


Abstract
We investigated the effect of variability and instability in aldehyde oxidase (AO) content and activity on the scaling of in vitro metabolism data. AO content and activity in human liver cytosol (HLC) and five recombinant human AO preparations (rAO) were determined using targeted proteomics and carbazeran oxidation assay, respectively. AO content was highly variable as indicated by the relative expression factor (REF; i.e., HLC to rAO content) ranging from 0.001 to 1.7 across different in vitro systems. The activity of AO in HLC degrades at a 10-fold higher rate in the presence of the substrate as compared with the activity performed after preincubation without substrate. To scale the metabolic activity from rAO to HLC, a protein-normalized activity factor (pnAF) was proposed wherein the activity was corrected by AO content, which revealed up to sixfold higher AO activity in HLC versus rAO systems. A similar value of pnAF was observed for another substrate, ripasudil. Physiologically based pharmacokinetic (PBPK) modeling revealed a significant additional clearance (CL; 66%), which allowed for the successful prediction of in vivo CL of four other substrates, i.e., O-benzyl guanine, BIBX1382, zaleplon, and zoniporide. For carbazeran, the metabolite identification study showed that the direct glucuronidation may be contributing to around 12% elimination. Taken together, this study identified differential protein content, instability of in vitro activity, role of additional AO clearance, and unaccounted metabolic pathways as plausible reasons for the underprediction of AO-mediated drug metabolism. Consideration of these factors and integration of REF and pnAF in PBPK models will allow better prediction of AO metabolism.
SIGNIFICANCE STATEMENT This study elucidated the plausible reasons for the underprediction of aldehyde oxidase (AO)-mediated drug metabolism and provided recommendations to address them. It demonstrated that integrating protein content and activity differences and accounting for the loss of AO activity, as well as consideration of extrahepatic clearance and additional pathways, would improve the in vitro to in vivo extrapolation of AO-mediated drug metabolism using physiologically based pharmacokinetic modeling.
Introduction
Over the past decade, metabolism by aldehyde oxidase (AO) has been gaining importance as drug design has evolved in the direction of making compounds that are more stable to cytochrome P450 (P450)-mediated metabolism (Argikar et al., 2016). AO is a highly promiscuous enzyme and has been implicated in the oxidative, reductive, and hydrolytic metabolism of drugs belonging to various therapeutic categories (Dalvie and Di, 2019). Failure to sufficiently characterize AO-mediated metabolism has led to inaccurate predictions of clearance resulting in either toxicity or poor exposure in humans, leading to the discontinuation of compounds such as carbazeran, BIBX1382, FK3453, LuAF09535, and RO1 (Kaye et al., 1984; Dittrich et al., 2002; Akabane et al., 2011; Zhang et al., 2011; Jensen et al., 2017). Recent reports such as methotrexate-induced liver toxicity due to inhibition of its metabolism following coadministration of an AO substrate and inhibitor, favipiravir (Demir et al., 2022), also indicate the possible role of AO in clinically significant drug-drug interactions. AO shows wide species differences, which makes the translation of toxicological data challenging. For example, dogs do not express functional AO, and the activity in rats is significantly lower than in humans and monkeys (Terao et al., 2016). Despite the increased incidence of AO-mediated metabolism, challenges associated with in vitro to in vivo extrapolation (IVIVE) and species differences continue to pose hurdles in drug discovery. This leads to drug discovery programs preferring to avoid advancement of new chemical entities (NCEs) with perceived AO liabilities. The AO protein content differences in preclinical models and humans can partly explain the species differences in AO-mediated metabolism (Basit et al., 2022); however, the reasons for the poor IVIVE of AO metabolism are not fully characterized.
The challenges of characterizing in vitro activity of AO include high interlot variability in hepatocytes, human liver cytosol (HLC) or S9 fractions, and loss of activity on long-term storage (Hutzler et al., 2012; Barr et al., 2013). The variability between lots has been attributed to differences in the method of preparation, handling, and decline in activity during storage and in postmortem tissues (Barr et al., 2013). Alterations in interindividual AO activity have also been reported due to the presence of allelic variants of the AOX1 gene, resulting in proteins with increased or decreased activity (Hartmann et al., 2012; Ueda et al., 2022). The in vitro activity of AO for most of its oxidative substrates is nonlinear with time due to concurrent inactivation (Abbasi et al., 2019), and the enzyme is also liable to substrate inhibition (Barr et al., 2013). However, there has been no documentation of the rates of decline in activity in the presence and absence of substrate.
Heterologously expressed recombinant systems have proven to be key tools in in vitro studies of drug-metabolizing enzymes such as P450s and UDP-glucuronosyl transferases. Several attempts have been made previously to prepare recombinant AO (rAO) in different expression systems (Hartmann et al., 2012; Coelho et al., 2015; Foti et al., 2016). However, the expression and purification of rAO have been proven to be difficult. Although rAO is available commercially in the form of enriched preparations containing the expressed enzyme, such as those from HEK293 cells (Corning and Origene) and Escherichia coli (Cypex/Xenotech, Hypha discovery), there are no commercially available purified rAO systems. AO is a complex homodimer, which makes the production of the catalytically active enzyme challenging (Cronin et al., 2021).
The differences between AO-specific activity in HLC and E. coli–expressed enzymes have been explained by possible incomplete incorporation of the molybdenum cofactor (MoCo) and iron, perturbed dimerization, and a lack of expression of MoCo sulfurase, the enzyme responsible for incorporating the terminal sulfido ligand to the molybdenum cofactor (Barr et al., 2013). As the native sulfurases present in E. coli are not capable of sulfurating MoCo effectively, chemical sulfuration techniques have been employed—for example, treatment with sodium sulfide under anaerobic conditions to convert MoCo to its active sulfurated form (Foti et al., 2016). Human AO dimers with high specific activity have been produced in baculovirus-infected insect cells without a requirement for postproduction in vitro sulfuration or for coexpression with MoCo sulfurase (Cronin et al., 2021).
