


Abstract
Predicting transporter-mediated in vivo hepatic drug clearance (CL) from in vitro data (IVIVE) is important in drug development to estimate first-in-human dose and the impact of drug interactions and pharmacogenetics on hepatic drug CL. For IVIVE, one can use human hepatocytes and the traditional milligrams of protein content per gram of liver tissue (MGPGL) approach. However, this approach has been found to consistently underpredict the observed in vivo hepatic drug CL. Therefore, we hypothesized that using transporter-expressing cells and the relative expression factor (REF), determined using targeted quantitative proteomics, will accurately predict in vivo hepatic CL of drugs. We have successfully tested this hypothesis in rats with rosuvastatin, which is transported by hepatic Organic anion transporting polypeptides (OATPs). Here, we tested this hypothesis for another drug and another transporter; namely, organic cation transporter (OCT)1-mediated hepatic distributional CL of metformin. First, we estimated the in vivo metformin hepatic sinusoidal uptake CL (CLh,s,in) of metformin by reanalysis of previously published human positron emission tomography imaging data. Next, using the REF approach, we predicted the in vivo metformin CLh,s,in using OCT1-transporter–expressing HEK293 cells or plated human hepatocytes. Finally, we compared this REF-based prediction with that using the MGPGL approach. The REF approach accurately predicted the in vivo metformin hepatic CLh,s,in, whereas the MGPGL approach considerably underpredicted the in vivo metformin CLh,s,in. Based on these and previously published data, the REF approach appears to be superior to the MGPGL approach for a diverse set of drugs transported by different transporters.
SIGNIFICANCE STATEMENT This study is the first to use organic cation transporter 1-expressing cells and plated hepatocytes to compare proteomics-informed REF approach with the traditional MGPGL approach to predict hepatic uptake CL of metformin in humans. The proteomics-informed REF approach, which corrected for plasma membrane abundance, accurately predicted the positron emission tomography–imaged metformin hepatic uptake CL, whereas the MGPGL approach consistently underpredicted this CL.
Introduction
Predicting transporter-mediated in vivo clearance of drugs from in vitro data (IVIVE) is important in drug development to estimate first-in-human dose and interindividual variability in drug clearance (CL) due to drug interactions and pharmacogenetics. For IVIVE of hepatic metabolic CL of a drug, the relative activity factor (RAF) (with or without a probe drug) is traditionally used. When used without a probe drug, this approach scales the in vitro intrinsic metabolic CL of the drug to that in vivo using hepatocellularity or milligrams of protein microsomal protein yield per gram of liver tissue (Obach et al., 1997; Andersson et al., 2001). When used with a probe drug, the probe drug must selectively measure the intrinsic CL (CLint) of drug by a single enzyme. An alternative approach, the relative expression factor (Rostami-Hodjegan and Tucker, 2007; Soars et al., 2007; Rowland et al., 2011), is used where in vitro metabolic CLint of the drug (e.g., in recombinant enzymes) is scaled to that in vivo using the ratio of the abundance of the enzyme in vivo (e.g., hepatic tissue) to that in vitro (e.g., in recombinant enzymes).
Although using RAF for IVIVE of metabolic CL has been successful (Rostami-Hodjegan and Tucker, 2007; Soars et al., 2007; Rowland et al., 2011), predicting transport-mediated CL using this approach results in underestimation of the observed in vivo CL of the drug (Jones et al., 2012). In addition, the magnitude of this misprediction is not uniform across drugs (Jones et al., 2012). Therefore, generalization across drugs cannot be made through the use of a “common” empirical scaling factor. Moreover, because the RAF requires selective probe substrate and primary human cells (e.g., hepatocytes), this approach cannot be used where selective transporter probe substrates are not available (e.g., OATP substrates) or when primary cells from the organ of interest are not available or validated (e.g., blood-brain endothelial cells/kidney epithelial cells). Therefore, we have hypothesized that the REF approach, coupled with quantitative targeted proteomics, can overcome many of these problems and be used for accurate IVIVE of transporter-mediated CL of drugs. However, like any other approach, the accuracy of REF-based predictions needs to be verified. Indeed, we along with others have shown that this approach can be used to successfully predict in vivo transporter-mediated CL (Bosgra et al., 2014; Vildhede et al., 2016; Ishida et al., 2018; Kumar et al., 2018) and hepatic concentrations of rosuvastatin (an OATP substrate) obtained through positron emission tomography (PET) imaging (Bosgra et al., 2014; Vildhede et al., 2016; Ishida et al., 2018; Kumar et al., 2018). However, this success needs to be tested with other drugs transported by other transporters, such as metformin, which is transported into the liver by OCT1 and possibly by OCT3 and THTR2 (Liang et al., 2015; Pakkir Maideen et al., 2017). Therefore, we first estimated the in vivo hepatic sinusoidal uptake CL (CLh,s,in) of [11C]metformin by reanalysis of previously published PET imaging data (Gormsen et al., 2016). This reanalysis was conducted to correct for hepatic blood [11C]metformin content not conducted in that publication. Next, using the REF approach, we predicted the in vivo metformin CLh,s,in using transporter-expressing cells and plated human hepatocytes (PHH). These predictions were compared with that obtained using the milligrams of protein content per gram of liver tissue (MGPGL) approach (i.e., RAF without probe drug). Our prediction of the in vivo metformin CLh,s,in was considered a success if it fell within 2-fold of the observed value.
