


Abstract
Whereas many cytochrome P450 enzymes are transcriptionally suppressed by inflammatory stimuli, down-regulation of CYP2B protein by the inflammatory cytokine interleukin (IL)-1β is nitric oxide (NO)-dependent and occurs via polyubiquitination and proteasomal degradation. Here, we used iTRAQ proteomic analysis to search for other proteins that are potentially down-regulated by cellular NO in cultured rat hepatocytes, and we identified CYP3A1 as one such protein. Therefore, we examined whether CYP3A proteins, like CYP2B, undergo NO- and proteasome-dependent degradation in response to cytokine treatment of rat hepatocytes. In cultured rat hepatocytes treated with phenobarbital, IL-1β stimulation failed to down-regulate CYP3A1 mRNA within 24 h of treatment, whereas CYP3A protein was down-regulated to 40% of control within 6 h, showing the post-transcriptional down-regulation of CYP3A1 protein. The down-regulation of CYP3A after 9 h of stimulation by IL-1β was attenuated by inhibitors of NO synthase (NOS) and of the proteasome, showing NO- and proteasome-dependent down-regulation at earlier time points. However, the down-regulation of CYP3A evoked by IL-1β measured 24 h after stimulation was not affected by the inhibition of NOS or by proteasomal inhibitors, showing that CYP3A1 down-regulation at later time points is NO- and proteasome-independent. IL-6, which did not evoke NO production nor affect CYP3A1 mRNA within 24 h, produced a delayed proteasome-independent down-regulation as well. Taken together, these observations show a novel dual mode of post-transcriptional CYP3A down-regulation by cytokines: NO- and proteasome-dependent at earlier time points and NO- and proteasome-independent at later times.
Cytochromes P450 (P450s) are heme-thiolate enzymes whose carbon monoxide-bound forms have peak spectral absorbances of 450 nm. Two thirds of the human CYP gene superfamily is important in the biosynthesis or catabolism of endogenous substances, such as steroid hormones, sterols, and fatty acids. The other third is involved in metabolism of xenobiotics, and most of them are mainly expressed in liver. However, a subset of P450s is also expressed to some degree in extrahepatic tissues (Ding and Kaminsky, 2003). Cancer cells also express various P450 enzymes (Rooney et al., 2004). The enzyme activities and the expression levels of many P450s are down-regulated during inflammation and infection, causing decreased metabolic clearance of P450 substrates, elevation of plasma drug levels, and drug toxicity (Aitken et al., 2006). Proinflammatory cytokine treatments of primary human and rodent hepatocytes recapitulate many of the in vivo effects of inflammation of hepatic P450 enzymes (Aitken et al., 2006; Aitken and Morgan, 2007).
Nitric oxide (NO) is a short-lived cellular messenger with important roles in cell signaling. Inducible NO synthase [(NOS) NOS2] is induced during infection and inflammation via nuclear factor κB (NFκB) activation, and NFκB activation is dependent on proteasomal activity (Palombella et al., 1994). Bacterial endotoxin [lipopolysaccharide (LPS)] or certain proinflammatory cytokine treatments cause NOS2 induction and NO production in vivo and in cultures of various cell lines (Geller et al., 1994). We reported that CYP2B1 is down-regulated in primary cultures of rat hepatocytes by two independent mechanisms in response to LPS: NO-independent mRNA suppression at lower concentrations and NO-dependent protein suppression at higher concentrations (Ferrari et al., 2001). More recently, we discovered that NO-dependent CYP2B1 degradation proceeds via polyubiquitination and the proteasomal degradation pathway (Lee et al., 2008).
Although the transcriptional regulation of CYP genes by inflammatory factors has been relatively well characterized, post-transcriptional regulation of P450 proteins is less well understood. Therefore, the purpose of this study was to identify other hepatic proteins that undergo NO-dependent degradation in a manner similar to that which we described for CYP2B proteins (Lee et al., 2008). Using the iTRAQ-based proteomic approach (Zieske, 2006), we identified CYP3A1 as a protein whose levels were increased in hepatocytes treated with interleukin (IL)-1β plus the NOS inhibitor NG-methyl-l-arginine (NMA) compared with hepatocytes treated with IL-1β alone. Subsequent experiments revealed that CYP3A protein, like CYP2B proteins, undergoes NO- and proteasome-dependent degradation in response to IL-1β treatment in phenobarbital (PB)-treated rat hepatocytes. However, these studies also revealed a slower mechanism of post-transcriptional down-regulation of CYP3A by IL-6, or IL-1β, that is both NO- and proteasome-independent.
Materials and Methods
Materials and Reagents. IL-1β and IL-6 were purchased from R&D Systems (Minneapolis, MN). l-N6-(1-iminoethyl)lysine (l-NIL), (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (NOC-18), and N-(N-l-γ-glutamyl-S-nitroso-l-cysteinyl)-glycine (GSNO) were purchased from Cayman Chemical (Ann Arbor, MI). Nω-Nitro-l-arginine methyl ester hydrochloride (l-NAME), NMA, PB, dexamethasone (Dex; water-soluble), Williams' E medium, Krebs-Ringer buffer, collagenase IV, and other general chemicals were acquired from Sigma-Aldrich (St. Louis, MO). Insulin solution was acquired from Invitrogen (Carlsbad, CA). Antibodies to CYP2B1 were provided by Dr. James Halpert (University of California, San Diego, CA), and anti-CYP3A2 antibody was purchased from Daiichi Pure Chemicals (Tokyo, Japan). 3,5-Dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine (DDEP) was provided by Dr. M. Almira Correia (University of California, San Francisco, CA).
Rat Hepatocyte Isolation and Treatment. Rat hepatocytes were isolated by a two-step in situ collagenase perfusion procedure as described previously (Lee et al., 2008). After male Sprague-Dawley rats (230–280 g) were anesthetized by ketamine/xylazine solution, the liver was perfused with Krebs-Ringer-bicarbonate buffer and then with 0.3 mg/ml collagenase type IV for 10 min. Cells were plated on collagen plates with plating medium containing 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA), and 3 h later cells were overlaid with new medium containing Matrigel (0.23 mg/ml; BD Biosciences, San Jose, CA). Next day, the medium was changed to CYP2B1 medium (Williams' E medium containing 10 mM Hepes, pH 7.4, 10 nM insulin, 25 nM Dex, and 10 mg/ml penicillin/streptomycin). Cultures were maintained for 6 to 7 days at 37°C in 5% CO2 with regular media changes. Beginning on day 4, cells were treated for 2 days with 1 mM PB to induce CYP3A expression, and PB was present for the rest of the experiment. At the conclusion of the experiment, media were removed and reserved for NO assay by the Griess reaction (Lee et al., 2008).
