


Visual Overview

Abstract
Cytochrome P450 1A2 (CYP1A2) is a known tumor suppressor in hepatocellular carcinoma (HCC), but its expression is repressed in HCC and the underlying mechanism is unclear. In this study, we investigated the epigenetic mechanisms of CYP1A2 repression and potential therapeutic implications. In HCC tumor tissues, the methylation rates of CYP1A2 CpG island (CGI) and DNA methyltransferase (DNMT) 3A protein levels were significantly higher, and there was a clear negative correlation between DNMT3A and CYP1A2 protein expression. Knockdown of DNMT3A by siRNA significantly increased CYP1A2 expression in HCC cells. Additionally, treating HCC cells with decitabine (DAC) resulted in a dose-dependent upregulation of CYP1A2 expression by reducing the methylation level of CYP1A2 CGI. Furthermore, we observed a decreased enrichment of H3K27Ac in the promoter region of CYP1A2 in HCC tissues. Treatment with the trichostatin A (TSA) restored CYP1A2 expression in HCC cells by increasing H3K27Ac levels in the CYP1A2 promoter region. Importantly, combination treatment of sorafenib with DAC or TSA resulted in a leftward shift of the dose-response curve, lower IC50 values, and reduced colony numbers in HCC cells. Our findings suggest that hypermethylation of the CGI at the promoter, mediated by the high expression of DNMT3A, and hypoacetylation of H3K27 in the CYP1A2 promoter region, leads to CYP1A2 repression in HCC. Epigenetic drugs DAC and TSA increase HCC cell sensitivity to sorafenib by restoring CYP1A2 expression. Our study provides new insights into the epigenetic regulation of CYP1A2 in HCC and highlights the potential of epigenetic drugs as a therapeutic approach for HCC.
SIGNIFICANCE STATEMENT This study marks the first exploration of the epigenetic mechanisms underlying cytochrome P450 (CYP) 1A2 suppression in hepatocellular carcinoma (HCC). Our findings reveal that heightened DNA methyltransferase expression induces hypermethylation of the CpG island at the promoter, coupled with diminished H3K27Ac levels, resulting in the repression of CYP1A2 in HCC. The use of epigenetic drugs such as decitabine and trichostatin A emerges as a novel therapeutic avenue, demonstrating their potential to restore CYP1A2 expression and enhance sorafenib sensitivity in HCC cells.
Introduction
Hepatocellular carcinoma (HCC) stands as the second deadliest cancer globally (Forner et al., 2018). Surgical resection and liver transplantation are the potential curative treatments for very early stage HCC. Unfortunately, most HCC patients are in the advanced stages at the time of initial diagnosis, precluding these surgical options (Forner et al., 2018; Heimbach et al., 2018). Sorafenib is a recognized first-line drug for advanced HCC patients (Llovet et al., 2021; Li et al., 2023). However, the high frequency of sorafenib resistance is a major obstacle in advanced HCC treatment, necessitating exploration of new strategies to enhance sorafenib sensitivity.
Cytochrome P450 1A2 (CYP1A2) plays a pivotal role in drug metabolism in the liver, with its activity closely tied to the metabolism of numerous drugs used in clinical settings (Guo et al., 2021). Beyond drug metabolism, CYP1A2 significantly influences HCC development and prognosis. An independent multi-institutional cohort study involving 211 patients revealed that lower CYP1A2 expression in paracancer tissue significantly correlates with HCC recurrence, indicating its potential as an independent predictor of early HCC recurrence (Tanaka et al., 2011). A subsequently retrospective multicenter validation study confirmed the aforementioned findings in patients with HCV-related HCC, highlighting the potential of CYP1A2 as a tool for stratifying HCC patients and tailoring personalized treatment plans (Sciarra et al., 2017). Recent research indicates decreased CYP1A2 expression in HCC tumor tissues, with high CYP1A2 expression associated with a favorable prognosis. Moreover, CYP1A2 overexpression suppressed HCC cell proliferation and tumorigenicity both in vitro and in vivo by inhibiting the hepatocyte growth factor/MET signal pathway (Yu et al., 2021b). Additionally, CYP1A2 overexpression enhances the inhibitory effect on HCC cells by promoting the metabolism of 17β-estradiol to produce cytotoxic 2-methoxyestradiol (Ren et al., 2016). Collectively, these findings suggest that CYP1A2 acts as a tumor suppressor in HCC and its expression is downregulated in this cancer. However, the mechanism of CYP1A2 silencing in HCC remains unclear.
Epigenetics plays a vital role in gene expression and regulation, including the CYP family, and abnormal epigenetic alterations have been implicated in the development of HCC (Wang et al., 2020). DNA methylation and histone modification are two key epigenetic mechanisms, with the former often associated with gene silencing (Grewal, 2023). Genome-wide integrative analysis revealed that some drug metabolizing enzyme genes, including CYP1A2 are regulated by DNA methylation (Habano et al., 2015). The low expression of CYP1A2 in human embryonic stem cell-derived hepatocytes is associated with hypermethylation of the CpG site of the second exon and increased inhibitory histone modification of H3K27me3, respectively (Park et al., 2015). In addition, the mRNA expression level of CYP1A2 in normal liver correlates with the degree of methylation of two core CpG sites near the transcription start site (Ghotbi et al., 2009). However, whether epigenetic modifications underlie the downregulation of CYP1A2 expression in HCC tissues and the underlying mechanism remain unknown.
A recent study reported that increasing CYP1A2 expression through omeprazole (a CYP1A2 inducer) can sensitize sorafenib resistance cells to sorafenib (Yu et al., 2021a). Epigenetic drugs, such as the DNA methyltransferase (DNMT) inhibitor decitabine (DAC) and the histone deacetylase inhibitor trichostatin A (TSA), have been investigated for their potential to treat solid tumors, including HCC (Zhang et al., 2022). We hypothesize that if the downregulation of CYP1A2 is regulated by epigenetic modifications, then restoring its expression with epigenetic drugs could be a potential strategy to overcome sorafenib resistance. Therefore, the current study aims to investigate: (1) the epigenetic mechanism underlying CYP1A2 downregulation in HCC and (2) whether epigenetic drugs can improve the sensitivity of HCC cells to sorafenib by increasing CYP1A2 expression.
Materials and Methods
Patients and Tissue Samples
A total of 64 paired tissue samples from patients with liver cancer were collected from the Henan Cancer Hospital (Zhengzhou, China) from September 2016 to June 2018. The samples contained 48 hepatitis B virus-related primary HCCs, eight hepatitis B-related recurrent HCCs, three primary HCCs, three intrahepatic cholangiocarcinomas (ICCs), and two metastatic liver cancers. The patient information is shown in Supplemental Table 1. The protocol was approved by the ethics committee of Zhengzhou University (Approved No: ZZUIRB-2022-139), and written informed consent was obtained from each patient. The study methodologies conformed to the standards set by the Declaration of Helsinki.