The goal of this study was to characterize the factors that affect the IVIVE of AO activity from rAO and HLC. Current practice to determine in vitro intrinsic clearance of AO-mediated metabolism are based on the normalization by the total protein amount and expressed as µL/min per mg protein. A caveat of using this approach is that the Vmax for a given reaction would differ based on the protein amount used in the incubation. In the present study, we used a quantitative proteomics approach to determine the content of AO in rAO from four different sources, HLC, and hepatocytes to obtain their relative expression factor (REF) values. Activity assays, with carbazeran as substrate, were carried out using a fixed amount of AO (pmol per incubation) across the different systems.
The substrate independence of protein-normalized activity factor (pnAF) was evaluated using another AO substrate, ripasudil. To investigate the loss of activity of AO during incubations, this was determined in HLC, and in the rAO preparation with the highest activity, in the presence and absence of the probe substrate, carbazeran. A proteomics-informed physiologically based pharmacokinetic modeling (PBPK) model was then developed to estimate the additional clearance required to address the underprediction of in vivo clearance for carbazeran from in vitro metabolism data. This model was used to assess the IVIVE of other AO substrates, i.e., O6 benzyl guanine, BIBX1382, zaleplon, and zoniporide.
Experimental Section
Materials
Methanol, DMSO, Mass spectrometry (MS)-grade acetonitrile, potassium di-hydrogen phosphate, dipotassium hydrogen phosphate, and formic acid were procured from Fisher Scientific (Fair Lawn, NJ). Acetone was purchased from Sigma Aldrich (St Louis, MO). Bicinchoninic acid (BCA) kit for total protein quantification was purchased from Pierce Biotechnology (Rockford, IL). Ammonium bicarbonate(ABC), dithiothreitol, iodoacetamide, (tris(2-carboxyethyl) phosphine) (TCEP), and MS-grade trypsin were procured from Thermo Fisher Scientific (Rockford, IL). Human serum albumin (HSA) and bovine serum albumin were purchased from Calbiochem (Billerica, MA) and Thermo Fisher Scientific (Rockford, IL), respectively. Synthetic unlabeled peptides (with amino acid analysis) and stable-labeled (heavy peptides) were purchased from New England peptides (Boston, MA) and Thermo Fisher Scientific, respectively. Carbazeran, ripasudil, and diclofenac (internal standard) were procured from Sigma Aldrich, whereas 4-oxo carbazeran was procured from Toronto Research Chemical (ON, Canada). The list of in vitro reagents used in this study is presented in Table 1. Hepatocyte-thawing media (INVITROGRO HT) and incubation media (INVITROGRO KHB) were gifted by BioIVT (Baltimore, MD).
In vitro reagents used for investigating AO content and activity
Quantification of AO Content and REF
We applied an optimized, targeted LC-MS/MS methodology (Ahire et al., 2021) to selectively quantify AO in rAO systems, HLC, and hepatocyte homogenate (Table 1). The cryopreserved adult hepatocytes (n = 8) were thawed using the manufacturer protocol, and the cells were counted and diluted using INVITROGRO KHB media. One million hepatocytes were mixed with 300 µL of solubilization buffer and incubated for 60 minutes at 300 rpm (4°C). rAOP2 (Table 1) was used as calibrators, and the stable-labeled (heavy) peptides served as internal standards. Briefly, total protein concentration of HLC and rAO systems was quantified using a BCA assay kit, and ∼80 µg protein was digested as described previously (Ahire et al., 2021). The calibration curve was constructed, ranging from 1.95 to 125 fmol/µL of AO protein, by serially diluting rAOP2 in ABC buffer (100 mM) containing 1 mg/mL HSA. The calibration curve samples (rAOP2), other rAO systems, HLC, and hepatocytes were digested using a previously optimized protocol (Ahire et al., 2021). Eighty microliters of each of the calibration standards, rAO systems, HLC (1 mg/mL total protein), or hepatocyte homogenate was mixed with ABC and bovine serum albumin, and proteins were denatured and reduced with dithiothreitol. The sample was cooled to room temperature for 10 minutes, and the denatured proteins were alkylated with iodoacetamide. Ice-cold acetone was added to precipitate proteins, followed by vortex mixing and centrifugation. The protein pellet was dried and washed with ice-cold methanol, followed by centrifugation. The pellet was dried and resuspended in ABC buffer before digestion by trypsin at 37°C for 16 hours. The reaction was quenched by the addition of the peptide internal standard, and the sample was centrifuged. The supernatant was transferred into an LC-MS vial for analysis. Two surrogate peptides of AO (LILNEVSLLGSAPGGK and MIQVVSR) were quantified using optimized LC-MS conditions (Supplemental Tables 1–3). LC-MS/MS data were acquired using an M-class Waters UPLC system coupled with Waters Xevo TQ-XS microflow mass spectrometer connected to a standard electrospray inoization (ESI) source using the parameters outlined (Supplemental Tables 1 and 2). The peptides were separated on an Acquity UPLC HSS T3 C18 column (1.8 µm, 1 mm × 100 mm) and a Vanguard precolumn (1.8 µm, 2.1 × 5 mm) using the multiple reaction monitoring (MRM) transitions specified (Supplemental Table 3). The content of AO in the rAO systems, HLC, and hepatocyte, was estimated using the standard curve of rAOP2.