Protein Extraction and Immunoblotting. Hepatocytes were harvested with a cell scraper, after which the cells were incubated on ice in phosphate-buffered saline with 1 mM EDTA for at least 20 min to remove Matrigel, and then were collected by centrifugation at 1000g for 5 min. To extract total protein, cells in lysis buffer (50 mM Tris, pH 7.5, 0.1% SDS, 0.5% Nonidet P-40, 1 mM EDTA, protease inhibitor mixture; Sigma-Aldrich) were sonicated briefly for 10 s and then centrifuged for 10 min at 11,000g. The supernatant (total cell lysate) was used for SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblotting. SDS-PAGE and Western blotting were carried out as described previously (Ferrari et al., 2001). Anti-CYP2B1 antibody (diluted 1:20,000) and anti-CYP3A2 antibody (diluted 1:10,000) were incubated overnight at 4°C, and then horseradish peroxidase-conjugated secondary antibody was incubated for 1 h at room temperature. Chemiluminescence was detected with enhanced chemiluminescence substrate (Pierce Chemical, Rockford, IL) on X-ray film. The protein loads used in the Western blot assays were chosen to be within the range that gave a linear correlation of band densities with amount of applied antigen, as determined in optimization experiments. The intensities of bands were measured by densitometry and Eastman Kodak imaging software (Carestream Health, Rochester, NY).
iTRAQ-Based Scan for NO-Regulated Proteins. Four-day-old primary rat hepatocytes cultured on 150-mm plates were treated for 2 days with 1 mM PB and then treated with 5 ng/ml IL-1β or IL-1β + NMA (300 μM) in the continued presence of PB for 9 h. Total cell lysates were prepared as described above, except that an additional wash step with phosphate-buffered saline/EDTA was included to remove any residual Matrigel from the cells. Total cell lysates were precipitated with ice-cold acetone for 2 h at –20°C, followed by centrifugation at 10,000g for 2 min. For each treatment condition, proteins from three 150-mm culture plates were pooled. After washing the precipitates, the lysates were redissolved in 20 mM potassium phosphate buffer, pH 7.4, containing 1% SDS. SDS was removed on BioGel P-6 desalting columns (Bio-Rad, Hercules, CA). Samples were analyzed by SDS-PAGE and Coomassie Blue staining to ensure that there were no gross differences between the samples before iTRAQ analysis. Two identical aliquots of each sample (IL-1 or IL-1 plus NMA) were subjected to iTRAQ analysis by the Emory Microchemical and Proteomics Facility. The samples (0.1 mg of protein/77 μl each) reconstituted in 0.1 M triethylammonium bicarbonate, pH 8.5, containing 1 mM EDTA, 0.1% SDS, and 0.1% Triton X-100 were reduced with 5 mM tris-2-carboxyethyl phosphine (60°C, 1 h) followed by oxidation with 10 mM methyl methanethiosulfonate (25°C, 10 min). The samples were then digested with 10 μg of sequencing grade trypsin (Promega, Madison, WI) for 16 h at 37°C and checked by SDS-PAGE to ensure adequate and equal digestion. The samples were dried in a SpeedVac evaporator and reconstituted in 30 μl of dissolution buffer followed by acylation with one of the four iTRAQ reagents having mass tags of 114, 115, 116, or 117 (1 h at 25°C) following the manufacturer's standard protocol (Applied Biosystems, Foster City, CA). The four iTRAQ-labeled samples were then diluted 10-fold with cation exchange loading buffer (10 mM potassium phosphate in 25% acetonitrile, pH 3.0), and the peptides were fractionated on a cation exchange column (POROS 50 HS, 50-μm particle size, 4.0 × 15 mm; Applied Biosystems) using step-wise elution with increasing concentrations of KCl in the loading buffer: 5, 10, 20, 40, 60, 120, and 350 mM KCl. Each of the seven cation exchange fractions was analyzed by nanoliquid chromatography-electrospray ionization-time of flight tandem mass spectrometry (MS/MS) using the Applied Biosystems model QSTAR-XL mass spectrometer interfaced with the Dionex (Sunnyvale, CA) model U3000 nanobore high-performance liquid chromatography equipped with an autosampler. The peptides were fractionated on a Dionex PepMap100 C18 reversed-phase column (100 Å, 3-μm particle size, 0.075 × 150 mm) with initial trapping using the matching phase Dionex trap cartridge (0.3 × 5 mm). The elution of peptides was effected using a linear gradient of acetonitrile (2–70% v/v in 140 min including 20-min equilibration) at a flow rate of 180 nl/min during the core of the run. The MS/MS spectra of the five most intense peaks with two to four charges in the MS scan were automatically acquired in information-dependent acquisition mode. MS/MS spectra were extracted and searched against rat nonredundant protein database (National Center for Biotechnology Information) using ProQuant (Applied Biosystems) and MASCOT (Matrix Science Ltd., London, UK) algorithms.
RNA Extraction and Real-Time Reverse Transcription-Polymerase Chain Reaction. Total RNA was extracted from six-well plates with TRIzol reagent (Invitrogen) according to the manufacturer's instruction. Two micrograms of total RNA was used for cDNA synthesis with the High-Capacity cDNA Archive Kit (Applied Biosystems), and real-time polymerase chain reaction (PCR) was carried out with SYBR Green PCR Master Mix (Applied Biosystems) using ABI 7300 real-time-polymerase chain reaction (RT-PCR) equipment. Glyceraldehyde-3-phosphate dehydrogenase mRNA was used as the normalization control. The primers described by Hoen et al. (2000) were used for CYP3A isoform measurements by real-time PCR; the glyceraldehyde-3-phosphate dehydrogenase primers used were TTCAACGGCACAGTCAAG and CACACCCATCACAAACAT. Analysis of real-time PCR was carried out by the ΔΔCt method (Livak and Schmittgen, 2001).
