed as means ± S.D. Differences among groups were tested by one-way ANOVA followed by Fisher’s least significant difference multiple comparison test and corrected by the Benjamini-Hochberg method to control the false discovery rate (performed by R project, version 3.5.3). A value of P < 0.05 was considered statistically significant. The bar plots were generated by GraphPad Prism (version 7.0).
Results
BBR and M1 Protected Mice against High-Fat Diet–Induced Obesity and Hyperglycemia.
There was no obvious difference in food intake among groups (Supplemental Fig. 2). Compared with mice on the control diet, the mice in the HFD group showed a significant increase in body weight (HFD group 31.79 ± 1.85 g vs. vehicle group 26.21 ± 1.68 g, P < 0.001) and blood glucose levels (HFD group 12.17 ± 1.70 mmol/l vs. vehicle group 5.22 ± 2.34 mmol/l, P < 0.001). BBR treatment protected the mice against diet-induced obesity (BBR group 28.16 ± 2.19 g vs. HFD group 31.79 ± 1.85 g, P = 0.011, Fig. 1A) and hyperglycemia (BBR group 9.35 ± 1.60 mmol/l vs. HFD group 12.17 ± 1.70 mmol/l, P = 0.015) (Fig. 1B). Oral administration of M1 also showed protection from diet-induced obesity and hyperglycemia at different doses (25 mg/ml group 28.89 ± 1.60 g, 50 mg/ml group 23.98 ± 1.91 g vs. HFD group 31.79 ± 1.85 g, P = 0.011, Fig. 1A) and hyperglycemia (25 mg/ml group 8.83 ± 2.03 mmol/l, 50 mg/ml group 6.31 ± 1.67 mmol/l vs. HFD group 12.17 ± 1.70 mmol/l, P = 0.015) (Fig. 1B). However, oral administration of M3 did not affect diet-induced obesity or hyperglycemia (Fig. 1, A and B).

BBR and M1 prevented high-fat diet–induced weight gain and hyperglycemia in C57BL/6J mice. (A) Weekly body weight changes. (B) Blood glucose. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, compared with high-fat diet group, n = 6.
GC-MS Profiles of Hepatic Metabolites and Differential Compound Analysis.
The representative chromatography of each group is shown in Fig. 2A. After deconvolution, identification, and quantitation, a total of 98 compounds were identified in the liver, including various amino acids, organic acids, amines, saccharides, purines and fatty acids. For an overview of the data set, six groups were separated by PLS-DA [R2X (cum) = 0.339, R2Y (cum) = 0.313 and Q2 (cum) = 0.0963], and samples in each group clustered close to each other (Fig. 2B). The orthogonal partial least-squares discriminant analysis of each treatment group and HFD group was performed. BBR and both doses of M1 showed a good effect on regulation of abnormal metabolism of the liver induced by HFD feeding (Fig. 3, A–C). Surprisingly, M3 also showed an obvious modulatory effect of metabolites in the liver, although it showed no obvious hypoglycemic effect (Fig. 3D). The SUS-plot and univariate statistical test revealed that the glycogenic amino acids, including alanine, glutamic acid, glutamine, isoleucine, valine, cysteine, proline, and threonine, are elevated in the high-fat diet group. Fatty acids, including arachidonic acid and palmitic acid, are also elevated in the high-fat diet group. The metabolites in the TCA cycle, including fumaric acid and malic acid, also increased. The level of lactic acid decreased in the high-fat diet group. BBR significantly reversed the increase of these amino acids and fatty acids and increased lactic acid, fumaric acid, and malic acid. The modulatory effect of M1 was weaker than that of BBR, and M3 treatment even showed an opposite trend (Fig. 3, E–H; Fig. 4). The concentrations of BBR and its metabolites in the liver were shown in Supplemental Fig. 3.