Quantification of AO Activity and Relative Activity Factor
The AO-mediated metabolite formation activity of carbazeran or ripasudil was determined in rAO systems from Corning (rAOC), Xenotech (rAOX), WSU (rAOW), and HLC in 0.1 M potassium phosphate buffer (pH 7.4) and for purified recombinant AO preparations 1 (rAOP1) and 2 rAOP2 in 25 mM Tris-HCl buffer (pH 7.4), 250 mM NaCl, and 0.25 mM TCEP. The content of AO in all incubations was maintained at 2 pmol, and the total protein content was adjusted to be 174 µg using HSA. All incubations were carried out in triplicate in a water bath shaker at 37°C, with incubation volumes of 100 µL each. The final concentration of DMSO in all the incubations was 0.5% v/v, which has been reported not to affect AO activity (Behera et al., 2014). All reactions were initiated by the addition of the substrate and terminated at the end of 5 minutes by adding 200 µL of acetonitrile containing diclofenac as internal standard (33.3 nM, final concentration). Samples were centrifuged at 10,000g for 5 minutes (4°C), and the supernatants were collected and transferred to an LC vial for analysis. Calibration standards were prepared by adding stock solutions (1–2000 µM in 25% DMSO) of 4-oxo carbazeran in buffer to give final concentrations of 1–2000 nM and were processed in the same manner as the incubation samples. In the case of ripasudil, the samples were quenched and processed as described above, and prior to analysis, the supernatants were diluted fourfold using water containing 0.1% formic acid and acetonitrile containing 0.1% formic acid (95:5 v/v). The formation of the hydroxy metabolite of ripasudil was monitored, and the peak area ratio of metabolite to internal standard was determined.
AO Stability in the Presence and Absence of Substrate
We investigated the loss of AO activity in the presence and absence of carbazeran as a substrate as described below:
1) Stability in presence of substrate: In the first experiment, 4-oxo carbazeran formation was monitored in HLC and rAOP2 following the addition of carbazeran (25 µM) at different time points (5, 15, 30, and 60 minutes) using the assay protocol described above.
2) Stability in absence of substrate: In the second experiment, HLC was preincubated without adding the substrate (carbazeran) at 37°C for 0, 0.5, 1, 3, and 6 hours. Carbazeran (25 µM) was added at the end of each of the preincubation times, and the formation of 4-oxo carbazeran was monitored for 5 minutes using the assay protocol described above.
The data for the formation rates of 4-oxo carbazeran from both of these experiments were analyzed using a one-phase decay model using GraphPad prism version 8.4.3 (La Jolla, CA).
Enzyme Kinetics of AO-Mediated 4-Oxo Carbazeran Formation
The incubation time of 5 minutes was selected to avoid significant decay in protein activity, whereas the AO content of 2 pmol was used for the reactions based off the literature (Xie et al., 2019). The incubation time of 5 minutes was found to be within the linear range for metabolite formation. Therefore, HLC and rAOP2 (2 pmol) were incubated with a range of concentrations of carbazeran (0.125–128 µM) at 37°C for 5 minutes. The reaction was stopped, and the samples were processed using the carbazeran formation activity protocol described above. The processed samples were transferred to LC-MS vials for analysis.
Determination of pnAF
The reported Michaelis Menten constant (Km) of carbazeran for AO-mediated metabolism is 5 µM (Xie et al., 2019). Therefore, the formation rate of 4-oxo carbazeran was evaluated at 1, 5, and 25 µM (fivefold lower to fivefold higher than Km) of the substrate in rAO, HLC, and suspended hepatocytes. Similarly, ripasudil was incubated at 25 µM (∼5 times the reported Km) (Isobe et al., 2016) with rAO and HLC. Hepatocytes in suspension (0.1 million cells) were incubated with 25 µM carbazeran in a total incubation volume of 300 µL. The incubation was carried out in a 5% CO2 incubator at 37°C. The incubation time for the hepatocyte assay was 10 minutes as compared with HLC (5 minutes) to allow equilibration of the substrate in the cell system so that all metabolic pathways of carbazeran can be captured. Samples were quenched with acetonitrile containing internal standard (diclofenac) and processed as described earlier, and the processed samples were transferred to LC-MS vials for analysis.
LC-MS/MS Analysis of 4-Oxo Carbazeran and Hydroxy Metabolite of Ripasudil
The samples were analyzed using an LC-MS/MS system consisting of microflow LC and Xevo-TQ-XS MS systems (Waters, Milford, MA). Acquity UPLC HSS T3 C18 column (1.8 µm, 1 mm × 100 x100 mm) equipped with a guard column (Vanguard precolumn, 1.8 µm, 2.1 × 5 mm) was used for the analysis. Fifty microliters per minute flow rate of mobile phase A (water containing 0.1% formic acid) and B (acetonitrile containing 0.1% formic acid) was run using the following gradient programs: 1) carbazeran: 0–1 minutes (5% B), 1–3.5 minutes (5%–65% B) 3.5–4 minutes (65% B), 4–9 minutes (65%–95% B), 9–9.5 minutes (95%–5% B), 9.5–12 minutes (5% B), and 2) ripasudil: 0–0.5 minutes (5% B), 0.5–1.5 minutes (5%–20% B), 1.5–4.5 minutes (20%–60% B), 4.5–9 minutes (60%–95% B), 9–9.5 minutes (95%–5% B), and 9.5–12 minutes (5% B). The mass spectrometer was operated in MRM and positive ionization (ESI+) mode with a cone voltage of 27 V. The MRM transitions were 4-oxo carbazeran (m/z 377.3 → 288.1; collision energy [CE], 25 eV), hydroxy metabolite of ripasudil (m/z 340.11→ 99.0; CE, 20 eV), and diclofenac (m/z 296.0 → 214.0; CE, 25 eV).
Data Analysis
The REF for the rAO was calculated using eq. 1.