Cellular CYP3A Activity Assay. CYP3A activity was assayed by using the P450-Glo CYP3A4 assay system with Luciferin-PPXE as a substrate of CYP3A following the manufacturer's direction (Promega). According to the supplier, Luciferin-PPXE shows good selectivity for rat CYP3As over other P450s, although its specificity has not been rigorously established. For cellular assays, hepatocytes were incubated with substrate (1 ml of media containing 25 μM Luciferin-PPXE/well in six-well plates or 0.5 ml/well in 12-well plates) for 1 h, and then media were collected for luciferin assay. Equal volumes of the supernatant and Luciferin Detection Reagent (Promega) were mixed, and 20 min later luciferin was measured on a SpectraMax L luminometer (Molecular Devices, Sunnyvale, CA).
Results
Identification of CYP3A1 as an NO-Regulated Protein in Cultured Rat Hepatocytes by iTRAQ Proteomic Scan. iTRAQ is a method to analyze the differential expression or regulation of proteins in cells. Cellular proteins from two to four different samples (e.g., control and treated) are digested with, for example, trypsin and then labeled on amine groups with tags of identical mass that, on fragmentation in MS/MS, generate four different reporter ions. The samples are mixed together, and then after purification and resolution by liquid chromatography, the differentially labeled peptide fragments can be separated and identified, and the relative amounts of the peptide in the four samples are quantified by MS/MS (Zieske, 2006). Because all the peptides are labeled, the ratios of the different mass tags can usually be obtained for several peptides from the same protein, increasing accuracy of the calculations and confidence of the predictions.
To identify proteins whose levels are rapidly affected by NO in response to cytokines, primary hepatocytes were treated with IL-1β or IL-1β plus the NOS inhibitor NMA for 9 h, and total cell lysates were used for iTRAQ analysis. Proteins whose levels differ in the two groups are candidates for being regulated by cellularly generated NO. From the iTRAQ data, we identified 229 proteins at a confidence level of 95% (data not shown). We then searched for proteins whose abundance differed between the two samples. One such protein was CYP2B, which is known to be regulated by NO (Lee et al., 2008). Its relative abundance, calculated from the peptides FSLATMR and YFPGAHR (contained in both CYP2B1 and CYP2B2), was increased by a factor of 1.6 by NMA treatment (data not shown). Another protein identified in this manner was CYP3A1 (Fig. 1). Peptides ALLSPTFTSGR (CYP3A1- and CYP3A2-specific) and GSVVMIPSYALHR (CYP3A1-specific) were identified and assigned to CYP3A1. The mass chromatograms for these peptides are shown in Fig. 1, in which the samples labeled with the higher mass tags (116 and 117, corresponding to cells treated with IL-1β + NMA) were almost twice as abundant as those labeled with the lower mass tags (114 and 115, IL-1β alone). These data suggested that CYP3A1 might be down-regulated by NO.
CYP3A Expression in PB-Treated Rat Hepatocytes. Because the proteomic scan identified CYP3A1 as a protein likely to be regulated by NO, we examined in detail the CYP3A1 regulation by NO in the primary cultures. In the rat, five CYP3A isoforms have been identified (Hoen et al., 2000): CYP3A1, CYP3A2, CYP3A9, CYP3A18, and CYP3A23. However, CYP3A23 encodes active CYP3A1 protein and was suggested to be the same gene (Hoen et al., 2000). CYP3A1 and CYP3A2, the major rat liver isoforms of CYP3A, are inducible via pregnane X receptor and/or constitutive androstane receptor activation (Guengerich, 1999). In cultured primary hepatocytes without an inducer, it was difficult to detect CYP3A protein in total cell lysates by immunoblotting (Fig. 2A). Therefore, to study the down-regulation of CYP3A proteins, we treated primary hepatocytes with 1 mM PB for 2 days to induce CYP3A mRNAs and proteins. Several studies have reported CYP3A regulation by PB treatments in rat primary cultures (Brown et al., 1997; Joannard et al., 2000), but the effect of PB on all four rat isoforms of CYP3A has not been reported in these cultures. CYP3A protein expression was greatly enhanced by PB treatment (Fig. 2A). In our system, CYP3A9 was not detectable in control or PB-treated samples. CYP3A2 showed the highest -fold increase (285-fold) by PB treatment (Fig. 2B), followed by CYP3A1 (98-fold) and CYP3A18 (14-fold). Even though CYP3A2 mRNA showed the highest -fold increase, the absolute Ct values indicated that CYP3A1 mRNA was 100- and 18-fold more abundant than CYP3A2 and CYP3A18, respectively, in the PB-treated cells (Supplemental Fig. 1A). Based on the Ct values and our iTRAQ data, we suggest that the CYP3A protein we detect in the PB-treated primary cultures is CYP3A1, even allowing for possible small differences in amplification of the different CYP3A transcripts in the PCR reaction. CYP3A activity in live cultures was measured using the P450 Glo assay system with luciferin-PPXE as a CYP3A substrate. Although the specificity of this substrate for rat CYP3A proteins has not been proven, PB treatment of the cells resulted in approximately a 20-fold increase in CYP3A activity (Supplemental Fig. 1B), which is well correlated with CYP3A protein level.

CYP3A1 peptide quantification by iTRAQ labeling and mass spectrometry. Duplicate aliquots of pooled lysates from hepatocytes treated with IL-1β or IL-1β + NMA were digested with trypsin, and the peptides were labeled with iTRAQ reagent 114 and 115 (for IL-1β) or 116 and 117 (for IL-1β/NMA) as described in the text. After cation exchange column separation, fractions were analyzed by nanoliquid chromatography-electrospray ionization-time of flight MS/MS as described under Materials and Methods. The graphs show the relative intensities of the mass tags on the two identified CYP3A1 peptides, indicating the relative abundance of CYP3A1 in the different samples.
Post-Transcriptional Down-Regulation of CYP3A. Rat hepatocytes treated with PB for 2 days were stimulated with IL-1β for various times. Hepatocyte CYP3A protein levels started to decline within 6 h to 40% of control level, and continued to decline to 20% of control levels at 24 h, whereas the mRNA of CYP3A1 was not significantly affected at any time point (Fig. 3, A and B). NO2 and NO3 (NOx) species were detectable in culture medium within 6 h and achieved their maximum levels at 24 h (Supplemental Fig. 2A). The data show that CYP3A protein is down-regulated post-transcriptionally by IL-1β stimulation because protein down-regulation occurs before mRNA down-regulation.