Materials and Methods
Chemicals and Reagents.
Specific synthetic peptides and corresponding stable labeled peptides for OCT1, Na+/K+ ATPase, and calreticulin were purchased form New England Peptides (Boston, MA). Bicinchoninic acid assay kit, dithiothreitol, iodoacetamide, mass spectrometry–grade trypsin, pierce cell-surface protein isolation kit, High-performance liquid chromatography-grade acetonitrile, Dulbecco’s phosphate-buffered saline, and Dulbecco’s modified Eagle’s medium high-glucose medium were purchased form Thermo Fisher Scientific (Rockford, IL). Metformin (hydrochloride salt), thiamine (hydrochloride salt), quinidine, potassium chloride, magnesium sulfate, d-glucose, HEPES, sodium chloride, sodium bicarbonate, calcium chloride dipotassium phosphate, and formic acid were purchased from Sigma-Aldrich (St. Louis, MO). [14C]Metformin hydrochloride (114 mCi/mmol) was obtained from Moravek Biochemicals, Inc. (Brea, CA). Collagen-coated 24-well plates were purchased from Corning (Kennebunk, ME). Human hepatocyte thaw medium, INVITROGRO HI medium, INVITROGRO CP medium, and TORPEDO antibiotic mix, as well as cryopreserved human hepatocytes (PHH), were generously provided by BioIVT (Westbury, NY). OCT1-HEK293 cells were generously provided by SOLVO Biotechnology (Hungary).
Determination of Metformin Uptake CLint into OCT1-HEK293 Cells and Human Hepatocytes (PHH).
OCT1-HEK293 cells were grown in T-75 cm2 flasks with 20 ml of high-glucose Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 IU/ml penicillin, and 100 mg/ml streptomycin. Medium was changed daily, and cells were kept in a humidified incubator with 5% carbon dioxide at 37°C. For uptake assays, 0.5 million HEK293 cells per well were plated in a 24-well plate for 24 hours. For uptake assays using PHH, cryopreserved hepatocytes were thawed and plated 0.35 million per well in a 24-well collagen-coated plate with 0.5 ml/well TORPEDO containing INVITROGRO CP medium (1:9 v/v) for 5 hours.
To determine [14C]metformin uptake, HEK293 cells or PHH were washed three times with HEPES-buffered Krebs-Ringer bicarbonate buffer (HK buffer) (4.8 mM KCl, 1.2 mM K2HPO4, 1.2 mM MgSO4, 5.6 mM glucose, 25 mM HEPES, 12 mM CaCl2, 125 mM NaCl, 25 mM NaHCO3) and incubated at 37°C. To initiate uptake, 500 μl of HK buffer containing 5 or 20 μM [14C]metformin (for HEK293 cells or PHH, respectively) with or without inhibitor (preincubated at 37°C) was added to the cells. After an appropriate time (over a period in which uptake was linear; 5 minutes for HEK293 cells and 15 minutes for PHH), [14C]metformin uptake into the cells was terminated by removing [14C]metformin-containing solution and immediately washing it three times with ice-cold HK buffer. Then, cells were lysed by adding 1 ml of 2% SDS solution to each well and incubating them on a shaker at 125 rpm for 1 hour at 37°C. After 1 hour, 700 μl of lysed solution from each well was used to measure radioactivity by Tri-Carb Liquid Scintillation Counters (PerkinElmer, Waltham, MA). In addition, 50 μl of lysed solution was used to measure protein content using the bicinchoninic acid assay method. [14C]Metformin uptake CL into the cells was calculated by the ratio of rate of [14C]metformin uptake and the [14C]metformin concentration in the media. Uptake CLint,in vitro was calculated by dividing the slope of [14C]metformin accumulation over time by [14C]metformin concentration in media. CLint calculated in absence and presence of inhibitor was considered as total CL (CLint,total) and passive CL (CLint,in vitro,passive), respectively. Active CL (CLint,in vitro,active) was calculated by subtracting CLint,in vitro,passive from CLint,in vitro,total.
To determine the contribution of OCT1 or thiamine transporter 2 (THTR2) to [14C]metformin transport into PHH, [14C]metformin uptake assays were conducted in the presence or absence of 500 μM quinidine or 400 mM thiamine [a THTR2 substrate with Km of 3.4 mM (Liang et al., 2015)], respectively.
Quantification of Total and Percent Plasma Membrane Abundance of OCT1 in HEK293 Cells.