The kinetic parameters for 4-oxo carbazeran formation, Km, and Vmax (pmol/min per pmol AO) were determined by fitting the Michaelis-Menten equation (eq. 2) to the data using GraphPad Prism (ver. 8.4.3) (La Jolla, CA):

Where Vmax was calculated as:
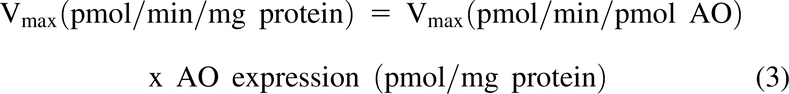 pnAF for the rAO preparations was calculated with respect to HLC and hepatocytes as:
pnAF for the rAO preparations was calculated with respect to HLC and hepatocytes as:
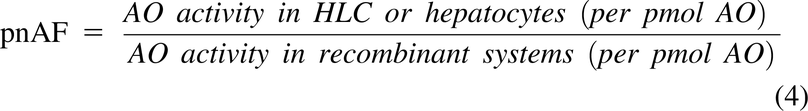
The intrinsic clearance (CLint) across all systems was calculated using eq. 5:

The CLint values obtained from eq. 5 were then integrated with their respective REF and pnAF values obtained from eqs. 1 and 4:
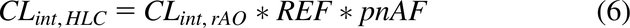
Metabolite Identification of Carbazeran in Human Hepatocytes
Carbazeran is considered to be a highly selective AO substrate, but for more accurate IVIVE analysis, a determination of fraction metabolized by AO (fm,AO) value was advisable. A comprehensive untargeted metabolite identification study was performed to identify any non–AO-mediated metabolic pathways in human hepatocytes (n = 8). The two sets of samples analyzed include control (hepatocytes without carbazeran) and treated (hepatocytes incubated with carbazeran). The samples were analyzed by a nano-flow LC coupled to high-resolution mass spectrometer (Thermo Q-Exactive-HF). The mass spectrometer was equipped with a standard easy spray ion source and reverse phase column (0.075 × 250 mm). LC conditions were set at 300 nL/min flow rate and 1 µL injection volume using mobile phase A: 0.1% formic acid in water and B: 0.1% formic acid in 80% acetonitrile. The LC and source parameters are provided (Supplemental Tables 4 and 5, respectively), and MS parameters are given (Supplemental Table 6). XCMS online platform (https://xcmsonline.scripps.edu) was used to process the MS raw data. The following screening criteria were employed to filter potential metabolites: 1) hepatocytes incubated with carbazeran with >10-fold response compared with the control, 2) a mass defect filter (milliDalton range from −50 to 50), 3) a retention time window of 15–60 minutes (considering carbazeran retention time was observed at about 34 minutes), and 4) an MS intensity of >106 in the carbazeran-treated hepatocyte samples and MS intensity of <106 in the control samples.
PBPK Model Development of Carbazeran
A PBPK model of carbazeran was developed using Simcyp software (version 21, Certara, NY) to estimate unaccounted clearance by in vitro scaling. The system-dependent parameters (e.g., organ weight, body composition, and blood flow rates) were already incorporated into the software, whereas drug-dependent parameters were added from literature (Supplemental Table 8). The simulated trial designs and virtual population were selected to match the reported clinical data (Kaye et al., 1984). The carbazeran intravenous PBPK disposition model was developed and validated using the published clinical data. A minimal PBPK model was built using the in vitro CLint obtained from the HLC experiment in this study and corrected using the unbound fraction obtained from literature (de Sousa Mendes et al., 2020).
Initially, a top-down fitting of the PBPK model was done using the reported systemic CL of 157.9 L/h and volume of distribution at steady state (Vss) value of 0.8 L/kg. By keeping the value of systemic CL constant, Vss values were optimized by visual inspection of simulated plasma concentration-time curve fitting with the literature reported pharmacokinetic (PK) profile. This generated an optimized value of 0.4 L/kg. The CLint obtained from HLC data were then scaled using a well stirred model to get the hepatic clearance.
Optimized additional CL versus adjusted additional CL: The scaled CL (from HLC) was around 66% lower (53.4 L/h) than the reported CL, even after considering the renal and UDP-glucuronosyl transferase–mediated CL, suggesting an unaccounted additional CL. A local sensitivity analysis was carried out using a range of 60–120 L/h to estimate the optimized value of the unaccounted additional CL. The effects of the additional systemic CL and Vss on the carbazeran PK profile were evaluated using a range of values with multiple virtual trials. For each rAO system, the additional CL values were further adjusted by dividing their respective CLint with that of HLC (Table 3) and termed as “adjusted additional CL.” The in vitro CLint values from HLC and the rAO systems were then integrated with the optimized value for additional CL and Vss.
Parameters (mean and 95% confidence interval in parenthesis) for curve fitting of time course of 4-oxo carbazeran formation in rAOP2 and in HLC in the presence and absence of carbazeran
Clearance calculations and IVIVE predictions for carbazeran
Contribution of AO-mediated metabolism toward total hepatic CL (CLH) for four AO substrates (O-benzylguanine, BIBX1382, zaleplon, and zoniporide) was estimated using the well stirred model by scaling the in vitro CLint data (de Sousa Mendes et al., 2020). To estimate total AO-mediated CLH for these four substrates, a ratio of the optimized additional CL to the AO-mediated hepatic clearance (CLAO,hepatic) of carbazeran was considered as shown in Supplemental Table 9. Finally, the reported fraction metabolized by AO (fmAO) values were used to estimate the total in vivo CL, which was compared with the reported data (de Sousa Mendes et al., 2020).