Effect of PB treatment on CYP3A expression. Four-day-old primary rat hepatocytes were cultured with fresh medium with (PB) or without (Con) 1 mM PB for an additional 2 days. Cells were cultured with an additional media change for 4 h and then harvested for RNA and protein extraction. A, total cell lysates (20 μgof protein) from three different culture plates were separated on SDS-PAGE, and immunoblotting was carried out with anti-CYP3A2 antibody. Twenty micrograms of postmitochondrial supernatants from untreated and PB-treated (50 mg/kg, 24 h) male rats (3 rats/group) is shown for comparison. B, levels of CYP3A mRNAs in hepatocytes were measured by real-time RT-PCR, comparing control with 1 mM PB treatments.

Time course of the effects of IL-1β on hepatic CYP3A proteins in primary rat hepatocyte cultures. After 48-h treatment with 1 mM PB, hepatocytes were treated with fresh medium containing PB without (Con) or with IL-1β (10 ng/ml). Cells were harvested at indicated time points from six-well plates and 60-mm cell culture dishes for RNA or protein extraction, respectively. A, Western blots of CYP3A proteins. B, levels of CYP3A1 mRNA were assayed by RT-PCR. CYP3A protein levels were quantified from the Western blots. Error bars represent S.E.M. *, significantly different from control group mean (p < 0.05; n = 3), t test.
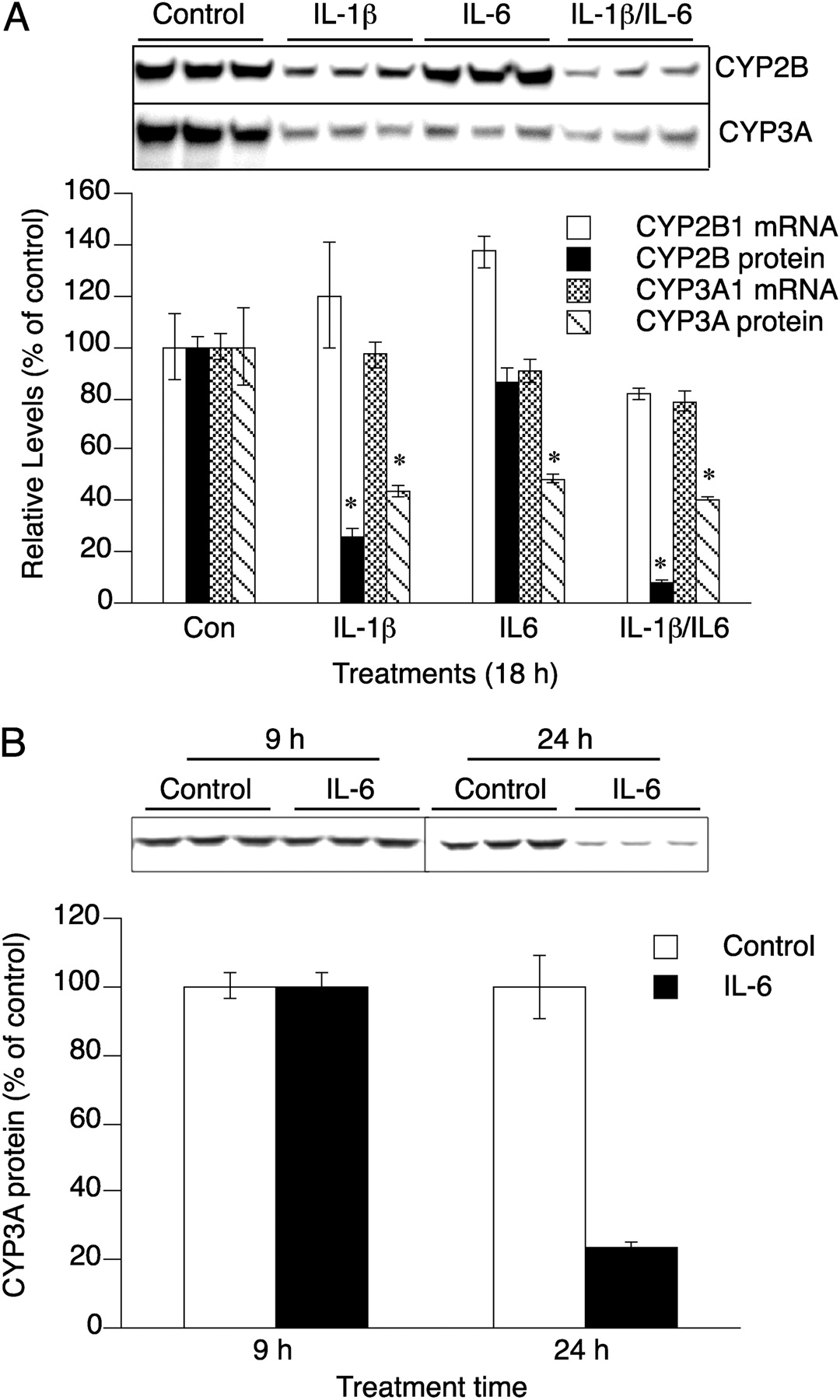
IL-6 is another proinflammatory cytokine known to regulate P450 expression, which does not induce NOS2. Treatment of hepatocytes with IL-1β, but not IL-6, stimulated NO production (Supplemental Fig. 2B). In agreement with our previous findings (Lee et al., 2008), CYP2B proteins were down-regulated only in IL-1β-stimulated cells or IL-1β- and IL-6-cotreated samples, which were producing NOx in the culture media (Fig. 4A). In sharp contrast, CYP3A protein was down-regulated by both cytokines (Fig. 4A), although their effects were not additive. CYP2B1 or CYP3A1 mRNAs were not down-regulated within 18 h of any treatment (Fig. 4A). IL-6 treatment did not down-regulate CYP3A protein within 9 h but resulted in CYP3A down-regulation to 25% of control at 24 h (Fig. 4B).

Down-regulation of hepatocyte CYP3A proteins by IL-6. After 48-h treatment with 1 mM PB, hepatocytes were treated with fresh medium containing 1 mM PB and the indicated treatments (Con, no addition; IL-1β, 5 ng/ml; IL-6, 10 ng/ml). After 18 h of treatment, cells were harvested for RNA and protein analysis. A, top, immunoblot of CYP3A and CYP2B1 from the indicated treatments. Bottom, quantification of immunoblots and steady-state mRNA levels of CYP3A1 and CYP2B1 determined by quantitative RT-PCR. B, after 48-h treatment with 1 mM PB, hepatocytes were treated with fresh medium containing 1 mM PB with IL-6 (10 ng/ml) for the indicated times. Cells were harvested and assayed by Western blotting. Error bars represent S.E.M. *, significantly different from control group mean (p < 0.05; n = 3), t test.