Data for the total and PMA of OCT1 in PHH were obtained from our previous publication (Kumar et al., 2019). OCT1 total and PMA in HEK293 cells was quantified using our previously published biotinylation approach (Kumar et al., 2017). HEK293 cells were grown in three independent experiments in a 75-cm2 flask until 80% confluency and incubated at 37°C with 10 ml of 0.78 mg/ml sulfo-NHS-SS-biotin. After 1 hour, cells were lysed, and 50 μl of cell lysate was separated to quantify total abundance. From the rest of the total cell lysate, the biotinylated plasma membrane fragments (to quantify %PMA) were separated from the total cell lysate and intracellular membranes using the neutravidin resin columns. Briefly, 0.2 mg of total protein homogenate and biotinylated fraction were denatured by adding 45 μl of 100 mM ammonium bicarbonate buffer (pH 7.8), 25 μl of 3% sodium deoxycholate (w/v), 12.5 μl of 250 mM dithiothreitol, and 5 μl of 10 mg/ml human albumin and incubated at 95°C for 5 minutes. After incubation, samples were returned to room temperature, 25 μl of 200 mM iodoacetamide was added, and samples were incubated in the dark for 30 minutes. Then, 500 μl of ice-cold methanol, 200 μl of chloroform, and 450 μl of water were added. Samples were mixed and centrifuged at 12,000g for 10 minutes. Precipitated protein samples were redissolved in 45 μl of 100 mM ammonium bicarbonate buffer and digested with 20 μl (∼3.2 μg) of trypsin for 18 hours at 37°C in a shaker (300 rpm). Finally, the digestion was terminated by adding to the sample 20 μl of ice-cold acetonitrile containing the internal standard. Then, the sample was centrifuged at 5000g for 5 minutes at 4°C. The supernatant was collected and analyzed on AB Sciex 6500 triple-quadrupole mass spectrometer (Sciex, Framingham, MA) coupled to the Water Acquity Ultra-Performance Liquid Chromatography (UPLC) system (Waters Corporation, Milford, MA) operated in electrospray positive ionization mode using a previously described approach (Prasad et al., 2016). An Acquity UPLC HSS T3 Column (1.8 mm, 2.1 100 mm) with 0.2-mm inlet frits (Waters) was used for chromatographic separation and resolution. The flow rate of the mobile phase was 0.3 ml/min using a gradient ranging from 3% to 90% acetonitrile/water containing 0.1% formic acid. Signature peptides and product ions as well as their respective Stable Isotope Labelled (SIL) peptides used to quantify proteins were obtained from our previous report (Kumar et al., 2019). The ratio of abundance of protein in biotinylated fractions and total lysate was used to determine %PMA.
Estimation of In Vivo [11C]Metformin CLh,s,in by Compartmental Modeling of PET Imaging Data.
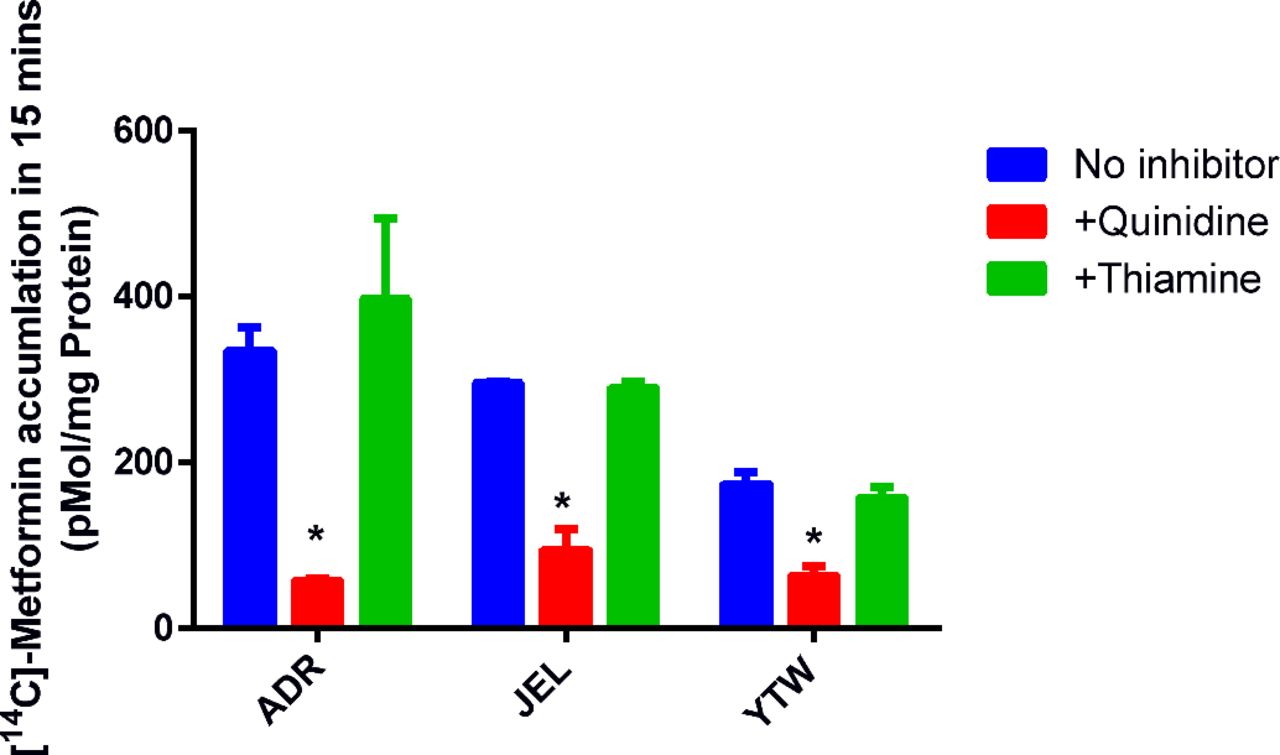
PET-imaged metformin hepatic radioactivity concentration-time data previously published by Gormsen et al. (2016) were reanalyzed using a blood input function and a one-tissue compartment model (Fig. 3A) with a fixed blood volume (SAAMII). This differed from the published analysis, in which radioactivity from the volume of blood within the liver tissue was not taken into account, plasma concentrations were used as input, and a two-compartment model was used (Gormsen et al., 2016).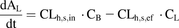 (1)
(1)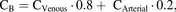 (2)where A, C, B, L,
(2)where A, C, B, L,  , and
, and 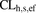 represent amount, concentration, blood, liver, sinusoidal uptake CL, sinusoidal efflux CL, biliary efflux CL, and metabolic CL respectively. Since no biliary efflux was observed and the majority of metformin is excreted renally unchanged, biliary clearance and metabolic clearance were not included in eq. 1 (Hardie, 2007; Graham et al., 2011; Gormsen et al., 2016). The hepatic input blood concentration is a sum of the input via the portal vein (80%) and the hepatic artery (20%) (Schenk et al., 1962), where
represent amount, concentration, blood, liver, sinusoidal uptake CL, sinusoidal efflux CL, biliary efflux CL, and metabolic CL respectively. Since no biliary efflux was observed and the majority of metformin is excreted renally unchanged, biliary clearance and metabolic clearance were not included in eq. 1 (Hardie, 2007; Graham et al., 2011; Gormsen et al., 2016). The hepatic input blood concentration is a sum of the input via the portal vein (80%) and the hepatic artery (20%) (Schenk et al., 1962), where 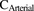 and