Cell Culture and Reagents
Human HCC cell line HepG2 were donated by Tingting Liu (Sino-American Hormelian Cancer Research, Zhengzhou, China), and HCC-LM3 was purchased from China Center for Type Culture Collection (Wuhan, China). HepG2 and LM3 cells were maintained in Dulbecco’s modified Eagle’s medium (BI, Shanghai) at 37°C in 5% CO2, containing 10% fetal bovine serum (Clark), 100 μg/ml streptomycin, and 100 U/ml penicillin. DNA methyltransferase inhibitor (DAC) and histone deacetylase inhibitor (TSA) was purchased from MCE.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
qRT-PCR was performed according to our previous study (Zhu et al., 2022) and the primers used are listed in Supplemental Table 2. Briefy, the RNAiso Plus kit and the PrimeScript RT reagent kit (Takara Bio, Inc.) were used to extract the total RNA in HCC tissues and cells and to synthesize the cDNA, respectively. The transcript levels of the target genes were measured by an ABI 7500 Fast Real-Time PCR system. Glyceraldehyde-3-phosphate dehydrogenase was used as an internal control.
Western Blotting
Cells or tissue samples were homogenized on ice in RIPA buffer containing protease inhibitor cocktail (CW2200s, CWBIO, Beijing, China) and phosphatase inhibitors to extract the total protein. Proteins were isolated by 10% SDS-PAGE and then transferred onto the polyvinylidene difluoride membrane. After blocking in 5% skim milk for 2 h, the membrane was incubated overnight at 4°C with the primary antibody against CYP1A2 (sc-53241), DNMT3A (sc-373905), DNMT3B (sc-81252), and GADPH (sc-47724), all obtained from Santa Cruz Biotechnology. Following incubation with the secondary antibodies (Goat Anti-Mouse IgG [H+L] HRP and Goat Anti-Rabbit IgG [H+L] HRP, Proteintech, Wuhan, China SA00001-2) for 2 hours at room temperature, the membrane was visualized using an ECL detection system (FluorChem E, USA).
Immunohistochemistry
Formalin-fixed, paraffin-embedded HCC tissues were sectioned and subjected to dewaxing. Antigen retrieval was performed, followed by incubation in 5% goat serum for 1 hour. Subsequently, the sections were treated with a primary antibody against CYP1A2 (dilution 1:125) overnight at 4°C. The following day, a secondary antibody (Goat Anti-Mouse IgG [H+L] HRP, dilution 1:250) was applied at room temperature. After tris-buffered saline with Tween washing, the tissue sections were stained using 3,3′-diaminobenzidine and counterstained with hematoxylin. For the negative control, the primary antibody was replaced with phosphate-buffered saline.
Methylation-Specific PCR Amplification (MSP)
Total DNA was extracted from tissues or cells using DNA extraction kits (Biomed, DL107-01). Bisulfite conversion of genomic DNA was performed following the instructions of the EZ DNA Methylation-GoldTM Kit (ZYMO RESEARCH, D5005). Briefly, 500 ng of DNA was incubated with sodium bisulfite at 64°C for 2.5 hours, and the converted DNA was collected using a Zymo-SpinTM IC column.
It has been reported that methylation of two specific core CpG sites (+368 bp and +384 bp) in the CYP1A2 CpG island (CGI), located close to the transcription start site, is strongly associated with CYP1A2 mRNA levels in human liver samples (Ghotbi et al., 2009). We designed MSP primers containing the above two sites by MethPrimer software and the primer sequences were listed in Supplemental Table 3. For MSP amplification, 1 μl of bisulfite-modified DNA templates was used. Two separate MSP reactions were performed: one for detecting unmethylated DNA sequences and the other for detecting methylated DNA sequences. The PCR products (4 μl) were analyzed by 1% agarose gel electrophoresis. The electrophoresis bands were then analyzed using Image J software. The methylation rate was calculated using the following formula: methylation rate = (Area of methylated band grayscale)/(Area of methylated band grayscale + Area of non-methylated band grayscale) × 100%.
Small Interfering RNA Transfection
The small interfering RNAs (siRNAs) targeting DNMT3A were obtained from GenePharma (Shanghai, China) and introduced into HCC cells using Lipofectamine 3000 transfection reagent following the manufacturer's instructions. As controls, cells were transfected with non-targeting siRNA. The DNMT3A siRNA sequences are listed in Supplemental Table 4.
Chromatin Immunoprecipitation (ChIP) Assay
Tissue samples and cells were subjected to crosslinking using 1% formaldehyde for 10 minutes. The crosslinking reaction was halted by adding 0.125 M glycine for 5 minutes. Following chromatin extraction, the chromatin was sonicated to generate fragments ranging from 100 to 500 base pairs. The fragmented chromatin was then incubated with 4 μl of antibodies overnight at 4°C. Subsequently, 25 μl of Pierce ChIP grade protein A/G magnetic beads was added to the solution and incubated at 4°C for 4 hours. The immunoprecipitated DNA was eluted using 20 μl of elution buffer for subsequent real-time PCR analysis.
Based on JASPAR predictions, we identified five potential binding sites of the transcription factor aryl hydrocarbon receptor (AHR) in the CYP1A2 promoter region, located within the first 2000 bp upstream of the transcription start site, specifically at positions -233 to -228, -606 to -601, -1128 to -1123, -1552 to -1547, and -1673 to -1668. To target these sites, we designed primers ChIP1-5. Additionally, previous literature (Sogawa et al., 2004) reported AHR binding to the XRE region of the CYP1A2 promoter, specifically at positions -2532 to -2423 and -2195 to -1987. Consequently, we designed primers ChIP6 and ChIP7 based on these reported AHR binding sites in the CYP1A2 promoter region. The primer sequences used are listed in Supplemental Table 5.
ChIP-grade antibodies used are as follows: anti-H3K4me3 (Abcam, ab8580), anti-H3K9Ac (CST, 9649), and anti-H3K27Ac (CST, 8173). Normal Rabbit IgG (CST, 2729) was used as a negative control and histone H3 (CST, 4620) as a positive control.
Cell Viability Assay
For the proliferation assay, 4 × 103 cells per well were seeded in 96-well plates. The cells were divided into two groups: the sorafenib group and the DAC-sorafenib combination group. Both groups were treated with varying concentrations of sorafenib (0 μM, 1 μM, 2 μM, 4 μM, 8 μM, 16 μM, 32 μM, 64 μM) for 48 hours. In the combination group, 2.5 μM DAC was added simultaneously. The same procedure was followed for the sorafenib-TSA combinations, with the addition of 25 nM TSA. At the designated time point, 10 μl of CCK-8 reagent (Dojindo Laboratories, Japan) was added to each well, and the absorbance at 450 nm was measured using a microplate spectrophotometer (BioTek, USA). The IC50 values were calculated using GraphPad Prism 5.0 software.