Results
AO Content in Recombinant Systems, Human Liver Cytosol, and Hepatocytes
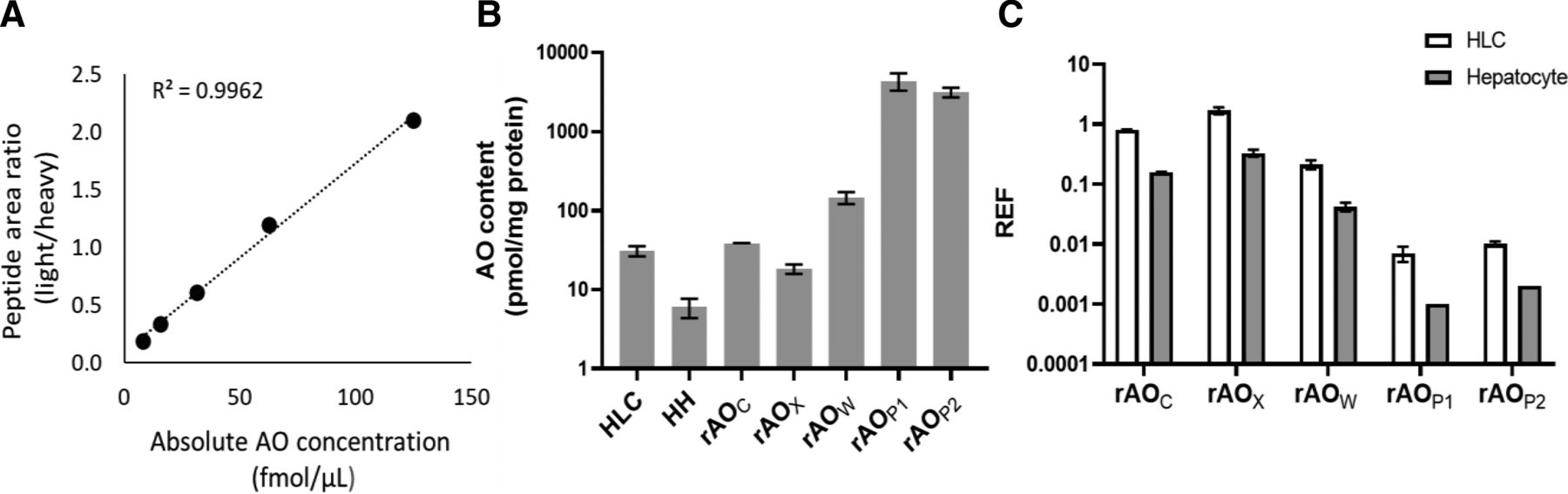
The contents of AO in recombinant systems, HLC, and hepatocytes were quantified using a calibration curve constructed using rAOP2 ranging from 1.95 to 125 fmol/µL (Fig. 1A). The mean content of AO (Fig. 1B) in rAOC (38.5 pmol/mg) and rAOX (18.4 pmol/mg) were within approximately twofold, whereas rAOW was approximately fivefold higher (146.1 ± 24.98 pmol/mg) as compared with HLC (30.9 pmol/mg protein). The contents of AO were highest in rAOP1 (4368 ± 1078 pmol/mg protein) and rAOP2 (3143 ± 438 pmol/mg). The content of AO in hepatocytes (n = 8) ranged from 4.3 to 7.6 pmol/mg (mean ± S.D.; 6.07 ± 1.67 pmol/mg hepatocyte homogenate).

(A) Calibration curve generated using purified recombinant human AO protein (rAOP2). Protein content of AO in HLC (pool of n = 50 donors), hepatocyte homogenate (n = 8), and recombinant systems from four sources (B) and REF across different recombinant systems (C). The recombinant human systems are the cytosolic extracts of AO-overexpressing HEK293 cells (rAOC) and E. coli (rAOX). The purified AO proteins were those purified from ecoAO paste (rAOW) and baculovirus expression vector systems, rAOP1 and rAOP2. The data are presented as mean and S.D. of triplicate analysis. ****P < 0.0001; one-way ANOVA. HH, hepatocyte homogenate.
Based on AO content, the REF values (Fig. 1C) for rAOC and rAOX were 0.2–1.6 with respect to HLC and hepatocytes, whereas, as expected, the REF values for rAOW, rAOP1 and rAOP2 ranged between 0.001 and 0.05.
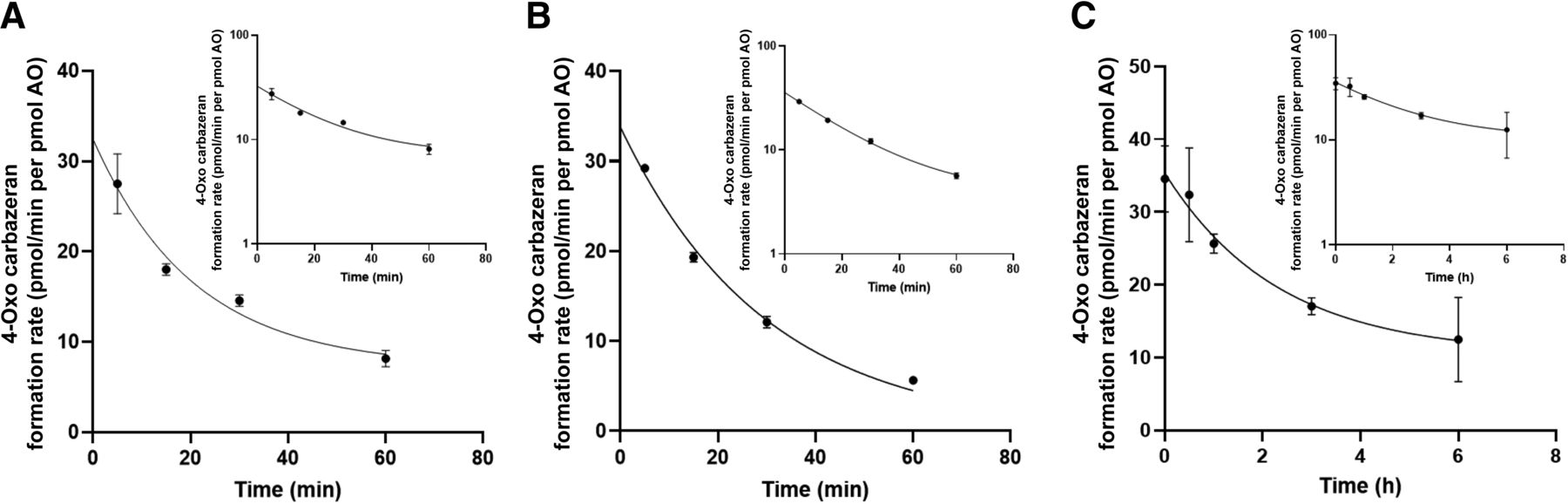
Stability of AO in the Presence and Absence of Carbazeran
The rates of formation of 4-oxo carbazeran in rAOP2 and HLC in the presence of carbazeran declined over time in a similar manner (Fig. 2, A and B) as indicated by the values of the rate constant, a constant relating the rate of the reaction to the substrate concentrations (Table 2), and the residual plots (Supplemental Fig. 1). The rate of 4-oxo carbazeran formation was highest at 5 minutes, hence all subsequent activity assays were carried out using a 5-minute incubation time.