and 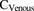 are the observed [11C]metformin arterial and portal vein concentrations, respectively (eq. 2). The volume of the liver was determined based on eq. 3 (Chan et al., 2006).
are the observed [11C]metformin arterial and portal vein concentrations, respectively (eq. 2). The volume of the liver was determined based on eq. 3 (Chan et al., 2006).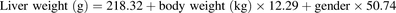 (3)For male and female, sex = 1 and 0, respectively. The volume of blood in the liver was fixed to 10% of total blood volume, estimated based on body weight (75 ml/kg for males and 65 ml/kg for females) (Lautt, 1977; Stabin and Siegel, 2003; Pham and Shaz, 2013). Goodness of fit of the model to the data was assessed by the weighted residual plots, visual inspection of the predicted and observed data, and the confidence (CV%) in the estimates of the parameters.
(3)For male and female, sex = 1 and 0, respectively. The volume of blood in the liver was fixed to 10% of total blood volume, estimated based on body weight (75 ml/kg for males and 65 ml/kg for females) (Lautt, 1977; Stabin and Siegel, 2003; Pham and Shaz, 2013). Goodness of fit of the model to the data was assessed by the weighted residual plots, visual inspection of the predicted and observed data, and the confidence (CV%) in the estimates of the parameters.
Prediction of the In Vivo Metformin Blood Hepatic Uptake CL from PHH Using the MGPGL Approach.
Metformin in vivo blood intrinsic hepatic sinusoidal uptake CL (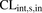 ) was predicted from PHH data using the MGPGL approach (eq. 4).
) was predicted from PHH data using the MGPGL approach (eq. 4).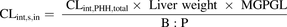 (4)Liver weight was assumed to be 1500 g, B:P is the blood-to-plasma ratio (0.6), and MGPGL was 120 mg of total protein per gram of liver based on a previous report (1.2 × 108 cells per gram of liver and 1 million cells ∼1 mg protein) (Jones et al., 2012; Gormsen et al., 2016; Kim et al., 2019).
(4)Liver weight was assumed to be 1500 g, B:P is the blood-to-plasma ratio (0.6), and MGPGL was 120 mg of total protein per gram of liver based on a previous report (1.2 × 108 cells per gram of liver and 1 million cells ∼1 mg protein) (Jones et al., 2012; Gormsen et al., 2016; Kim et al., 2019).
Then, metformin sinusoidal uptake clearance was computed using the well stirred model (WSHM). (5)where fu is the fraction of metformin unbound in plasma (assumed to be 1), and Q is the hepatic blood flow (1500 ml/min) (Scheen, 1996).
(5)where fu is the fraction of metformin unbound in plasma (assumed to be 1), and Q is the hepatic blood flow (1500 ml/min) (Scheen, 1996).
Prediction of In Vivo Metformin Hepatic Uptake CL from OCT1- HEK293 Cells and PHH Using the REF Approach.
In vivo blood metformin hepatic intrinsic sinusoidal active uptake CL (CLint,s,active in) was determined from in vitro metformin uptake CL in OCT1-HEK293 cells or PHH using the REF approach, corrected for OCT1 PMA in these cells or PHH (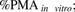 eq. 6). In doing so, the following assumptions were made: 1) OCT1 is the only transporter responsible for metformin hepatic uptake (see Results for justification) and 2) because it is impossible to use the biotinylation method for quantification of OCT1 PMA in liver tissue, the PMA abundance of OCT1 in liver tissue
eq. 6). In doing so, the following assumptions were made: 1) OCT1 is the only transporter responsible for metformin hepatic uptake (see Results for justification) and 2) because it is impossible to use the biotinylation method for quantification of OCT1 PMA in liver tissue, the PMA abundance of OCT1 in liver tissue 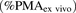 was assumed to be 100%.
was assumed to be 100%.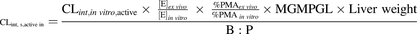 (6)
(6) is OCT1 abundance in HEK293 cells or PHH.
is OCT1 abundance in HEK293 cells or PHH.  is total OCT1 abundance in human liver tissue, and MGMPGL is milligrams of total membrane protein per gram of liver from individual control liver (n = 36) obtained from our previous report (Wang et al., 2016).
is total OCT1 abundance in human liver tissue, and MGMPGL is milligrams of total membrane protein per gram of liver from individual control liver (n = 36) obtained from our previous report (Wang et al., 2016).