Typical GC-MS chromatograms of all groups and the overview of the metabolomics data. (A) Typical GC-MS chromatograms of mouse liver from all groups. (B) The scores plot of PCA showed that samples from the vehicle group (Vehicle), HFD group, 150 mg/kg of BBR group (BBR), 25 mg/kg of M1 group (M1-25), 50 mg/kg of M1 group (M1-50), and 180 mg/kg of M3 group (M3-180) were clustered separately.

The scores plot of PLS-DA analysis and SUS-plot of metabolite profile in the liver of C57BL/6J mice from all groups. (A) Scores plot of PLS-DA analysis of HFD, vehicle, and BBR group; (B) scores plot of PLS-DA analysis of HFD, vehicle, and M1-25 group; (C) scores plot of PLS-DA analysis of HFD, vehicle, and M1-50 group; (D) scores plot of PLS-DA analysis of HFD, vehicle, and M3-180 group; (E) SUS-plot of metabolite profile in HFD, vehicle, and BBR group; (F) SUS-plot of metabolite profile in HFD, vehicle, and M1-25 group; (G) SUS-plot of metabolite profile in HFD, vehicle, and M1-50 group; (H) SUS-plot of metabolite profile in HFD, vehicle, and M3-180 group. M1-25, 25 mg/kg of M1 group; M1-50, 50 mg/kg of M1 group; M3-180, 180 mg/kg of M3 group. MEMY, methyl myristate; TMS, trimethyl chlorosilane.

Change of endogenous metabolites in the liver of C57BL/6J mice after BBR, M1, and M3 treatment for 8 weeks. Gluconeogenesis pathway: lactic acid; TCA cycle: fumaric acid, malic acid; amino acids: aspartic acid, glutamic acid, glutamine, tryptophan, phenylalanine, proline, alanine, cysteine, leucine, isoleucine, valine; fatty acids: arachidonic acid. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001, compared with high-fat diet group, n = 6. M1-25, 25 mg/kg of M1 group; M1-50, 50 mg/kg of M1 group; M3-180, 180 mg/kg of M3 group.
BBR and Its Metabolites Changed the Composition of Bile Acids.
Bile acids are the metabolites of cholesterol and physiologic ligands of FXR. The bile acid profiles in serum, liver, and feces were analyzed by the LC-MS/MS method (shown in Fig. 5). The composition of conjugated bile acids and free bile acids in the serum, liver, and feces was changed by HFD feeding and medical treatment. Specifically, in the high-fat diet group, the serum levels of conjugated bile acids showed a trend of decrease, and the levels of free bile acids significantly decreased (Fig. 5, A and B). The levels of conjugated bile acids and free bile acids in the liver were both reduced after high-fat feeding (Fig. 5, C and D). In the feces, the levels of conjugated bile acids and free bile acids were both elevated (Fig. 5, E and F). BBR treatment increased the conjugated bile acids and reduced the free bile acids in serum and feces (Fig. 5, A–F), which is consistent with our previous study (Sun et al., 2017). The high dose of M1 increased free bile acids in the liver (Fig. 5D) and the conjugated bile acids in the feces (Fig. 5E). The low dose of M1 (25 mg/kg) and M3 (180 mg/kg) showed no noticeable effect on the total conjugated and free bile acids. In conclusion, BBR and M1 both modulated the composition of system bile acids, and BBR showed a far stronger modulatory effect than its metabolite (Fig. 5).