NO-Dependent and NO-Independent CYP3A Down-Regulation. To confirm our iTRAQ data suggesting that CYP3A1 is an NO-regulated protein, primary hepatocytes were treated with IL-1β for 9 or 24 h with or without the NOS inhibitor NMA. NMA (100 μM) treatment resulted in blockade of NO production in media (data not shown). As shown previously (Lee et al., 2008), IL-1β-stimulated CYP2B down-regulation was prevented by NMA treatment at both 9 and 24 h (Fig. 5). IL-1β stimulation caused CYP3A down-regulation to less than 30% of control at 24 h; this down-regulation was not blocked by NMA cotreatment and therefore is NO-independent (Fig. 5). However, at 9 h CYP3A was down-regulated by IL-1β to 40% of control levels, and this down-regulation was partially inhibited by NMA cotreatment (Fig. 5), showing that IL-1β-stimulated CYP3A down-regulation is at least partially NO-dependent at earlier time points. Other NOS inhibitors, l-NIL and l-NAME (100 μM each), each effectively blocked NO production and also blocked the down-regulation of CYP3A protein caused by 9 h of IL-1β stimulation (Supplemental Fig. 3). Furthermore, treatment with the chemical NO donors GSNO and NOC-18 mimicked the cellular effect of NO, resulting in 50% down-regulation of CYP3A protein within 8 h in the primary hepatocytes (Supplemental Fig. 4). Taken together, our data indicate that IL-1β-stimulated CYP3A down-regulation is NO-dependent at earlier time points (9 h) but not at later times (24 h).
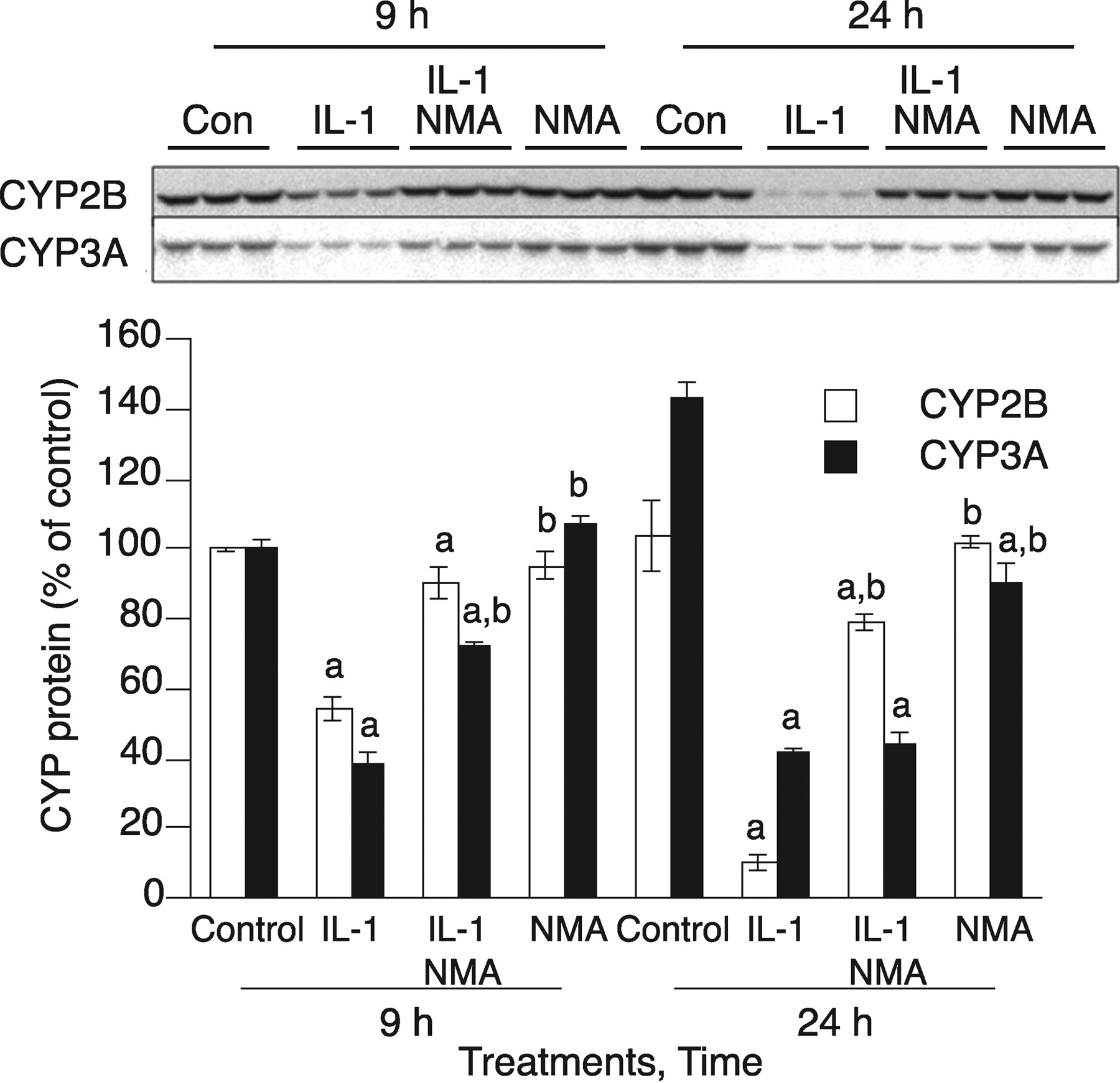
Temporal switch of the mechanism of CYP3A down-regulation by IL-1. After treating cells for 2 days with 1 mM PB, they were treated with IL-1β (5 ng/ml) with or without NMA (300 μM). P450 protein levels were measured by immunoblotting 9 or 24 h later as shown. Top, immunoblots of CYP3A and CYP2B1. Bottom, quantification of immunoblots. Error bars represent S.E.M. a, significantly different from control group mean; b, significantly different from IL-1β-treated group (p < 0.05; n = 3), one-way analysis of variance (ANOVA) and Tukey's post hoc test.
We also measured CYP3A activity in the cultures using luciferin-PPXE. IL-1β stimulation in the hepatocytes resulted in down-regulation of CYP3A activity to 35 and 22% of control within 9 and 24 h, respectively (Supplemental Fig. 5). These data are well correlated with the above data on CYP3A protein levels, suggesting that protein down-regulation is causing the reduced CYP3A activity in the hepatocytes (with the caveat that this substrate may not be specific for CYP3As in rat).