Also, the blood hepatic intrinsic sinusoidal passive uptake CL of metformin ( ) was scaled based on liver weight and adjusted for the sinusoidal surface area (SSA) (37%; eq. 7) since the entire plasma membrane of hepatocytes is not exposed to blood (Esteller, 2008). The correction for SSA was not made for the active uptake of metformin into cells since OCT1 is thought to be present only on the sinusoidal membrane of the hepatocytes.
) was scaled based on liver weight and adjusted for the sinusoidal surface area (SSA) (37%; eq. 7) since the entire plasma membrane of hepatocytes is not exposed to blood (Esteller, 2008). The correction for SSA was not made for the active uptake of metformin into cells since OCT1 is thought to be present only on the sinusoidal membrane of the hepatocytes.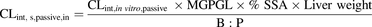 (7)Finally, the total blood intrinsic hepatic sinusoidal uptake CL (
(7)Finally, the total blood intrinsic hepatic sinusoidal uptake CL ( ) was computed (eq. 8) to estimate the sinusoidal hepatic uptake clearance (
) was computed (eq. 8) to estimate the sinusoidal hepatic uptake clearance (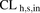 ) using the WSHM (eq. 5).
) using the WSHM (eq. 5).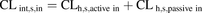 (8)Data are presented as means of REF-predicted
(8)Data are presented as means of REF-predicted 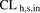 based on 36 control livers, whereas the mean MGPGL-predicted
based on 36 control livers, whereas the mean MGPGL-predicted 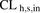 was computed as a mean of four different lots of PHH.
was computed as a mean of four different lots of PHH.
Results
[14C]Metformin Uptake into Hepatocytes Is Mediated by OCT1 and Not THTR2.
Since [14C]metformin uptake was linear in PHH over a period of 15 minutes (Fig. 2), this was the time used to determine [14C]metformin uptake into PHH in the presence and absence of OCT (quinidine) or THTR2 (thiamine) inhibitor (Fig. 1). Quinidine, but not thiamine, significantly inhibited metformin uptake by 83%, 68.1%, and 63.5% in ADR, JEL, and YTW PHH, respectively.

[14C]Metformin uptake into three lots of PHH in the absence and presence of an OCT1 or a THTR2 inhibitor. Quinidine (500 μM), an OCT inhibitor, significantly inhibited hepatocyte uptake of metformin, whereas thiamine (400 mM), a THTR2 inhibitor, did not. Data are presented as means ± S.D. of triplicate determinations. Data were analyzed by one-way analysis of variance followed by Tukey multiple comparison post hoc test. *P < 0.05 compared with the respective control.
Estimation of [14C]Metformin CLint Uptake into HEK293 Cells or PHH.
[14C]Metformin uptake into HEK293 cells and PHH was linear over 5 and 15 minutes, respectively (Fig. 2). The mean [14C]metformin  or
or 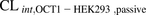 was similar to
was similar to 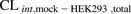 or
or 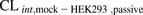 . These data further confirm that [14C]metformin uptake into hepatocytes is mediated primarily by OCT1 (Table 1).
. These data further confirm that [14C]metformin uptake into hepatocytes is mediated primarily by OCT1 (Table 1).

Estimation of [14C]metformin uptake CLint into HEK 293 cells or PHH. [14C]Metformin uptake into OCT1-expressing HEK293 and mock cells (A) or into four different lots of PHH (B–E) was linear over a period of 5 and 15 minutes, respectively. Data in (A–E) are presented as means ± S.D. of experiments conducted in triplicate.

Estimate of the in vivo [11C]metformin hepatic uptake clearance after correcting for blood [11C]metformin content in the liver. [11C]Metformin hepatic concentrations calculated from PET imaging and corresponding plasma [11C]metformin concentrations were obtained from a previous publication (Gormsen et al., 2016). A one-compartment pharmacokinetic model (A), which incorporated correction for blood [11C]metformin content in the liver, was fitted to the hepatic [11C]metformin concentration vs. time data in five subjects. Arterial plus image–estimated portal venous blood [11C]metformin concentrations were used as the input function using the forcing function (FF) approach. Observed (diamonds) and model-fitted hepatic [11C]metformin concentration vs. time profile in a representative subject demonstrates relatively good fit of the model to the data (B).Where CLh,s,in is hepatic sinusoidal uptake clearance and CLh,s,ef is hepatic sinusoidalefflux clearance
Total and passive [14C]metformin (500 μM) uptake clearances ( ) into PHH and HEK293 cells estimated in the absence and presence of quinidine, respectively
) into PHH and HEK293 cells estimated in the absence and presence of quinidine, respectively
Plasma and Total Membrane Abundance of OCT1 in OCT1-HEK293 Cells and PHH.