Changes in bile acid composition in the serum, liver, and feces of C57BL/6J mice after BBR, M1, and M3 treatment. Bile acids in serum, liver, and feces were measured using LC-MS/MS. (A) Change in total conjugated bile acids in the serum. (B) Change in total free bile acids in the serum. (C) Change in total conjugated bile acids in the liver. (D) Change in total free bile acids in the liver. (E) Change in total conjugated bile acids in the feces. (F) Change in total free bile acids in the feces (n = 6). (G) the production of d4-CA by the gut microbiota of each group. *P < 0.05; **P < 0.01; ***P < 0.001, compared with high-fat diet group, n = 6. M1-25, 25 mg/kg of M1 group; M1-50, 50 mg/kg of M1 group; M3-180, 180 mg/kg of M3 group. DCA, deoxycholic acid; F-BA, free bile acids; GCA, glycocholic acid; HDCA, hyodeoxycholic acid; T-BA, conjugated bile acids; TCDCA, tauro-chenodeoxycholic acid; THDCA, tauro-hyodeoxycholic acid; TLCA, tauro-lithocholic acid; TUDCA, tauro-ursodeoxycholic acid; TαMCA, tauro-α-muricholic acid; TβMCA, tauro-β-muricholic acid; ωMCA, ω-muricholic acid; αMCA, α-muricholic acid; βMCA; β-muricholic acid.
BBR Inhibited the Activity of BSH, whereas M1 and M3 Showed No Effect.
BSH activity indicates the capacity of gut microbiota for hydrolyzing conjugated bile acids to unconjugated bile acids. Consistent with our previous study (Sun et al., 2017), a significant decrease of d4-CA production was observed after BBR treatment. However, M1 and M3 treatment had no apparent effect on the hydrolyzation of d4-TCA to d4-CA (Fig. 5G).
BBR and M1 Activated the Intestinal FXR Signaling Pathway and Repressed Hepatic Gluconeogenic Gene Expression.
The intestinal FXR target genes of Fgf15 and Shp were upregulated after BBR administration, but without significance. The expression of organic solute transporter-α (Ostα) was upregulated significantly by M1. M3 had no apparent effect on the expression of Fgf15, whereas it increased the expression of Shp (Fig. 6, A–D). In the liver, BBR or its metabolites did not change the expressions of hepatic FXR target genes. These results suggested that BBR and M1 might activate the intestinal but not hepatic FXR signaling pathway. BBR and M1 treatment both upregulated the expression of Ldlr and inhibited the expression of G6pase and Pepck in the liver, whereas M3 only reduced the expression of Pepck (Fig. 6, E–L).

BBR and M1 activated intestinal FXR signaling pathway but did not activate the hepatic FXR signaling pathway in C57BL/6J mice. Relative mRNA levels of Fxr (A), Fgf15 (B), Shp (C), and Ostα (D) in the distal ileum. Relative mRNA levels of Fxr (E), Fgfr4 (F), Shp (G), Cyp7a1 (H), bile salt export pump (Bsep) (I), Ldlr (J), G6pase (K), and Pepck (L) in the liver. *P < 0.05; **P < 0.01, compared with high-fat diet group, n = 6. M1-25, 25 mg/kg of M1 group; M1-50, 50 mg/kg of M1 group; M3-180, 180 mg/kg of M3 group.
BBR and M1 Acted as Agonists of FXR.