Proteasome-Dependent and -Independent Down-Regulation of CYP3A. We previously showed that NO-dependent CYP2B down-regulation occurs via proteasomal degradation (Lee et al., 2008). Therefore, we examined whether NO-dependent CYP3A down-regulation is proteasome-dependent. In a control experiment, we ascertained that MG132, a proteasome inhibitor, effectively inhibited CYP3A degradation caused by the suicide substrate DDEP (Supplemental Fig. 6A) (Wang et al., 1999). We also determined that that MG132 effectively blocked the proteasome activity in our cultures using Suc-LLVY-luciferin as a proteasome substrate (Supplemental Fig. 6B).

Role of the proteasome in the NO-dependent and -independent pathways. Cells were treated with 1 mM PB for 2 days to increase CYP3A expression and then treated for various times with the indicated agents. CYP3A protein levels were measured by immunoblotting. In each panel, the graphs represent the quantification of the immunoblotting data. A, cells were treated for 9 h with IL-1β (5 ng/ml) ± MG132 (10 μM). MG132 (10 μM) was added 3 h after IL-1β. B, cells were treated with NOC-18 (500 μM), NOC-18/bortezomib [(Bort) 10 μM], or Bort (10 μM) for 12 h. Error bars represent S.E.M. a, significantly different from control group mean; b, significantly different from IL-1β group (p < 0.05; n = 3), one-way ANOVA and Tukey's post hoc test.
Next, we stimulated the hepatocytes with IL-1β to allow for NO production and added MG132 3 h later. Cells were harvested 9 h after IL-1β treatments for measurement of CYP3A protein levels. This 3-h delay in MG132 treatment is necessary because NOS2 induction requires proteasome activity for its transcriptional activation via NFκB. As shown previously (Lee et al., 2008), this design permitted NO production to a level that was 60% of that of cells treated with IL-1β alone (Fig. 6A). MG132 treatment completely blocked IL-1β-evoked down-regulation of CYP3A (Fig. 6A), suggesting that the NO-dependent CYP3A down-regulation by IL-1 stimulation is proteasome-dependent. Treatment of the hepatocytes for 12 h with the NO donor NOC-18 (500 μM) resulted in the down-regulation of CYP3A to 25% of control levels, and this was blocked by cotreatment with bortezomib (Fig. 6B), a proteasome inhibitor used to treat cancer patients with multiple myeloma. Bortezomib effectively inhibited proteasome activity in the hepatocyte cultures (Supplemental Fig. 6B). Taken together, our data show that NO-evoked CYP3A down-regulation is proteasome-dependent.
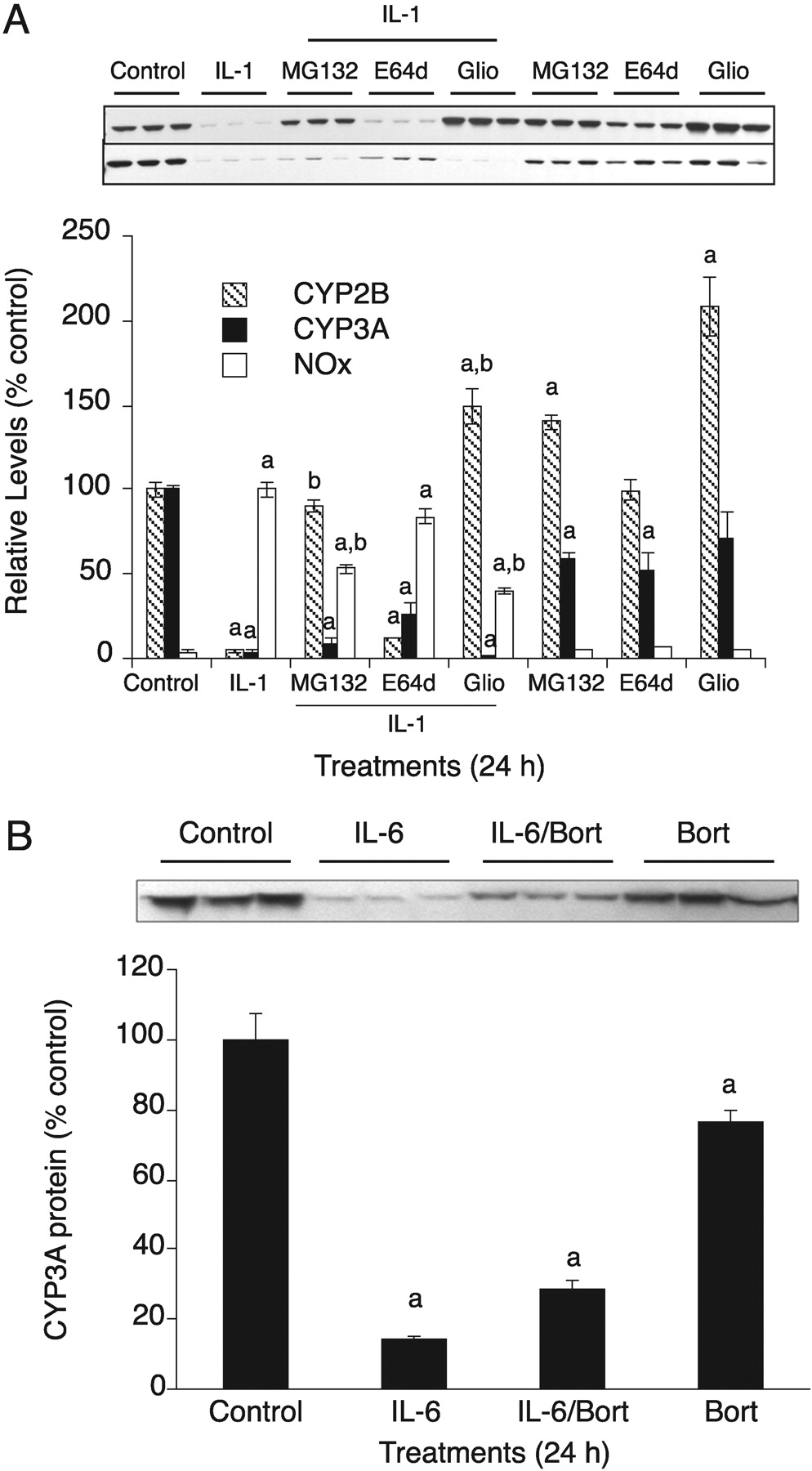
Proteasome independence of the NO-independent pathway. A, cells were treated for 24 h with indicated treatments: IL-1β (5 ng/ml), MG132 (10 μM); E64-d (10 μM); gliotoxin [(Glio) 1 μM]; MG132 or Glio was added 3 h after IL-1β to provide time for activation of NFκB in cotreated cells. CYP3A and CYP2B proteins were assayed by Western blotting. B, cells were treated with IL-6 (10 ng/ml), IL-6/bortezomib [(Bort) 10 μM], or Bort (10 μM) for 24 h. In each panel, the graphs represent the quantification of the immunoblotting data. Error bars represent S.E.M. a, significantly different from control group mean; b, significantly different from IL-1β group (p < 0.05; n = 3), one-way ANOVA and Tukey's post hoc test.