Colony Formation Assay
Three thousand cells were plated in 6-well plates and cultured overnight. HepG2 cells were then treated with three concentrations of sorafenib (0 μM, 2.5 μM, 5 μM) alone, as well as with the respective sorafenib concentrations combined with 2.5 µM DAC or 25 nM TSA. The same procedure was followed for LM3 cells, except the sorafenib concentrations used were 0 μM, 5 μM, and 10 μM. After 48 hours of drug treatment, the cells were switched to regular medium and allowed to grow for 13 days. Colonies were then stained with 0.1% crystal violet.
Statistical Analysis
Paired data were assessed using the two-tailed paired parametric Student’s t test, while unpaired data underwent independent sample two-tailed Student’s t test. For comparisons involving three or more groups, one-way ANOVA with Tukey’s multiple comparisons tests was used. Spearman's rank correlation coefficient was employed for correlation analyses. GraphPad Prism 5.0 software was used for statistical analyses and graphical representation. P < 0.05 was considered to represent statistical significance.
Results
CYP1A2 is Repressed in Liver Cancer
The expression of both mRNA and protein of CYP1A2 was analyzed in 64 paired human liver tissue samples. The results showed that CYP1A2 mRNA expression levels were reduced in 90.6% (58/64) of the tumor tissues (Fig. 1A). Furthermore, weak expression of CYP1A2 was observed in tumor tissues compared with paracancer tissues through IHC staining (Fig. 1B). The liver samples were obtained from patients with different histologic types or different stages of liver cancer and were categorized into five groups (Supplemental Table 1). In hepatitis B virus-related primary HCC cases, the protein expression of CYP1A2 was lower in 91.7% (42/48) of tumor tissues (Fig. 1C), while CYP1A2 protein expression was completely downregulated in tumor tissues of all other four groups (Fig. 1, D–G). These findings suggest that CYP1A2 repression is a prevalent phenomenon across various types and stages of liver cancer.

CYP1A2 level is repressed in liver tumor tissues. (A) The expression levels of CYP1A2 mRNA in tumor tissues and adjacent paracancer tissues were evaluated by qRT-PCR. The data represent the mean ± S.D. of n = 64 samples. Paracancer-tumor difference was analyzed using a two-tailed parametric paired Student’s t test. (B) Representative images of CYP1A2 immunohistochemistry in paired paracancer and tumor tissues. Scale bars: 50 µm. C–G Representative images of the protein levels of CYP1A2 measured by western blot assay in paired paracancer (P) and respective tumor (T) tissues of various liver tumor types: hepatitis B virus-related primary HCC (C, n = 48), hepatitis B-related recurrent hepatocellular carcinoma (D, n = 8), HCC (E, n = 3), intrahepatic cholangiocarcinoma (F, n = 2), and metastatic liver cancer (G, n = 3).
DNA Hypermethylation is Involved in CYP1A2 Repression in HCC
The downregulation mechanism of CYP1A2 in HCC remains unclear, prompting us to explore the potential involvement of epigenetic regulation. One prominent epigenetic event associated with gene silencing is DNA methylation. In a study involving 48 normal human liver samples (Ghotbi et al., 2009), Roza Ghotbi et al. identified a CpG island located near the transcription start site of CYP1A2, encompassing 17 CpG sites (+134 bp → +556 bp). Remarkably, the methylation status of two specific core CpG sites (+368 bp and +384 bp) exhibited a strong correlation with CYP1A2 mRNA levels. To investigated whether DNA methylation of CGI contributes to CYP1A2 repression in HCC, we designed primers that covered a region of 14 CpG sites (174bp→386bp), including two core CpG sites to detect the DNA methylation status of CYP1A2 in HCC samples (Fig. 2A).

DNA hypermethylation is related to CYP1A2 repression in HCC. (A) Schematic of the CYP1A2 promoter region adjacent to transcription start site and the location of the CpG island (CGI). (B) Representative electrophoretic images of CYP1A2 CGI detected by MSP in paired HCC and paracancer tissues. (n = 23) U, un-methylated products; M, methylated products. (C) Methylation rates of CYP1A2 CGI in paired HCC (T) and paracancer samples (P). The data are the mean ± S.D. of n = 23 samples. (D) Upper panel: Representative images of depicting the protein levels of DNMT3A, CYP1A2, DNMT3B, and glyceraldehyde-3-phosphate dehydrogenase in paired HCC (T) and paracancer samples (P). Lower Panel: Relative protein quantification of DNMT3A, CYP1A2, and DNMT3B in paired HCC (T) and paracancer samples (P). The data represent the mean ± S.D. of n = 20 samples. The difference between paracancer and tumor samples was analyzed using a two-tailed paired parametric Student’s t test. (E) Negative correlation between CYP1A2 and DNMT3A protein levels in HCC tumor tissues. The relative protein levels of CYP1A2 and DNMT3A in 20 HCC tumor tissues (data from Fig. 2D) were analyzed using Spearman's rank correlation coefficient.
We analyzed 23 paired HCC samples with low mRNA expression of CYP1A2 and three paired HCC samples with high mRNA expression of CYP1A2 using MSP. The results showed that the methylation rates of CYP1A2 CGI in tumor tissues were significantly increased compared with paracancer tissues (Fig. 2, B and C). Specifically, the increased methylation rate of CGI was observed in 18 out of 23 (78.2%) tumor tissues with low CYP1A2 expression, while none of the tumor tissues with high CYP1A2 expression exhibited CGI hypermethylation (Fig. 2, B and C). These findings strongly suggest a potential association between CGI hypermethylation and the repression of CYP1A2 in HCC.
Additionally, we examined the expression levels of DNMTs, enzymes responsible for DNA methylation. Our investigation revealed a significant increase in the protein expression of DNMT3A in 14 out of 20 (70%) HCC tissues, whereas DNMT3B showed elevated expression in only six out of 20 (30%) HCC tissues (Fig. 2D). Importantly, a clear negative correlation between DNMT3A expression and CYP1A2 protein expression was observed (Fig. 2, D and E). Based on these results, we speculate that DNA hypermethylation, potentially mediated by the upregulation of DNMT3A expression, may play a role in the repression of CYP1A2 in HCC.