Time-dependent formation of 4-oxo carbazeran in purified rAOP2 (A) and HLC (B). Stability of HLC in the absence of carbazeran (C).
The initial value of CLint at 0 minutes was estimated using the equation of one phase decay model, i.e., [Y= (Y0 – Plateau) *exp(-K*X) + Plateau], which performed better than constraining the plateau to 0. The stability of AO in HLC in the absence of carbazeran (Fig. 2C; Table 2) showed a 10-fold higher decay (rate constant value 0.4 versus 0.047).
Enzyme Kinetic Parameters for 4-Oxo Carbazeran Formation
A Michaelis-Menten plot was fitted to the formation rate of 4-oxo carbazeran in rAOP2 and HLC (Fig. 3, A and B). The Km value of 4-oxo carbazeran formation in rAOP2 (15.26 ± 5.9 µM) was twofold higher as compared with HLC (7.76 ± 2.8 µM); however, the Vmax and the resultant CLint values were not statistically different (Vmax = 25.41 ± 2.8 versus 22.4 ± 2 and CLint = 1.67 versus 2.88 mL/min per pmol AO, respectively).

Michaelis-Menten plot of 4-oxo carbazeran formation kinetics in rAOP2 (A) and HLC (B).
Determination of pnAF and Clearance Calculations
The AO content-normalized activity in HLC using carbazeran was approximately sixfold higher as compared with that in rAOC and rAOX, at 1, 5, and 25 µM, and approximately fourfold higher compared with the rAOW, whereas the activities in the rAOP1 and rAOP2 were within twofold of the HLC values (Fig. 4A), with rAOP2 showing the highest activity comparable to that seen in HLC. The pnAF values for the rAO ranged from 1 to 6.57 (Fig. 4B). The pnAF values were substrate independent as both carbazeran and ripasudil showed similar values (Fig. 4C).

AO activity (4-oxo carbazeran formation rate) in HLC and the recombinant human systems using three carbazeran concentrations (1, 5, and 25 µM). The data are presented as mean and S.D. of triplicate analysis. *, **, and *** indicate significant difference with P values < 0.05, < 0.01, 0.0001, and ****P < 0.0001, respectively, utilizing, one-way ANOVA (A). pnAF for the recombinant systems with respect to HLC (1, 5, and 25 µM substrate concentration) and hepatocyte homogenate (25 µM substrate concentration) (B). Comparison of pnAF values for carbazeran and ripasudil in three different recombinant systems, i.e., rAOW, rAOP1, and rAOP2 (C).
Table 3 shows the scaling of the in vitro CLint of carbazeran to CLH from HLC and rAO. The integration of respective REF and pnAF values resulted in improved scaling, with CLint values comparable to those obtained in HLC.
Metabolite Identification Studies with Carbazeran
Differences in the metabolomic profiles in hepatocytes with and without carbazeran were evaluated using XCMS software. The output Excel file was first processed to shortlist the potential metabolites using the criteria described above. The shortlisted features were compared against the theoretical metabolite list of carbazeran to identify drug metabolites. The comparison confirmed the presence of 4-oxo carbazeran (m/z 377), carbazeran glucuronide (m/z 537), O-desmethyl carbazeran (m/z 347), N-desethyl carbazeran (m/z 333), decarbamylated carbazeran (m/z 290), and decarbamylated 4-oxo carbazeran (m/z 306). These metabolites were further confirmed by their unique MS2 fragmentation patterns (Supplemental Table 7). Based on the MS intensity, 4-oxo carbazeran was found to be the major metabolite (∼84%). The direct glucuronide of carbazeran was detected to be around 12%, demethylated carbazeran was 4%, and the other three metabolites were less than 1% of the sum of all metabolites.
Estimated Additional CL of Carbazeran and Its Translation to Other AO Substrates
The estimated values for additional CL and Vss using sensitivity analysis were 91.6 L/h and 0.42 L, respectively (Supplemental Fig. 2A). The observed plasma concentration versus time profiles fell within 95% confidence intervals of the predicted profiles obtained from the in vitro CLint in HLC (Supplemental Fig. 2B) and rAO systems (Supplemental Fig. 3) integrated with the optimized additional CL. However, when the in vitro CLint from rAO systems were integrated with their respective adjusted additional CL, the observed concentration time profiles did not fall within the 95% confidence intervals of the predicted profiles in the cases of rAOC, rAOX, and rAOW (Supplemental Fig. 4). The plots in Supplemental Fig. 4 represent the simulated profiles for scaled CLint from rAO systems without integration of pnAF, whereas those in Supplemental Fig. 3 represent the simulated profiles with correction of pnAF. A linear correlation was obtained between the area under the curve (AUC) fold ratio (predicted AUC in rAO/AUC in vivo) and the pnAF values (Fig. 5).

Correlation of ratio of predicted AUC in rAO versus in vivo AUC and pnAF values.
Using the optimized value of additional CL from the carbazeran model, IVIVE predictions for the four AO substrates (O-benzyl guanine, BIBX1382, zaleplon, and zoniporide), as illustrated in Supplemental Table 9, showed an improvement from severalfold underprediction (Fig. 6A) to within twofold (Fig. 6B).