We then examined whether the NO-independent (slower) down-regulation of CYP3A by IL-1β is proteasome-dependent. PB-treated primary hepatocytes were treated for 24 h with IL-1β with or without addition (3-h delay) of the proteasome inhibitors MG132 and gliotoxin. NOx production in cells receiving the delayed proteasome inhibitor treatment was only approximately half of that with IL-1β alone (Fig. 7A), but this may not be important because in this experiment we are studying an NO-independent mechanism. IL-1β treatment resulted in the down-regulation of CYP2B and CYP3A (Fig. 7A). The down-regulation of CYP2B, which is NO-dependent even at 24 h, was completely blocked by the proteasome inhibitors MG132 and gliotoxin. However, CYP3A down-regulation was not blocked by these treatments (Fig. 7A), suggesting that NO-independent CYP3A down-regulation is also proteasome-independent. E64-d, a membrane-permeable calpain inhibitor, did not inhibit the IL-1β-evoked CYP2B down-regulation (Fig. 7A). Although E64-d alone suppressed the expression level of CYP3A protein to some extent, IL-1β-stimulated CYP3A down-regulation was partially inhibited by E64-d treatment, suggesting that calpain might be involved in the NO-independent down-regulation of CYP3A by cytokines. The NO-independent down-regulation of CYP3A proteins by IL-6 is also proteasome-independent because it was not blocked by bortezomib (10 μM) cotreatment (Fig. 7B).
Discussion
This work shows that iTRAQ proteomic analysis can be used to identify novel modes of hepatic P450 regulation. Using this technique, we identified CYP3A1 as a protein whose expression was higher in hepatocytes treated with IL-1β and a NOS inhibitor than without the inhibitor, suggesting that it might be regulated by NO. The evidence for NO-dependent down-regulation of CYP3A at earlier time points of IL-1β stimulation includes the following: 1) NOS2 inhibitors, NMA, l-NAME, and l-NIL, all block the effect; 2) IL-6, which did not produce NO within 24 h, caused much slower CYP3A down-regulation; and 3) NO donors, NOC-18 and GSNO, down-regulated CYP3A, mimicking the effect of cellular NO. NO-dependent CYP3A down-regulation is proteasome-dependent in that proteasome inhibitors block the IL-1β- or NO donor-evoked down-regulation. However, IL-6 or IL-1β at later time points act to down-regulate CYP3A by slower post-transcriptional pathways that are not blocked by NOS inhibitors. This NO-independent CYP3A down-regulation by IL-1β or IL-6 was also insensitive to proteasome inhibitors. This was not a result of a lack of efficacy of the inhibitors because CYP2B down-regulation was blocked. Thus, CYP3A protein exhibits novel dual regulation mechanisms by cytokines.
How cellular NO can trigger the degradation of CYP3As remains to be determined. Proteasome-dependent degradation stimulated by cellular NOS-derived NO or chemical NO donors has been reported for several other proteins, e.g., neutral ceramidase in rat renal mesangial cells (Franzen et al., 2002), iron regulatory protein 2 (IRP2) in RAW 264.7 cells (Kim et al., 2004), and insulin receptor substrate-1 in murine myotubes (Sugita et al., 2005). NO (or reactive nitrogen species) also can modify amino acid residues, cysteine or tyrosine, of proteins by S-nitrosylation and tyrosine nitration. Moreover, S-nitrosylation of IRP2 triggers its polyubiquitination and proteasomal degradation, and mutation of cysteine (C178S) of IRP2 prevents NO-mediated degradation (Kim et al., 2004). We showed that CYP2B1 can be S-nitrosylated by GSNO treatment in vitro and that this treatment increases polyubiquitination of the protein (Lee et al., 2008). We have confirmed that CYP3A can also be S-nitrosylated by GSNO treatment in vitro (data not shown). It is not clear yet how NO can stimulate proteasome-dependent protein down-regulation of P450, but we speculate that S-nitrosylation or tyrosine nitration of CYP3A1 and CYP2B1, like IRP2, is necessary for their recognition by specific ubiquitin E3 ligases that target them for proteasomal degradation. However, NO can bind P450 heme reversibly and irreversibly (Wink et al., 1993), and Hollenberg's laboratory showed that tyrosines of CYP2B1 and CYP2E1 and heme of CYP2B1 are modified in vitro by peroxynitrite treatment, resulting in reduced activity of CYP2B1 but not CYP2E1 (Lin et al., 2007). These modifications also deserve mechanistic interrogation for their roles in P450 degradation.
However, unlike CYP2B1, CYP3A is degraded by a second, novel proteasome- and NO-independent mechanism. Both cytokine IL-1 and IL-6 treatments showed CYP3A down-regulation that was not blocked by proteasome inhibitors. We are currently working to elucidate the common downstream mechanisms that are responsible for the down-regulation. The observation that CYP3A degradation was attenuated by a calpain inhibitor deserves further study into the role of calpain proteases. Correia and coworkers (Wang et al., 1999; Correia et al., 2005; Faouzi et al., 2007) have elegantly documented the CYP3A degradation pathway in the normal or suicide substrate-modified states, showing that both occur via ubiquitin-dependent proteasomal degradation through the endoplasmic reticulum-associated degradation pathway in primary rat hepatocytes. However, our observation of a proteasome-independent down-regulation by cytokines was not as result of a lack of efficacy of proteasome inhibition because CYP2B1 degradation was clearly inhibited in the same cultures, and we showed that rapid DDEP-mediated CYP3A down-regulation was suppressed by MG132 in our cells. Cells cotreated with IL-6 and bortezomib showed a higher level of CYP3A compared with the IL-6 alone-treated sample, but the difference was not significant (p < 0.05) statistically. This tendency might reflect the degradation of the native CYP3A through the proteasomal pathway.