DNA Hypermethylation Mediated by DNMT3A Inhibits CYP1A2 Expression in HCC Cells
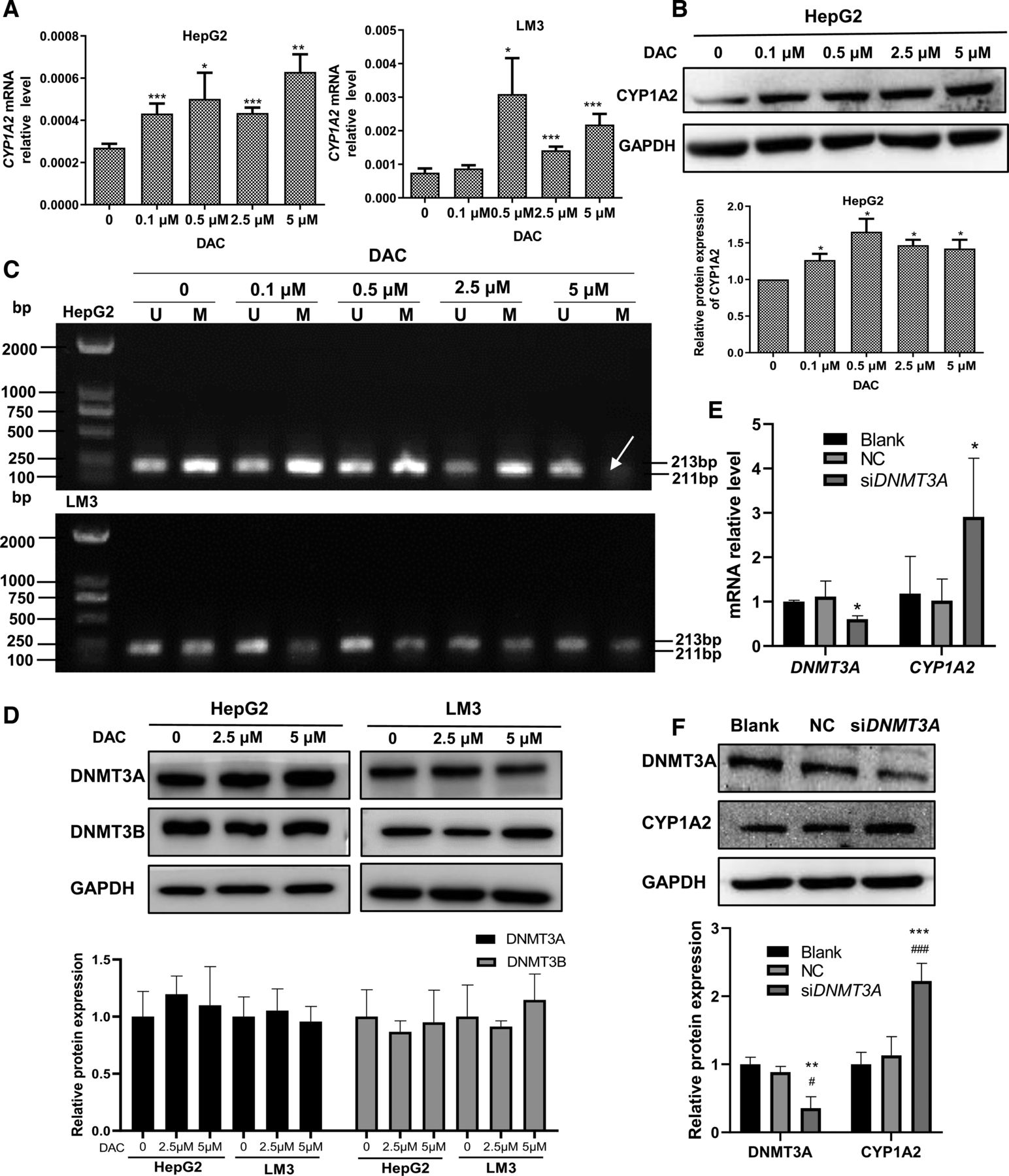
To explore whether DNA methylation directly regulates the expression of CYP1A2, we treated HCC cells with different concentrations of DAC, a DNA methyltransferase inhibitor, for 72 hours and detected changes in the expression of CYP1A2 and DNA methylation. The transcript level of CYP1A2 was significantly upregulated by different concentrations of DAC in both HepG2 and LM3 cells, whereas restored protein expression was observed only in HepG2 cells (Fig. 3A). Consistently, the methylation level of CYP1A2 CGI was significantly reduced in HepG2 cells and modestly in LM3 cells in a dose-dependent manner (Fig. 3B). With 5μmol/L DAC treatment of 72 hours, HepG2 cells showed complete demethylation (Fig. 3B), which is in line with the dramatically enhanced expression of CYP1A2 at both mRNA and protein levels.

DNA hypermethylation mediated by DNMT3A inhibits CYP1A2 expression in HCC cells. (A) CYP1A2 mRNA level in HCC cells treated with various concentrations of DAC for 72h. Each bar represents the mean ± S.D. of four independent experiments. One-way ANOVA with Tukey’s multiple comparisons test was used to analyze differences among the groups. *P < 0.05, **P < 0.01, *** P < 0.001 versus Con. (B) CYP1A2 protein level in HepG2 cells treated with various concentrations of DAC for 72h. Each bar represents the mean ± S.D. of four independent experiments. One-way ANOVA with Tukey’s multiple comparisons test was used to analyze differences among the groups. *P < 0.05, *** P < 0.001 versus Con. (C) Methylation status of CYP1A2 CGI in HCC cells treated with various concentrations of DAC for 72 h. The arrow indicates completely demethylated. (D) The effect of DAC on the protein expression of DNMT3A, DNMT3B in HCC cells. No significant difference was seen in the mean values among the 0 μM, 2.5 μM, and 5 μM groups by one-way ANOVA. (E) DNMT3A knockdown by siRNA activated CYP1A2 mRNA expression in HepG2 cells. Each bar represents the mean ± S.D. of three independent experiments. One-way ANOVA with Tukey’s multiple comparisons test was used to analyze differences among the groups. *P < 0.05 versus Blank and NC. (F) DNMT3A knockdown by siRNA activated CYP1A2 protein expression in HepG2 cells. Each bar represents the mean ± S.D. of three independent experiments. One-way ANOVA with Tukey’s multiple comparisons test was used to analyze differences among the groups. *P < 0.05, ***P < 0.01 versus Blank; #P < 0.05, ###P < 0.01 versus NC.
Next, we examined the role of DNMT3A in DNA methylation of CYP1A2 in HCC cells. DAC treatment had no effect on the protein expression level of DNMT3A in HCC cells (Fig. 3D), which is consistent with previous reports that DAC functions as an irreversible DNMT inhibitor (Zhou et al., 2018). Knockdown of DNMT3A by siRNA significantly increased the mRNA and protein expression of CYP1A2 in HepG2 cells (Fig. 3, E and F). These findings collectively indicate that DNA hypermethylation, mediated by DNMT3A, suppresses the expression of CYP1A2 in HCC.