Predicted intravenous clearance (CLIV) compared with mean observed CLIV before (A) and after scaling with additional clearance from carbazeran PBPK model (B). Dotted lines indicate twofold bias across the line of unity.
Discussion
In vivo CL of compounds cleared by the P450 family of enzymes is reasonably well predicted using both human liver microsomes and hepatocytes. However, IVIVE for AO-mediated metabolism using HLC, liver S9 fractions, and hepatocytes shows an underprediction of in vivo clearances (Table 4). For example, an underprediction of 13- to 15-fold for IVIVE of AO substrates was reported (Zientek et al., 2010; Akabane et al., 2012), and a scaling factor of 6.5 was recommended to improve predictions within twofold (de Sousa Mendes et al., 2020). The reasons postulated for the underprediction include instability of AO enzyme, nonlinearity of AO-mediated reactions in vitro, and contribution of extrahepatic AO toward clearance. Moreover, rAO systems have not been used for the IVIVE of drug metabolism. Given this background, we aimed at better understanding the reasons for the observed underprediction of AO-mediated drug clearance from HLC and hepatocyte data and explore the utility of rAO systems for the IVIVE of AO metabolism. We observed five important reasons that should be considered in the IVIVE of AO metabolism, which include 1) differential AO content between rAO systems and HLC, 2) compromised stability of in vitro reagents, 3) faster decay of AO activity during incubation, 4) the role of additional AO-mediated (extrahepatic) clearance, and 5) substrate-dependent unaccounted metabolic pathways (fm,AO <1).
Compilation of literature reports of IVIVE for AO-mediated metabolism
rAO from different sources have been isolated and evaluated for determining in vitro intrinsic clearance and fraction metabolized of an NCE, though most of these preparations have shown to exhibit lower activity than HLC (Barr et al., 2013). Recently, by comparison with known AO substrates, an approximation of NCE turnover using rAO in a plate-based MS assay has been described (Cronin et al., 2021).
In the present study, up to a 100-fold difference was seen in AO content between HLC and rAO systems (i.e., REF). Carbazeran is extensively metabolized by AO to 4-oxo carbazeran, and this oxidation has been reported to be an AO-selective catalytic marker (Xie et al., 2011), and hence was used as a probe for the activity assays. The metabolite identification study of carbazeran also confirmed that 4-oxo carbazeran formation was the major metabolic pathway, consistent with reported literature (Kaye et al., 1984). The activity assays carried out using the normalized content of AO across rAO from different sources and HLC revealed that the AO protein content differences alone did not account for the observed activity differences. Since AO activity involves protein-protein (MoCo sulfurase) and protein-cofactor interactions (MoCo, Fe, and FAD), the disconnect between the activity and content across rAO systems and HLC could partly be due to variability in these constituents of the reaction affecting the overall stability of the complex. We propose that pnAF should be used for comparing the protein activity of a recombinant human enzyme system (e.g., rAO) versus human tissue-derived reagents (e.g., HLC). Therefore, both REF and pnAF should be included in scaling AO-mediated CLint from rAO systems. Collectively, these two factors for non-P450 enzymes are sometimes referred to as intersystem extrapolation factor. However, intersystem extrapolation factor cannot tease out if the difference in overall activity is due to the differences in expression or intrinsic activity (per pmol). Our results show that the pnAF values for two AO substrates, carbazeran and ripasudil, were similar. This substrate independence suggests that while evaluating AO-mediated metabolism for an NCE, laboratories can establish pnAF for AO probe substrates, which then can be applied to scaling in vitro clearances of any other AO substrate. This method is particularly important in estimating the fractional contribution of AO toward metabolism of NCEs (fm,AO) using recombinant systems and for delineating fraction metabolized by P450 (fmCYPs) versus fmAO, as shown for other enzymes, (Parvez et al., 2021), which is otherwise not possible using conventional assays with HLC, human liver microsomes, or hepatocytes.
The loss of AO activity during the processes of tissue isolation, homogenization, and storage has been frequently discussed as one of the probable reasons for poor IVIVE predictions (Zientek et al., 2010; Kozminski et al., 2021). Our study shows, for the first time, that AO enzyme degrades ∼10 times faster in the presence of the substrate (carbazeran), which could possibly be triggered by certain catalytic events and an inability of the enzyme to regenerate its active state as suggested elsewhere (Abbasi et al., 2019). During each catalytic cycle, the molybdenum in MoCo is reoxidized to regenerate fully oxidized enzyme by electron transfer, producing hydrogen peroxide and superoxide anions (Beedham, 2020). These anions are hypothesized to cause loss of AO activity (Abbasi et al., 2019). Degradation of AO activity may be substrate dependent, with slow turnover substrates showing less degradation of AO activity. The in vivo activity of AO would likely depend on the dynamic changes in substrate concentration over time and degradation of activity. These estimates are unknown, but it can be presumed that regeneration of activity is more efficient than in in vitro systems. Nevertheless, we recommend carrying out time-course experiments for each substrate of interest and back calculate the activity to obtain the initial rate of reaction at 0 minutes. Thus, the conventional substrate depletion approach to scale in vitro intrinsic clearances for IVIVE, using longer incubation time—for example, >10 minutes for carbazeran (Zientek et al., 2010; Hutzler et al., 2012; de Sousa Mendes et al., 2020)—may not be ideal for AO substrates. A modified activity model that accounts for the loss of activity with time is also recommended by others for estimating CLint for AO substrates (Abbasi et al., 2019; Kozminski et al., 2021).