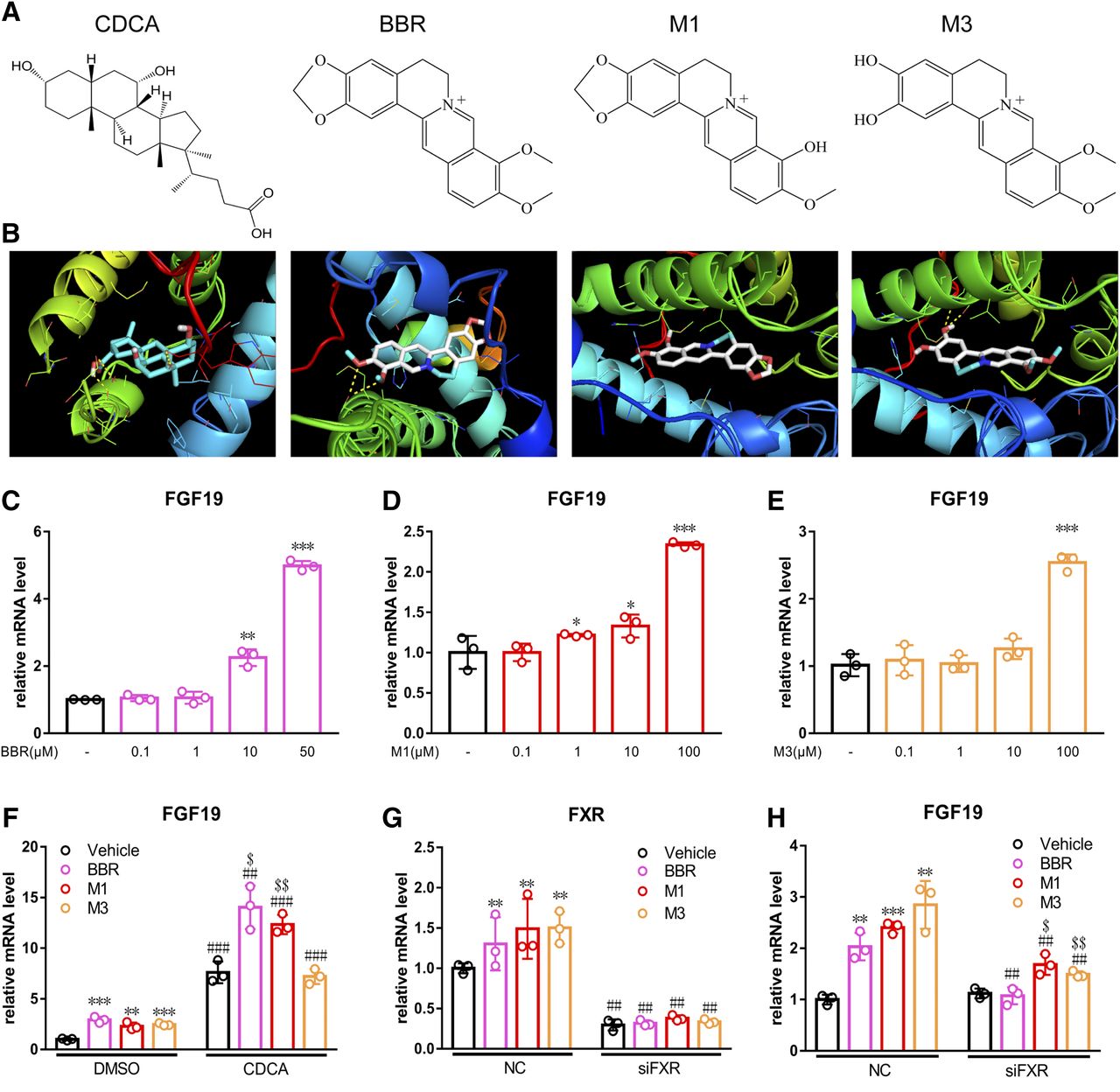
Computational molecular docking was used to predict the interaction between BBR and its metabolites with human FXR. BBR (−8.6 kcal/mol), M1 (−9.0 kcal/mol), and M3 (−8.3 kcal/mol) showed similar affinities as CDCA (−9.7 kcal/mol) (Fig. 7, A and B). FGF19 is the human ortholog of mouse FGF15. In LS174T cells, BBR induced the expression of Fgf19 at concentrations of 10 and 50 μM by 2.25-fold (P < 0.01) and 4.99-fold (P < 0.001), respectively (Fig. 7C). M1 induced the expression of Fgf19 at the concentrations of 1, 10, and 100 μM by 1.22-fold (P < 0.05), 1.33-fold (P < 0.05), and 2.34-fold (P < 0.001), respectively (Fig. 7D). M3 induced the expression of Fgf19 only at the concentration of 100 μM by 2.54-fold (P < 0.001) (Fig. 7E). To test the FXR antagonism effects for BBR, M1, and M3, cells were treated with physiological FXR ligands of CDCA followed by BBR and its metabolites. When used simultaneously, the two compounds of BBR and M1 both synergistically activated FXR (Fig. 7F). CDCA alone induced the expression of Fgf19 by 7.61-fold, whereas cotreatment with BBR (25 μM) and M1 (100 μM) induced Fgf19 gene expression by 14.04- and 12.34-fold, respectively. However, M3 showed no synergistic effects on the expression of Fgf19 (Fig. 7F). The induced expression of Fgf19 was weakened after BBR, M1, and M3 treatment and FXR was silenced (Fig. 7, G and H).