Histone Hypoacetylation Inhibits the Expression of CYP1A2 in HCC Cells
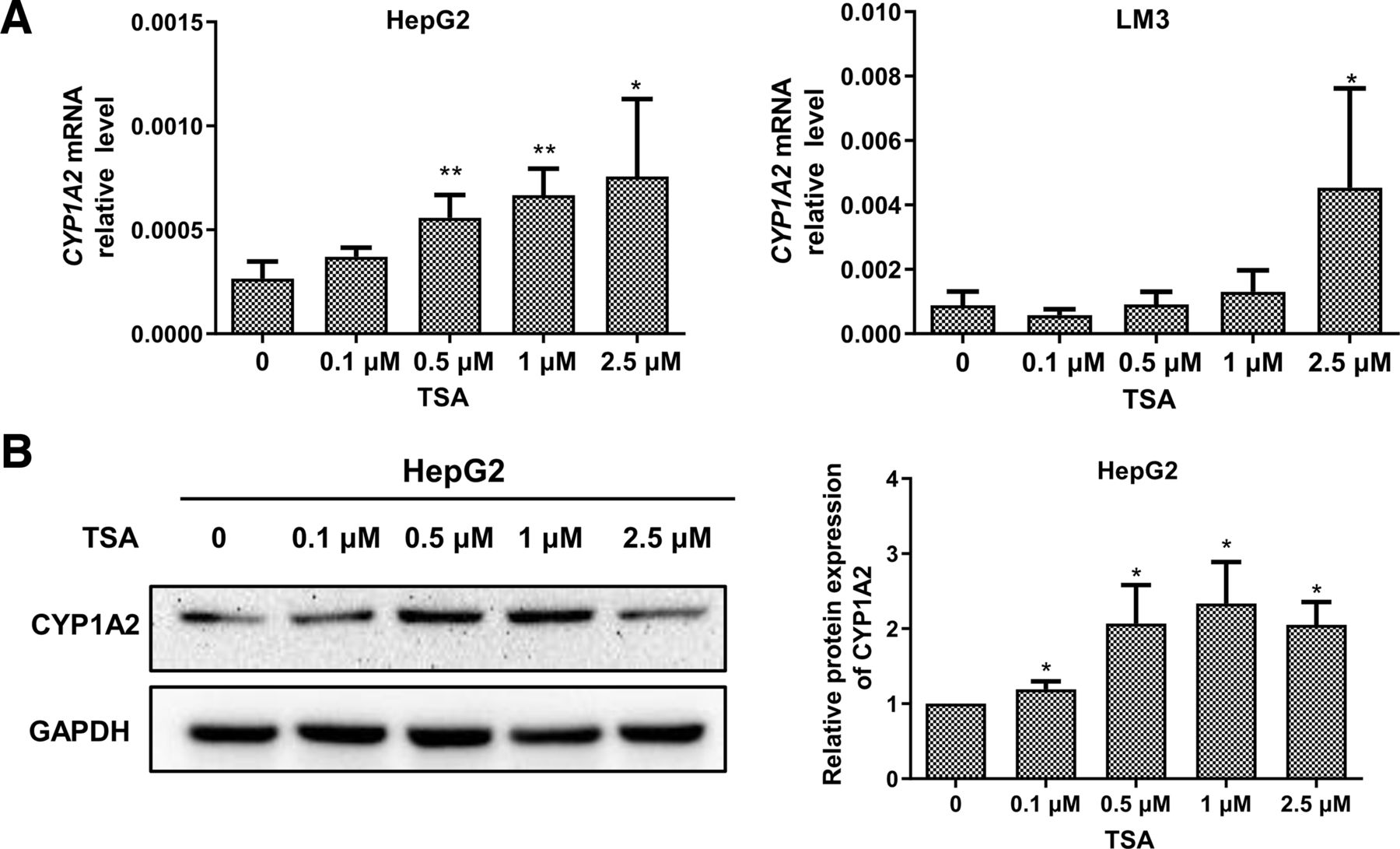
Histone acetylation also plays a crucial role in gene expression and we next examined the effect of histone acetylation on CYP1A2 expression in HCC. Following treatment with different concentrations of TSA (histone deacetylase inhibitors) for 24 hours, we observed a concentration-dependent restoration of both mRNA and protein expression of CYP1A2 in HepG2 cells (Fig. 4, A and B). In LM3 cells, only 2.5 μmol/L TSA exhibited a robust induction of CYP1A2 mRNA expression and CYP1A2 protein expression was not detected (Fig. 4A). These findings indicate that histone hypoacetylation is involved in the regulation of CYP1A2 expression in HCC cells.

TSA enhances CYP1A2 expression in HCC cells. (A) CYP1A2 mRNA level in HCC cells treatment with various concentrations of TSA for 24h. Each bar represents the mean ± S.D. of four independent experiments. One-way ANOVA with Tukey’s multiple comparisons test was used to analyze differences among the groups. *P < 0.05, **P < 0.01 versus Con. (B) CYP1A2 protein level in HepG2 cells treated with various concentrations of TSA for 24 h. Each bar represents the mean ± S.D. of three independent experiments. One-way ANOVA with Tukey’s multiple comparisons test was used to analyze differences among the groups. *P < 0.05 versus Con.
Inhibition of CYP1A2 Expression is Associated with Low H3K27Ac Levels in AHR Binding Site of CYP1A2 Promoter Region in HCC
Histone modifications, such as H3K27Ac, H3K9Ac, and H3K4me3, are known to be activating signals for gene expression and AHR is a crucial transcription factor that regulates CYP1A2 expression. To investigate the role of these histone modifications in CYP1A2 expression, we designed seven pairs of ChIP primers targeting the predicted AHR binding site in the CYP1A2 promoter region and examined the enrichment of H3K27Ac, H3K9Ac, and H3K4me3. In HepG2 cells treated with TSA, we observed a significant increase in the enrichment of H3K27Ac in the CYP1A2 promoter region compared with control cells. In contrast, the levels of H3K9Ac and H3K4me3 remained unchanged (Fig. 5A). These results indicate that the induction of CYP1A2 expression by TSA in HepG2 cells is associated with increased enrichment of H3K27Ac at the AHR binding site.

Histone acetylation of CYP1A2 promoter region in HCC. (A) ChIP-qPCR analyses of H3K27Ac/H3K9Ac/H3K4me3 at AHR binding site of CYP1A2 promoter region in HepG2 cells after treatment with 2.5 μM TSA. Each bar represents the mean ± S.D. of n = 3-5 independent experiments. Differences between DMSO and 2.5 μM TSA treatments were analyzed using a two-tailed unpaired parametric Student’s t test. *P < 0.05 versus DMSO. (B) H3K27Ac/H3K9Ac occupancy at AHR binding site of CYP1A2 promoter region in HCC samples. Each bar represents the mean ± S.D. of n = 6 samples. Paracancer-tumor differences were analyzed by a two-tailed paired parametric Student’s t test. *P < 0.05 versus P. P: paracancer tissues; T: HCC tissues.