The percent PMAs of OCT1, Na+/K+ ATPase (plasma membrane marker), and calreticulin (endoplasmic reticulum marker) in HEK293 cells were 36.04% ± 2.46%, 58.73% ± 2.79%, and 6.63% ± 0.39%, respectively. The low presence of calreticulin in the plasma membrane fraction demonstrated the fidelity of the biotinylation method. The percent PMA of OCT1 in PHH used in this study (ADR: 63.8%, FEA: 55.3%, JEL: 51.9%, YTW: 65.8%) was obtained from our previous report (Kumar et al., 2019). These PMA data, together with the total abundance of OCT1 in these cells, were used (eq. 5) to predict the in vivo metformin hepatic uptake clearance (see below and Table 4).
Estimating In Vivo [11C]Metformin Hepatic Uptake CL from PET Imaging Data after Correcting for Blood [11C]Metformin Content in the Liver.
The mean estimate of [11C]metformin CLh,s,in in four subjects was 441 ± 76 ml/min (range 361–552 ml/min), with each subject’s CLh,s,in estimated with a high degree of confidence (low %CV of the estimates) (Table 2).
Estimate of [11C]metformin hepatic uptake clearance from the PET imaging study
IVIVE of Metformin Hepatic Sinusoidal Uptake CL from HEK293 Cells and PHH Using REF Versus the MGPGL Approach.
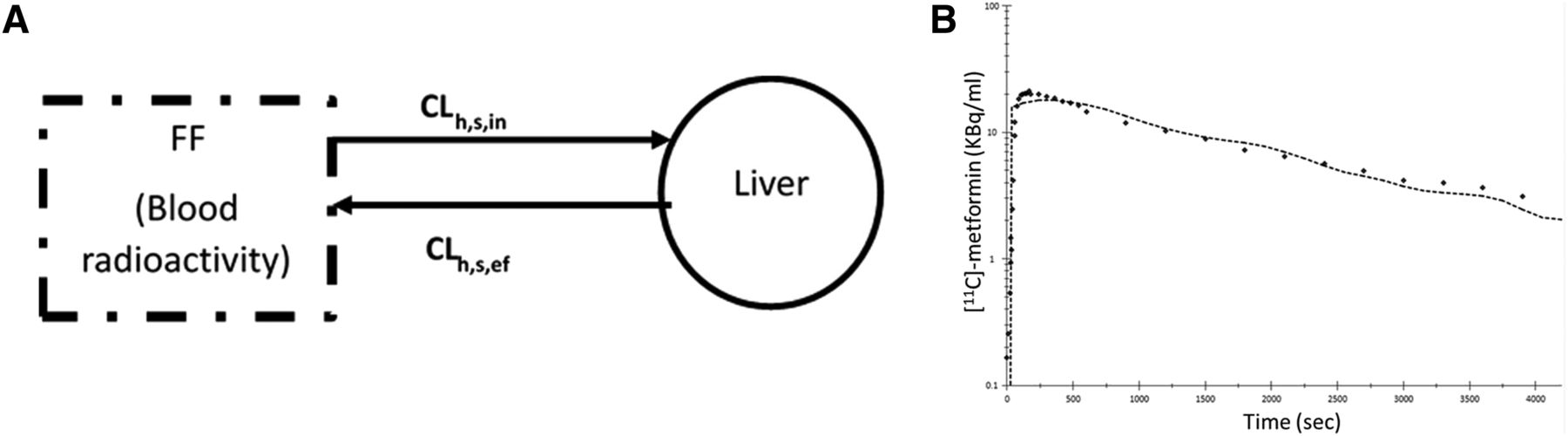
IVIVE of metformin CLh,s,in based on the MGPGL approach significantly underpredicted (by more than 56%) the observed value (Fig. 4A; Table 3). Likewise, IVIVE of metformin CLh,s,in based on total abundance of OCT1 (i.e., without correcting for PMA) in HEK293 cells or PHH underpredicted the observed value (Fig. 4B; Table 4). In addition, except for PHH JEL and YTW, the predicted CLh,s,in based on HEK293 cells or PHH (average or ADR or FEA) was below the predefined acceptance criteria for prediction of the observed in vivo CLh,s,in. In contrast, after correcting for PMA, the predicted CLh,s,in based on HEK293 cells or the average PHH was within predefined acceptance criteria, with the latter underpredicting the observed value much more than the former.

In vitro to in vivo extrapolation of metformin 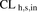 based on HEK293 cells or PHH. For all four lots of PHH, the predicted
based on HEK293 cells or PHH. For all four lots of PHH, the predicted 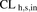 based on the MGPGL approach underpredicted the observed value (continuous line) and also fell outside the 2-fold predefined acceptance criteria (dashed line, A). Likewise, the predicted metformin
based on the MGPGL approach underpredicted the observed value (continuous line) and also fell outside the 2-fold predefined acceptance criteria (dashed line, A). Likewise, the predicted metformin 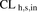 based on total abundance of OCT1 (i.e., without correcting for PMA) in HEK293 cells or PHH underpredicted the observed value (B). In contrast, after correcting for PMA, the predicted metformin
based on total abundance of OCT1 (i.e., without correcting for PMA) in HEK293 cells or PHH underpredicted the observed value (B). In contrast, after correcting for PMA, the predicted metformin  based on HEK293 cells or the average PHH was within the predefined acceptance criteria, with the latter underpredicting the observed value more than the former. Data shown are means ± S.D.
based on HEK293 cells or the average PHH was within the predefined acceptance criteria, with the latter underpredicting the observed value more than the former. Data shown are means ± S.D.