BBR and M1 activated FXR signaling pathway in LS174T cells. (A) Structures of CDCA, BBR, M1, and M3. (B) Molecular docking results of CDCA, BBR, M1, and M3 with human FXR. PCR analysis of LS174T cells after treatment with BBR (C), M1 (D), and M3 (E). (F) the expression of Fgf19 after cotreatment with CDCA. The expression of Fxr (G) and Fgf19 (H) after silencing by siRNA. *P < 0.05; **P < 0.01; ***P < 0.001, comparison between BBR-, M1-, and M3-treated groups with the corresponding control group. ##P < 0.01; ###P < 0.001, comparison between CDCA-cotreated or siRNA-silenced group with the corresponding untreated group. $P < 0.05; $$P < 0.01, comparison between BBR-, M1-, and M3-treated groups with the corresponding groups cotreated with CDCA or silenced by siRNA.
Discussion
BBR has been reported to have glucose- and lipid-lowering effects in humans and different animal models, and several studies have demonstrated that the mechanism involves the alteration of metabolic pathways in the liver (Xing et al., 2011; Li et al., 2014; Pirillo and Catapano, 2015; Yu et al., 2015; Zou et al., 2017). However, in our previous study using animal models, we found that BBR accumulated in the intestine with much excretion in feces. In contrast, its primary metabolites, M1 and M3, reached a high concentration of 100 ng/ml in the liver. This finding was also confirmed by another study (Tan et al., 2013). Therefore, we hypothesized that BBR or its metabolites might affect the expression of genes and pathways in the liver and/or intestine to lower serum glucose levels and reduce body weight.
The metabolomics of serum/plasma, urine, gut content/feces, and local tissues/organs had been studied after BBR treatment in obesity or hyperglycemia and hyperlipidemia models in different species, and these studies showed that orally administered BBR leads to a global change in metabolism, including glycolysis, amino acid metabolism, and lipid metabolism (Jiang et al., 2012; Li et al., 2015; Dong et al., 2016; Li et al., 2016). Because gluconeogenesis mainly takes place in the liver, and liver was regarded as the primary target organ for BBR in previous studies, the metabolomics of the liver was profiled to determine the modulatory effects of BBR and its metabolites on metabolism. After treatment with BBR or M1, the elevated levels of lactic acid and tricarboxylic acid cycle intermediates revealed acceleration in energy metabolism and glucose consumption. In contrast, decreased levels of amino acids involved in glucogenesis (glutamic acid and branched-chain amino acids) were also observed. The reduced fatty acid levels suggest inhibition of synthesis or enhanced metabolism of lipids.
BBR can improve hepatic glucose and lipid metabolism in various metabolic diseases through multiple pathways. The underlying mechanisms may involve activation of adenosine monophosphate–activated protein kinase, improvement of insulin sensitivity, and upregulation of LDLR (Kong et al., 2004; Zhang et al., 2011, 2012; Wei et al., 2016; Zhao et al., 2017a). BBR could also increase glucose utilization in adipocytes and myocytes and decrease glucose transportation in intestinal cells by inhibiting α-glucosidase. Moreover, the expression of gluconeogenic genes and several lipid metabolism genes were modulated by BBR (Xia et al., 2011). A significant reduction in Pepck and G6pase expression and a significant elevation in Ldlr expression in the livers of mice treated with BBR were observed, with the results consistent with previously published reports (Kong et al., 2004; Xia et al., 2011). Furthermore, in the livers of M1-treated mice, a similar trend of the expression of these genes was observed showing that M1 also modulated gluconeogenesis, even at a lower dose. We consider that these pathways may be the primary pathways responsible for the glucose-lowering effects of BBR and M1. However, whether these effects were the direct or indirect function of BBR and M1 still needs to be investigated further.