To validate this finding, we examined the occupancy of H3K27Ac and H3K9Ac at the AHR binding site in the CYP1A2 promoter region in HCC samples. We found a decrease in H3K27Ac levels at the promoter region in HCC tissues with reduced CYP1A2 expression, while the levels of H3K9Ac remained unchanged (Fig. 5B). These findings suggest that hypoacetylation of H3K27 at the AHR binding site of CYP1A2 contributes to the inhibition of CYP1A2 expression in HCC.
Effect of TSA on Enrichment of Transcription Factor AHR in the CYP1A2 Promoter Region of HCC ells
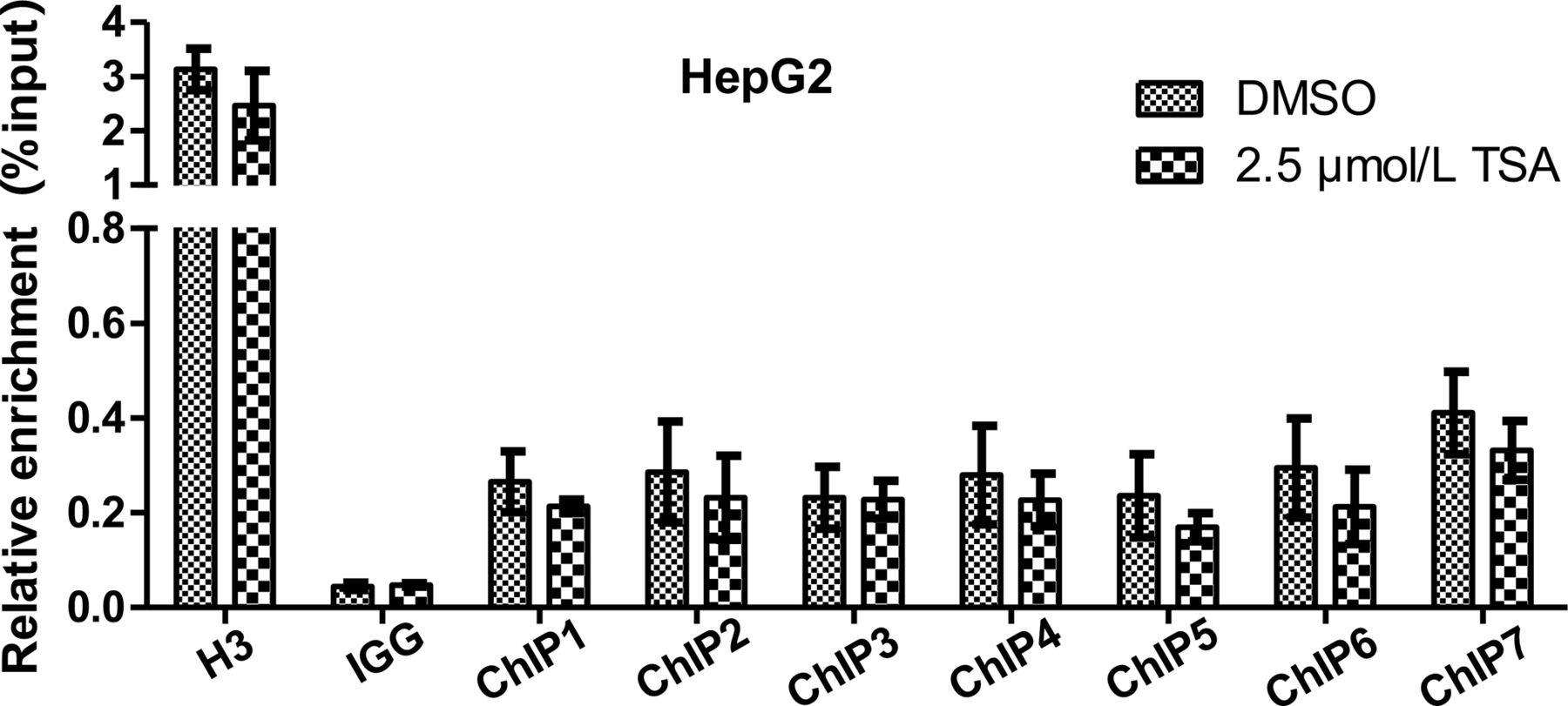
To further elucidate the involvement of AHR in CYP1A2 expression, we aimed to determine if AHR is enriched in the CYP1A2 promoter region following TSA treatment. However, our findings revealed no significant change in the enrichment of AHR across seven different binding regions of the CYP1A2 promoter region in HCC cells treated with 2.5μM TSA, compared with the control group (Fig. 6). These results suggest that AHR may not exert a significant role in the TSA-induced upregulation of CYP1A2 in HCC cells.

AHR enrichment in seven different binding sites of CYP1A2 promoter region in HepG2 cells after treatment with 2.5 μM TSA. Data are shown as mean ± S.D. of four independent experiments. No significant difference was observed in the mean values between DMSO and 2.5 μM TSA treatments by a two-tailed unpaired parametric Student’s t test.