IVIVE of metformin CLh,s,in based on the traditional MGPGL approach
IVIVE of metformin CLh,s,in based on the REF approach
Discussion
The major findings of this study are as follows: 1) metformin is transported into human hepatocytes predominantly by OCT1 with no (or little) contribution from THTR2; 2) PET imaging data should be corrected for hepatic blood metformin content to correctly estimate the in vivo metformin 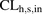 ; and 3) the REF approach is superior to the MGPGL approach in accurately predicting the in vivo CLh,s,in of metformin.
; and 3) the REF approach is superior to the MGPGL approach in accurately predicting the in vivo CLh,s,in of metformin.
In humans, hepatic uptake of metformin (the site of its pharmacological effect) is purely a distributional CL because metformin is not metabolized or excreted into the bile by the liver and is primarily (99%) eliminated unchanged in the urine (Pentikainen et al., 1979). The latter was confirmed in the human PET imaging study, in which no metformin radioactivity could be detected in the gallbladder (Gormsen et al., 2016). Various in vitro studies have confirmed that metformin is transported into the liver primarily by OCT1 (Km 1.2 mM) (Kimura et al., 2005). Although OCT3 can transport metformin (Km 1.1 mM) (Chen et al., 2010), because the hepatic protein abundance of OCT3, relative to OCT1, is negligible (Drozdzik et al., 2019), we assumed that it did not contribute to the total uptake of metformin either in vivo or in vitro. THTR2, a thiamine transporter, has also been found to transport metformin (Km 1.15 mM) (Liang et al., 2015). Therefore, we determined its contribution in the transport of metformin into PHH using a sufficiently high concentration (400 mM) of thiamine to completely inhibit THTR2. We found that THTR2 does not significantly contribute to metformin transport into PHH (Fig. 1). This was confirmed by our findings that metformin uptake CL into PHH was similar to that in OCT1-HEK293 cells when OCT1 was completely inhibited by quinidine in both systems (Fig. 2; Table 1).
Prior to predicting metformin CLh,s,in by scaling 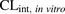 in OCT1-HEK293 cells or PHH, we estimated the in vivo metformin CLh,s,in by reanalysis of previously published PET imaging data. Reanalysis was performed because estimates of hepatic uptake CL (whether in vivo or in vitro into hepatocytes) of a drug are primarily derived from data in the initial uptake phase. In the case of PET imaging, because the liver has a significant amount of blood (10% of total blood volume) and the radioactivity content in hepatic tissue during the initial uptake phase may not be significantly larger than that in the blood, the drug content in the blood in the liver will upwardly bias the true hepatic uptake CL of the drug. Indeed, this was the case for metformin: the previously reported [11C]metformin plasma CLh,s,in was 0.55 ± 0.15 ml/min per milliliter (or 0.55 ± 0.15 ml/min per gram; assuming density of liver tissue as 1.0) of hepatic tissue (or blood CLh,s,in 1375 ± 375 ml/min assuming 1.5 kg as liver weight by PET imaging) (Gormsen et al., 2016). On reanalysis of these PET imaging data, in which we took into consideration the significant presence of blood in the liver, the estimated
in OCT1-HEK293 cells or PHH, we estimated the in vivo metformin CLh,s,in by reanalysis of previously published PET imaging data. Reanalysis was performed because estimates of hepatic uptake CL (whether in vivo or in vitro into hepatocytes) of a drug are primarily derived from data in the initial uptake phase. In the case of PET imaging, because the liver has a significant amount of blood (10% of total blood volume) and the radioactivity content in hepatic tissue during the initial uptake phase may not be significantly larger than that in the blood, the drug content in the blood in the liver will upwardly bias the true hepatic uptake CL of the drug. Indeed, this was the case for metformin: the previously reported [11C]metformin plasma CLh,s,in was 0.55 ± 0.15 ml/min per milliliter (or 0.55 ± 0.15 ml/min per gram; assuming density of liver tissue as 1.0) of hepatic tissue (or blood CLh,s,in 1375 ± 375 ml/min assuming 1.5 kg as liver weight by PET imaging) (Gormsen et al., 2016). On reanalysis of these PET imaging data, in which we took into consideration the significant presence of blood in the liver, the estimated 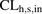 was found to be 441 ml/min, i.e., ∼32% of the previously reported values (Table 2).
was found to be 441 ml/min, i.e., ∼32% of the previously reported values (Table 2).