According to our previous study and several other studies, BBR may function through modulating the composition of gut bacteria, bile acids, and intestinal signaling pathways (Zhang et al., 2012; Gu et al., 2015; Sun et al., 2017). Bile acids are endogenous ligands of FXR, and individual bile acids have differential effects on bile acid signaling (Song et al., 2015). Bile acids activate FXR with different efficacy [CDCA > deoxycholic acid > cholic acid (CA)], and the conjugated bile acids are less potent, whereas tauro-β-muricholic acid, ursodeoxycholic acid (UDCA), and lithocholic acid (LCA) showed FXR antagonism activity (Yu et al., 2002; Mueller et al., 2015). We found that the concentration of most FXR agonists, including CA and taurocholic acid (TCA), in the feces were significantly increased and that the concentration of FXR antagonists, including LCA and UDCA, were reduced considerably. In contrast, the concentrations of bile acids in the serum and liver were rarely changed, except elevated TCA in the serum; this was in accordance with our previous study (Sun et al., 2017). However, the effects of BBR metabolites were far weaker than that of BBR. The overall impact of BBR and M1 treatment trended toward intestinal FXR activation, which suggests BBR and M1 may activate the intestinal FXR signaling pathway by their FXR activation effect directly or a combined activation of FXR with endogenous bile acids. It has been reported that FXR agonists could repress the expression of G6pase and Pepck in the liver (Zhang et al., 2006), and the inhibited expression of G6pase and Pepck by BBR and M1 may be due to activation of intestinal the FXR signaling pathway.
FXR activation can improve glucose and lipid metabolism, and FXR, especially intestinal-specific agonism of FXR, may be a potential target for treating obesity and NASH (Zhang et al., 2006; Potthoff et al., 2011; Adorini et al., 2012). The intestinal FXR-FGF15/19 pathway can regulate hepatic glucose metabolism, mainly by modulating the cAMP-response element binding protein-peroxlsome proliferator-activated receptor-γ coactivator-1α pathway and gluconeogenesis (Potthoff et al., 2011; Trabelsi et al., 2017). A phase III study of obeticholic acid (INT-747) for treating NASH has been finished, with the results showing that the primary endpoint for improving liver fibrosis without worsening of NASH was reached (Zhang et al., 2009; Mudaliar et al., 2013; Neuschwander-Tetri et al., 2015). For natural compounds, epigallocatechin gallate and its analogs are found to be able to activate FXR specifically and dose-dependently in vivo and in vitro (Li et al., 2012; Sheng et al., 2018). In this study, from molecular docking assay and target gene analysis in C57BL/6J mice and LS174T cells, we demonstrate that BBR and M1 may activate the FXR signaling pathway both in vivo and in vitro. In BBR- and M1-treated mice, the intestinal but not the hepatic FXR signaling pathway was activated, shown by increased expression of Fgf15 and Shp in the ileum and inhibition of Shp in the liver. Molecular docking analysis suggests that BBR and its metabolite M1 had similar binding energy with that of CDCA. We further validated the activation function of BBR and its metabolites on FXR by targeting gene analysis. These results all suggest that BBR and M1 may be agonists of the intestinal FXR signaling pathway. What is more, BBR and M1 can also enhance the activation effect of CDCA on FXR. We previously found that BBR can modulate the composition of bile acids, and the elevated TCA in the intestine activates the intestinal FXR signaling pathway and lowers lipids in the liver (Sun et al., 2017). Here, we found that BBR treatment increases the levels of bile acids that were FXR agonists and decreases the levels of bile acids that were FXR antagonists. In contrast, the modulatory effect of M1 and M3 on bile acid metabolism is far weaker than that of BBR. These results suggest that the intestinal-specific activation of FXR signaling pathway by BBR and M1 may be responsible for their glucose-lowering effect. BBR showed an agonism effect on FXR in vitro directly, and the modulated composition of bile acids mediated by gut bacteria may enhance these effects. M1 showed a weaker agonism effect on FXR than BBR in vitro at the same dose, whereas it showed a stronger effect on antiobesity and hypoglycemia in C57BL/6J mice at a lower dose; this difference may be due to the fact that the bioavailability of M1 is better than that of BBR, as shown in our previous study (Yang et al., 2017; Zhao et al., 2017b). The role of M1 itself may have been dominant for its hypoglycemic effect independent of bile acids. Our findings may provide new, natural, and intestinal-specific FXR agonists that have potential for the treatment of hyperglycemia and NASH.