Zangar et al. (2003) reported that basal CYP3A protein levels are slightly suppressed by proteasome inhibitors lactacystin (20 μM) and MG132 (200 μM) in primary cultured rat hepatocytes. Noreault-Conti et al. (2006) found that proteasome inhibitors reduced Dex-inducible CYP3A1 expression via both pretranslational and translational mechanisms. Consistent with these findings, our data also showed that CYP3A levels were slightly suppressed by 24 h of proteasome inhibitor treatments alone, such as MG132 and gliotoxin (Fig. 7A), and bortezomib (Fig. 7B). The reason why these agents affect CYP3A and not CYP2B enzymes is not known. Likewise, we do not know how NMA alone down-regulates CYP3A protein after 24 h in culture (Fig. 5). Nevertheless, no effect of NMA or the other inhibitors l-NAME and l-NIL on CYP3A protein occurred at 9 h, when these agents blocked down-regulation by IL-1β.
If CYP3A degradation is NO-dependent at early time points after IL-1β stimulation, how does it later become NO-independent? NOx measurement by the Griess reagent represents accumulation of NO2 plus NO3 in the media. We found that NOx started to be detected within 6 h and continuously accumulated up to 24 h, but the rate of accumulation was reduced after 18 h (data not shown), suggesting the NO radical production by NOS2 in the culture by IL-1β stimulation peaks at earlier time points. Thus, we hypothesize that CYP3A is degraded via the NO-dependent pathway when NO radicals are produced at higher rates (earlier time point, within 9 h), and then the degradation signal is changed to the NO-independent mode when the NO radical production is reduced, and cells produce antioxidants such as glutathione and thioredoxin (Haendeler et al., 2004) to maintain homeostasis in the cell. That both proteasomal and calpain pathways can participate in the degradation of the same protein and that degradation pathways can change under different conditions have been shown for several proteins, e.g., hypoxia-inducible factor 1α (Zhou et al., 2006), c-Fos, c-Jun, and p53 (Salvat et al., 1999).
Hepatic CYP3A enzymes are down-regulated by infection and inflammation in vivo. LPS injection caused the down-regulation of CYP3A1 mRNA by 80% in rat liver (Fang et al., 2004). We also reported that LPS injection down-regulated CYP3A2 mRNA more rapidly than CYP3A protein in rat liver (Sewer and Morgan, 1998), and these effects were not blocked by the NOS inhibitor aminoguanidine. This is in apparent opposition to the results we present here. The model we used, of primary rat hepatocytes cultured with Matrigel, is accepted as the best cell model to study P450 regulation in rat liver, and Dex-treated hepatocytes have been used to show the proteasome-dependent degradation of native and inhibitor-modified CYP3As (Faouzi et al., 2007). The reported down-regulation of CYP3A1 and CYP3A2 mRNAs in vivo could reflect direct effects of LPS and/or additive or synergistic effects of cytokines in vivo. The NO-independent down-regulation of CYP3A mRNAs in vivo could be obscuring the NO-dependent down-regulation of the protein. In any case, the lack of effect of the cytokines on CYP3A1 mRNA allowed us to study the regulation of the protein against a relatively stable mRNA background. This allowed us to validate the iTRAQ approach for identification of proteins regulated in a specific way and to uncover a novel proteasome-independent mode of CYP3A regulation as well. It is interesting to note that in the absence of cytokine stimulation, the levels of CYP3A protein tended to increase in the cultures during the 24 h after the medium change, whereas CYP3A1 mRNA declined slightly. The reason for this is unclear, but it may reflect different stabilities of the CYP3A proteins and mRNAs.
The relevance of these findings to human CYP3As is uncertain. CYP3A4 mRNA is down-regulated by proinflammatory cytokine treatments in primary human hepatocytes (Aitken and Morgan, 2007; Aitken et al., 2008), but neither its mRNA nor protein down-regulation is NO-dependent. The NO-responsiveness of human CYP3A5 and CYP3A7 remains to be determined.
In summary, we have validated iTRAQ technology as an approach to study multiple hepatocyte proteins regulated by similar pathways in hepatocytes, and thereby we showed that NO-regulated proteasomal degradation of P450 proteins is not limited to CYP2B1 but also pertains to CYP3A proteins as well. Moreover, the mechanism of cytokine-evoked degradation of rat CYP3As undergoes a temporal switch from NO- and proteasome-dependent to NO- and proteasome-independent. Our results suggest that yet more P450 enzymes, involved in metabolism of drugs or physiological substrates, may undergo NO-dependent degradation during inflammatory disease states or treatment with NO-releasing drugs, with obvious consequences for drug efficacy and/or toxicity.
Acknowledgments
We thank Dr. Almira Correia (University of California, San Francisco) for the gift of DDEP, Malik Raynor for expert technical assistance, and Dr. Keith Wilkinson (Department of Biochemistry, Emory University) for helpful discussions.
Footnotes
-
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant GM069971]; the National Institutes of Health National Institute of Environmental Health Sciences [Grant T32-ES01287]; and the National Institutes of Health National Center for Research Resources [Grants RR02878, RR12878, RR13948].
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.026187.
-
ABBREVIATIONS: P450, cytochrome P450; NO, nitric oxide; NOS, NO synthase; NFκB, nuclear factor κB; LPS, lipopolysaccharide; IL, interleukin; NMA, NG-methyl-l-arginine; PB, phenobarbital; l-NIL, l-N6-(1-iminoethyl)lysine; NOC-18, (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate; GSNO, N-(N-l-γ-glutamyl-S-nitroso-l-cysteinyl)-glycine; l-NAME, Nω-nitro-l-arginine methyl ester hydrochloride; Dex, dexamethasone; DDEP, 3,5-dicarbethoxy-2,6-dimethyl-4-ethyl-1,4-dihydropyridine; PAGE, polyacrylamide gel electrophoresis; MS/MS, tandem mass spectrometry; PCR, polymerase chain reaction; RT-PCR, reverse transcription-polymerase chain reaction; Ct, cycle threshold; NOx, nitrate plus nitrite; IRP2, iron regulatory protein 2; ANOVA, analysis of variance.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. - Received December 12, 2008.
- Accepted January 23, 2009.
- U.S. Government work not protected by U.S. copyright.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}