Epigenetic Activation of CYP1A2 by DAC/TSA Sensitizes HCC Cells to Sorafenib
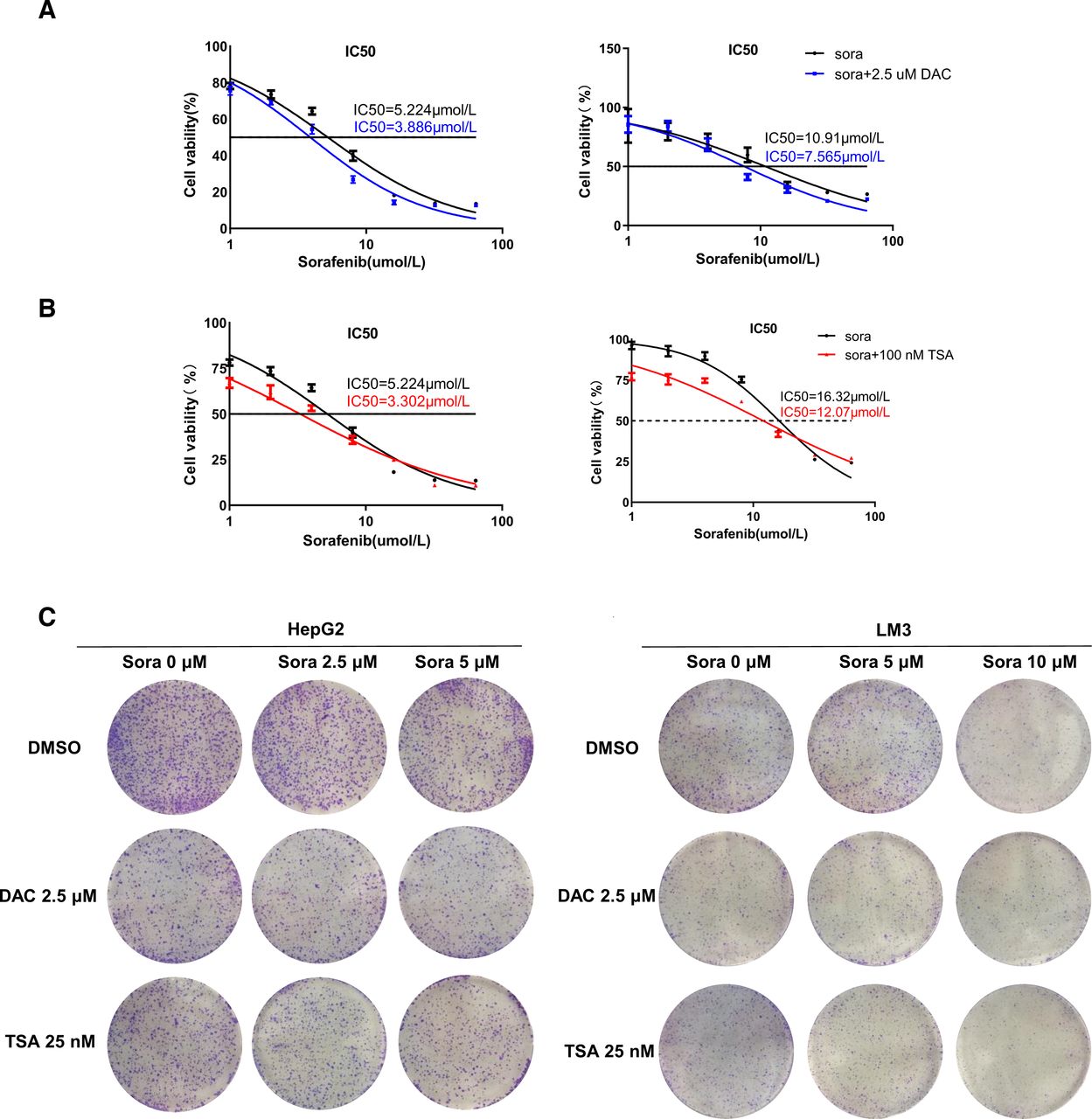
A recent study reported that omeprazole can induce CYP1A2 expression to overcome sorafenib resistance in HCC. Given that CYP1A2 expression can be restored by DNMT inhibitor DAC and histone deacetylase inhibitor TSA in HCC cells, we investigated whether the combination treatment of DAC/TSA with sorafenib could sensitize HCC cells to sorafenib. As shown in Fig. 7, A and B, the combination treatment shifted the dose-response curve to the left in HepG2 and LM3 cells, indicating an increased anti-cancer effect of sorafenib. The combination of DAC with sorafenib lowered the IC50 values for sorafenib from 5.224 μmol/L to 3.886 μmol/L in HepG2 cells and from 10.91 μmol/L to 7.565 μmol/L in LM3 cells (Fig. 7A). Similarly, the combination of sorafenib with TSA decreased the IC50 values for sorafenib from 5.224 μmol/L to 3.302 μmol/L in HepG2 cells and from 16.32 μmol/L to 12.07 μmol/L in LM3 cells (Fig. 7B). We also used a colony formation assay to evaluate cell proliferative capability. As shown in Fig. 7C, mono-therapy of sorafenib, DAC, and TSA moderately suppressed the growth of both HepG2 and LM3 cells, while co-treatment of DAC-sorafenib/TSA-sorafenib displayed a synergistic effect by significantly inhibiting the colony formation of HCC cells.

Epigenetic activation of CYP1A2 by DAC/TSA sensitizes HCC cells to sorafenib. (A) Dose-effect curve of sorafenib and DAC-sorafenib combination in HepG2 (left panel) and LM3 (right panel) cells. Cells were exposed to different concentration of sorafenib (0 μM,1 μM, 2 μM, 4 μM, 8 μM, 16 μM, 32 μM, 64 μM) with or without 2.5 μM DAC for 48 h and subsequently analyzed by CCK8 assay. (B) Dose-effect curve of sorafenib and TSA-sorafenib combination in HepG2 (left panel) and LM3 (right panel) cells. Cells are treated in the same way as (A). (C) Colony formation capability of HCC cells after sorafenib, DAC, TSA, DAC-sorafenib, or TSA-sorafenib treatment, respectively. Cells were initially treated with sorafenib, DAC, TSA, DAC-sorafenib, or TSA-sorafenib for 48 h, respectively, and then switched to regular medium and allowed to grow for 13 days until visible colonies formed.
Discussion
CYP1A2, as a key member of CYP family, is highly expressed in the liver, participating in the metabolism of various drugs and toxins (Zhang et al., 2016). Prior studies indicate downregulation of CYP1A2 in HCC, correlating with poorer prognosis (Tanaka et al., 2011; Yu et al., 2021b). Our study reveals that CYP1A2 was downregulated not only in various histologic types of liver cancer but also in different phases of HCC (primary HCC and recurrent HCC), suggesting that this is a common phenomenon in liver cancer. However, the mechanism underlying CYP1A2 silencing in HCC remains unclear.
DNA methylation involves the addition of a methyl group to cytosine residues in CpG dinucleotides, leading to gene expression silencing. In human embryonic stem cell-derived liver cells, the low expression of CYP1A2 correlates with hypermethylation of CpG sites in the second exon (Park et al., 2015). Other studies show that CYP1A2 mRNA expression in normal liver relates to methylation levels of core CpG sites near the transcription start site (Ghotbi et al., 2009). Our investigation found significantly higher methylation rates of CYP1A2 CGI in HCC tumor tissues with low CYP1A2 expression, but not in those with high expression. Additionally, treatment of HCC cells with decitabine, a DNA methyltransferase inhibitor, resulted in significant CYP1A2 upregulation and reduced methylation levels of CYP1A2 CGI in a dose-dependent manner. These findings suggest that CYP1A2 repression in HCC may correlate with CGI hypermethylation.
DNA methylation is facilitated by DNA methyltransferases. Studies indicate that aberrant DNA methylation patterns in HCC are associated with changes in DNMT expression and activity (Lai et al., 2019). Notably, HCC tissues exhibit elevated levels of DNMT1, DNMT3A, and DNMT3B compared with adjacent non-tumor tissues (Lin et al., 2001; Lai et al., 2019). This upregulation likely contributes to the hypermethylation of tumor suppressor genes, leading to their silencing and promoting HCC progression. Our investigation found significantly increased DNMT3A protein expression in the majority of HCC tissues, with DNMT3B elevated in a smaller subset. The inverse relationship between DNMT3A and CYP1A2 protein expression suggests their potential involvement in CYP1A2 regulation. Further investigation in HCC cells demonstrated that the knockdown of DNMT3A significantly increased both mRNA and protein levels of CYP1A2. These findings strongly suggest that DNA hypermethylation of the CpG island, mediated by the upregulation of DNMT3A expression, contributes to the repression of CYP1A2 in HCC.