In conclusion, we found that BBR and M1 could ameliorate obesity and hyperglycemia induced by high-fat diet feeding, and M1 was even more effective than BBR. BBR and M1 could modulate several metabolic pathways in the liver of C57BL/6J mice. The combined in vivo and in vitro data showed BBR and its metabolite M1 are FXR agonists. The activation of the intestinal FXR signaling pathway by BBR and the modulated composition of bile acids may work together to achieve its pharmacological effects. However, the impact of M1 may be bile acid–independent; it activated the intestinal FXR signaling pathway directly and repressed the expression of hepatic gluconeogenesis genes with higher systemic exposure. Overall, BBR and M1 could activate the intestinal FXR signaling pathway, inhibiting the expression and function of G6pase and Pepck in the liver, thus reducing hepatic gluconeogenesis and, finally, gaining their glucose-lowering and antiobeisty effects.
Authorship Contributions
Participated in research design: Sun, C. S. Yang, Guo, Wang, Aa.
Conducted experiments: Sun, Kong, N. Yang, Cao, D. Feng, Yu, Ge, S. Feng, Fei, Lu.
Contributed new reagents or analytic tools: Sun, N. Yang.
Performed data analysis: Sun, Huang.
Wrote or contributed to the writing of the manuscript: Sun, Kong, Xie, C. S. Yang, Guo, Wang, Aa.
Footnotes
- Received August 17, 2020.
- Accepted December 1, 2020.
This study was financially supported by the Key Technology Projects of China “Creation of New Drugs” [Grant 2017ZX09301013], the National Natural Science Foundation of the People’s Republic of China [Grants 81573495, 81530098, 81703601, 81773814, 81421005], the Natural Science Foundation of Jiangsu Province [BK20190122], and “Double First-Class” university project of China Pharmaceutical University [CPU2018GF01].
The authors declare that there is no conflict of interest.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- BBR
- berberine
- BSH
- bile salt hydrolase
- CA
- cholic acid
- cum
- cumulative
- d4-CA
- cholic-2,2,4,4-d4 acid
- d4-TCA
- taurocholic-2,2,4,4-D4 acid
- FGF
- fibroblast growth factor
- FXR
- farnesoid X receptor
- GC-MS
- gas chromatography-mass spectrometry
- G6pase
- glucose-6-phosphatase
- HFD
- high-fat diet
- LCA
- lithocholic acid
- LDLR
- low-density lipoprotein receptor
- M1
- berberrubine
- M3
- demethyleneberberine
- NAFLD
- nonalcoholic fatty liver disease
- NASH
- nonalcoholic steatohepatitis
- Ostα
- organic solute transporter-α
- PCA
- principal component analysis
- PCR
- polymerase chain reaction
- Pepck
- phosphoenolpyruvate carboxykinase
- PLS-DA
- partial least-squares discriminant analysis
- SHP
- small heterodimer partner
- SUS-plot
- shared and unique structure plot
- UDCA
- ursodeoxycholic acid
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}