Another plausible reason for the underprediction of AO-mediated metabolism was the unaccounted additional CL. AO is ubiquitously expressed in various extrahepatic tissues in humans, including the endocrine tissues, adrenal gland, reproductive tissues, adipose tissues, skin, etc. (de Sousa Mendes et al., 2020). Though the content of AO in the extrahepatic tissues may be low, the content may not necessarily correlate with the activity, and the combined contribution of these tissues toward CL of an AO substrate may be significant. However, for IVIVE predictions, the scaled in vitro CLint of carbazeran integrating data from kidney, lung, vasculature, and intestinal S9 fractions along with hepatic CLint did not show a significant role of extrahepatic tissues in in vivo clearance (Kozminski et al., 2021). This discrepancy can be explained by the fact that the stability of AO in S9 fractions may be compromised, leading to the underprediction of AO-mediated metabolism. Indeed, the stability of AO enzymes has been reported to be affected during the processes of isolation and handling (Barr et al., 2013). A substantial decline in AO activity has been observed within 24 hours of isolation of hepatocytes (Hutzler et al., 2012). Thus, the altered activity of AO in isolated hepatocytes and liver fractions may be a confounding factor in the extrapolation of in vitro data. This conclusion was also supported by another study done in PXB mice, which showed that the CLint for four AO substrates scaled from hepatocytes isolated from chimeric mice showed an underprediction of CL, whereas a good correlation was observed between human in vivo intrinsic clearance (CLint, in vivo) and that of PXB mice in vivo (Sanoh et al., 2012). These results suggest that loss of AO activity during isolation has an impact on the prediction of in vivo clearance. In a more recent report, in vivo assessments and single-species scaling for AO-specific substrates were done in chimeric PXB mice, nonhuman primates, and rats to predict steady-state clearance in humans (Miyamoto et al., 2017). The highest correlation was seen with PXB mice (r2, 0.84), followed by nonhuman primates (r2, 0.7) and rats (r2, 0.4). This report indicates that the role of extrahepatic AO in the metabolic clearance is limited, and the compromised activity of AO in in vitro systems is a likely reason for the underprediction.
The underprediction of in vivo CL could be related to substrate-dependent factors. For example, in the clinical study of carbazeran, two aglycones were detected in the urine samples following treatment with glucuronidase enzyme (Kaye et al., 1984). The structures of these aglycones were elucidated based on the m/z values and were found to be hydrolysis products of 1) a direct glucuronide of carbazeran and 2) glucuronide derived from the 4-oxo metabolite. This suggests that a direct glucuronidation is a competing metabolic pathway for carbazeran; however, the mass balance study suggests that direct glucuronidation plays a minor role in vivo (∼5% of dose). In contrast, in a study by Sharma et al. (2012), carbazeran glucuronide was detected as a major metabolite (approximately twofold more intense UV absorbance than the 4-oxo metabolite) in human hepatocytes. Our investigations in understanding the contribution of non–AO-mediated hepatic clearance of carbazeran indicated around 12% (based on MS intensity) contribution of glucuronidation pathway toward its elimination in the liver, which was added to further refine the PBPK model for carbazeran. Therefore, compound-specific factors, especially fmAO, should be estimated for accurate IVIVE. Indeed, incorporation of additional CL from the present study and consideration of the fraction metabolized value successfully predicted (within twofold) the in vivo CL of O-benzyl guanine, BIBX1382, zaleplon, and zoniporide.
In conclusion, we recommend integrating differences in AO content (REF) and activity (pnAF) for extrapolating CLint data from rAO systems to HLC. We demonstrate that accounting for the loss of AO activity and the incorporation of additional clearance would result in improved predictions of IVIVE for AO substrates. PBPK modeling of AO substrates should consider these factors for improved PK predictions.
Supplemental Material
Chromatographic conditions and instrument parameters for quantitation of AO peptides and metabolite identification study, PBPK input parameters for carbazeran, and IVIVE calculations for AO-mediated substrates using additional clearance are provided in the Supplemental Material. Residual plots for stability of AO in HLC in the presence and absence of substrate and predicted versus observed concentration time profiles for carbazeran are also provided.
Acknowledgments
The authors would like to thank Jeffery Jones and Dmitri Daydov, Department of Chemistry, Washington State University, Pullman, WA, for providing the purified AO paste and Scott Obach, Pfizer, Groton, CT, for helping in procurement of purified AO proteins used in this study.
Data Availability
The authors declare that all the data supporting the findings of this study are contained within the paper.
Authorship Contributions
Participated in research design: Subash, Khojasteh, Murray, Zientek, Jones, Smith, Prasad.
Conducted experiments: Subash, Singh, Ahire.
Contributed reagents: Heyward, Cronin.
Performed data analysis: Subash, Prasad.
Wrote or contributed to the writing of the manuscript: Subash, Singh, Ahire, Khojasteh, Murray, Zientek, Jones, Kulkarni, Smith, Heyward, Cronin, Prasad.
Footnotes
- Received April 27, 2023.
- Accepted July 7, 2023.
This work was supported by PRINCE consortium (funded by Genentech, Gilead, and Takeda) with significant intellectual contributions by the industry members.
B.P. is a cofounder of Precision Quantomics Inc. and recipient of research funding from Bristol Myers Squibb, Genentech, Gilead, Merck, Novartis, Takeda, and Generation Bio. All other authors declared no competing interests for this work.
↵1Current affiliation: Terminal Phase Consulting LLC, Colorado Springs, Colorado.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ABC
- ammonium bicarbonate
- AO
- aldehyde oxidase
- AUC
- area under the curve
- CE
- collision energy
- CLH
- hepatic CL
- CLint
- intrinsic clearance
- fmAO
- fraction metabolized by AO
- HSA
- human serum albumin
- HLC
- human liver cytosol
- IVIVE
- in vitro to in vivo extrapolation
- MoCo
- molybdenum cofactor
- NCE
- new chemical entity
- P450
- cytochrome P450
- PBPK
- physiologically based pharmacokinetic modeling
- PK
- pharmacokinetic
- pnAF
- protein-normalized activity factor
- rAO
- recombinant aldehyde oxidase
- REF
- relative expression factor
- Copyright © 2023 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}