Distinct CYP1A2 expression patterns were observed in HepG2 and HCCLM3 cell lines after DAC treatment, indicating potential pharmacological discordance. HepG2 cells, originating from a low metastatic potential hepatoblastoma in Argentina, are well-differentiated and possess intact metabolic enzymes, including the CYP family (Aden et al., 1979). Conversely, HCCLM3 cells, derived from a highly metastatic liver metastasis in a Chinese HCC patient, are poorly differentiated and lack metabolic enzyme expression (Li et al., 2003). Consequently, these differences likely underlie the varied response to DAC treatment, as shown in Fig. 3. Considering the heterogeneous responses to anticancer therapies in HCC patients, tailoring DAC dosages for specific subgroups may optimize therapeutic outcomes.
Histone acetylation is also an important epigenetic mechanism that regulates gene expression in HCC. Treatment of MCF-7 and HeLa cells with TSA led to a 2- to 3-fold increase in constitutive CYP1A2 expression (Nakajima et al., 2003). In contrast, an earlier study demonstrated that TSA initially had no effect on CYP1A2 expression in isolated hepatocytes up to 96 hours post-plating, but induced CYP1A2 at 144 hours post-plating, suggesting a time-dependent role of histone deacetylation in CYP1A2 regulation (Jin and Ryu, 2004). Our study demonstrated that TSA treatment restored CYP1A2 expression in HCC cells in a concentration-dependent manner, correlated with increased H3K27Ac enrichment in the CYP1A2 promoter region. Intriguingly, this mechanism diverges from cigarette smoke condensate, which induces CYP1A2 expression in lung cancer cells by enhancing H3K4me3 and H4K16Ac enrichment in the CYP1A2 promoter region (Xie et al., 2017). This suggests that distinct inducers regulate CYP1A2 expression through different histone modification mechanisms in various cell types. Furthermore, we confirmed that CYP1A2 repression in HCC samples was associated with a decrease in H3K27Ac at the promoter region, while H3K9Ac remained unchanged. These findings suggest that hypoacetylation of H3K27 in the promoter region of CYP1A2 inhibits its expression in HCC.
Enrichment of transcription factors is pivotal in gene expression regulation. In HepG2 cells, TSA augments CYP2E1 expression by elevating acetylated histone H3 levels in the CYP2E1 promoter region, thereby recruiting hepatic nuclear factor-1 and hepatic nuclear factor-3β to the same site (Yang et al., 2010). AHR is the primary transcription factor that regulates CYP1A2 expression (Schuran et al., 2021). Studies on AHR-deficient mice have demonstrated that AHR is essential for the inducible expression of CYP1A2 (Schmidt et al., 1996; Mimura et al., 1997). We postulate that following TSA-induced H3K27Ac elevation, AHR may enrich the CYP1A2 promoter region. However, our TSA treatment of HepG2 cells yielded no change in AHR enrichment across seven distinct AHR binding regions of the CYP1A2 promoter, as evidenced by ChIP assay, compared with the control group. Our findings align with a prior study indicating that histone acetylation regulates CYP1A2 in hepatocytes independently of AHR pathways (Jin and Ryu, 2004). Hence, we speculate that AHR may act on other sites within the CYP1A2 promoter, or alternate transcription factors may enrich the region to induce CYP1A2 expression post-TSA treatment. Nevertheless, further experimental validation is warranted to affirm this hypothesis.
Sorafenib serves as a frontline therapy for advanced HCC patients, yet its efficacy is hampered by drug resistance. In our study, we uncovered that combining sorafenib with DAC or TSA markedly bolstered its anticancer potential. This enhancement was evidenced by the leftward shift of the dose-response curve, decreased IC50 values, and diminished colony formation in both HepG2 and LM3 cells. Sorafenib resistance stems from various mechanisms, including its direct action on the electron transfer chain complex, triggering reactive oxygen species (ROS) production and subsequent HCC cell death (Xu et al., 2021). Insufficient ROS levels in tumor cells have emerged as a critical facet of sorafenib resistance. Intriguingly, augmenting ROS levels through hydrogen peroxide or phosphatase and tensin homolog overexpression has been demonstrated to facilitate sorafenib-induced apoptosis in HCC cells (Wang et al., 2021; Xu et al., 2021). CYP1A2, a prominent ROS-producing member of the CYP family (Cornelis et al., 2010; Yue et al., 2018), was also found in our study to heighten ROS production in HCC cells (unpublished data). Therefore, we speculate that epigenetic drugs DAC and TSA may increase HCC cell sensitivity to sorafenib by restoring CYP1A2 expression and boosting ROS production.
Conclusions
Our study is the first to reveal the epigenetic regulation mechanism of CYP1A2 in HCC, which is clinically meaningful as CYP1A2 dysregulation plays a key role in HCC hepatogenesis and prognosis. Our findings suggest that hypermethylation of the CpG island at the promoter, mediated by the high expression of DNMT3A, and hypoacetylation of H3K27 in the AHR binding site of the CYP1A2 promoter region, leads to CYP1A2 repression in HCC. Epigenetic drugs DAC and TSA increase HCC cell sensitivity to sorafenib by restoring CYP1A2 expression, indicating that co-treatment of DAC-sorafenib/TSA-sorafenib could be a potential strategy for treating HCC.
Data Availability
The authors declare that all the data supporting the findings of this study are available within the paper and its Supplemental Material.
Authorship Contributions
Participated in research design: Mao, Zhang HF.
Conducted experiments: Zhang Y, Feng, Fan, Qin.
Performed data analysis: Zhang Y, Feng, Mi, Mei, Jin, Zhang HF.
Wrote or contributed to the writing of the manuscript: Zhang Y, Feng, Zhang HF.
Footnotes
- Received January 23, 2024.
- Accepted March 12, 2024.
This study was supported by the Scientific and Technological Breakthroughs of Henan Province (No. 222102310173) and Zhengzhou University Professor Team Supports Special Program for Promoting Innovation-Driven Development of Enterprises (No. JSZLQY2022057). The authors sincerely appreciate the assistance provided by the Experiment Center of the School of Basic Medical Sciences, Zhengzhou University.
No author has an actual or perceived conflict of interest with the contents of this article.
1 Y.Z. and J.F. contributed equally to this work.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AHR
- aryl hydrocarbon receptor
- CGI
- CpG island
- ChIP
- chromatin immunoprecipitation
- CYP1A2
- cytochrome P450 1A2
- DAC
- decitabine
- DNMT
- DNA methyltransferase
- HCC
- hepatocellular carcinoma
- MSP
- methylation-specific PCR
- qRT-PCR
- quantitative real-time polymerase chain reaction
- ROS
- reactive oxygen species
- TSA
- trichostatin A
- Copyright © 2024 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}