When we used the REF approach to predict CLh,s,in based on the assumption that 100% of OCT1 was present on the plasma membrane of both HEK293 cells or PHH (average abundance), the CLh,s,in was underpredicted by ∼60% for both in vitro systems (Fig. 4; Table 4). The predicted  by the individual lots of hepatocytes was also underpredicted, but that by the JEL and YTW was within 2-fold of the acceptance criteria. Since only OCT1 present in the plasma membrane is functional, and not all the OCT1 transporters are present in the plasma membrane of HEK293 cells or PHH, we asked whether correcting for PMA of OCT1 improved our prediction of CLh,s,in. This question was also motivated by our previous finding that such a correction allowed for better prediction of in vivo metformin secretory renal CL in humans (Kumar et al., 2018). Indeed, after correcting for PMA, OCT1-HEK293 cells predicted a value for CLh,s,in (405 ± 122 ml/min) that was not significantly different from that observed from PET imaging (441 ± 76 ml/min). Likewise, after correcting for PMA, data from the PHH, on average, predicted a value for CLh,s,in (230 ± 78 ml/min) that was within the lower limit of 2-fold predefined acceptance criteria (221 ml/min). In addition, except for ADR and FEA, the remaining lots of PHH predicted metformin
by the individual lots of hepatocytes was also underpredicted, but that by the JEL and YTW was within 2-fold of the acceptance criteria. Since only OCT1 present in the plasma membrane is functional, and not all the OCT1 transporters are present in the plasma membrane of HEK293 cells or PHH, we asked whether correcting for PMA of OCT1 improved our prediction of CLh,s,in. This question was also motivated by our previous finding that such a correction allowed for better prediction of in vivo metformin secretory renal CL in humans (Kumar et al., 2018). Indeed, after correcting for PMA, OCT1-HEK293 cells predicted a value for CLh,s,in (405 ± 122 ml/min) that was not significantly different from that observed from PET imaging (441 ± 76 ml/min). Likewise, after correcting for PMA, data from the PHH, on average, predicted a value for CLh,s,in (230 ± 78 ml/min) that was within the lower limit of 2-fold predefined acceptance criteria (221 ml/min). In addition, except for ADR and FEA, the remaining lots of PHH predicted metformin 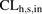 within the predefined acceptance criteria. In contrast, the traditional MGPGL approach (on average) underpredicted
within the predefined acceptance criteria. In contrast, the traditional MGPGL approach (on average) underpredicted 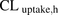 by more than 56% (ranging from 47% to 64%) (Fig. 4A; Table 3). This observation is consistent with previous reports that PHH underpredicts the hepatic uptake CL of several drugs (Jones et al., 2012). One possible reason proposed for such underprediction is the protein-mediated effect on hepatic uptake CL (Miyauchi et al., 2018; Bteich et al., 2019). However, this is unlikely to explain our findings since metformin is not bound to plasma proteins. Another potential explanation is that the value of MGPGL used is incorrect. Taken together these results clearly demonstrate that the PMA-corrected REF approach is superior to the traditional MGPGL approach when predicting
by more than 56% (ranging from 47% to 64%) (Fig. 4A; Table 3). This observation is consistent with previous reports that PHH underpredicts the hepatic uptake CL of several drugs (Jones et al., 2012). One possible reason proposed for such underprediction is the protein-mediated effect on hepatic uptake CL (Miyauchi et al., 2018; Bteich et al., 2019). However, this is unlikely to explain our findings since metformin is not bound to plasma proteins. Another potential explanation is that the value of MGPGL used is incorrect. Taken together these results clearly demonstrate that the PMA-corrected REF approach is superior to the traditional MGPGL approach when predicting 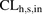 of metformin. We have also demonstrated that the REF approach is superior to the MGPGL approach when predicting
of metformin. We have also demonstrated that the REF approach is superior to the MGPGL approach when predicting 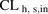 of rosuvastatin (an OATP substrate) in rat (Ishida et al., 2018). Thus, the REF approach appears to be superior to the MGPGL approach for a diverse set of drugs that are transported by different transporters. Additional studies need to be conducted to determine whether this conclusion can be generalized across a larger number of drugs transported by a variety of transporters.
of rosuvastatin (an OATP substrate) in rat (Ishida et al., 2018). Thus, the REF approach appears to be superior to the MGPGL approach for a diverse set of drugs that are transported by different transporters. Additional studies need to be conducted to determine whether this conclusion can be generalized across a larger number of drugs transported by a variety of transporters.
Acknowledgments
The authors thank Tot Bui Nguyen for her support in cell-surface biotinylation experiments and LC-MS/MS proteomics.
Authorship Contributions
Participated in research design: Sachar, Kumar, Unadkat.
Conducted experiments: Sachar.
Performed data analysis: Sachar.
Wrote or contributed to the writing of the manuscript: Sachar, Munk, Gormsen, Unadkat.
Footnotes
- Received June 20, 2020.
- Accepted August 20, 2020.
This research was supported by University of Washington Research Affiliate Program on Transporters (UWRAPT), funded by Genentech, Biogen, Gilead, Merck, and Takeda.
Abbreviations
- CL
- clearance
- CLh,s,in
- hepatic sinusoidal uptake clearance
- CLint
- intrinsic clearance
- CLint,in vitro
- intrinsic in vitro uptake clearance
- HK HEPES-buffered
- Krebs-Ringer bicarbonate
- OATP
- organic anion transporting polypeptides
- OCT
- organic cation transporter
- IVIVE
- in vitro to in vivo extrapolation
- MGPGL
- milligrams of protein content per gram of liver tissue
- PET
- positron emission tomography
- PHH
- plated human hepatocyte
- PMA
- plasma membrane abundance
- REF
- relative expression factor
- THTR2
- thiamine transporter 2
- WSHM
- well stirred model
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}