


Abstract
To explore the mechanism of the interaction between gemfibrozil and cerivastatin, the enzyme mapping of the oxidative metabolism of cerivastatin and the effect of gemfibrozil on cerivastatin metabolism were studied using human liver microsomes and expressed cytochrome P450 (P450) CYP2C8 and 3A4 isoforms. Based on studies with isoform-selective chemical inhibitors and expressed enzymes, CYP2C8 and CYP3A4 were equally important in the formation of desmethylcerivastatin (M-1), whereas the formation of the quantitatively most important hydroxy metabolite (M-23) was predominantly mediated via CYP2C8; other P450 isoforms played a negligible role. In human liver microsomes, gemfibrozil markedly inhibited M-23 formation, with aKi (IC50) value of 69 (95) μM, whereas inhibition of M-1 formation was weaker with aKi (IC50) value of 273 (>250) μM. The inhibitory effect of gemfibrozil was attributable to inhibition of CYP2C8 rather than CYP3A4, as evidenced by potent inhibition of the formation of M-23 (IC50 = 68 μM) and M-1 (IC50 = 78 μM) in recombinant CYP2C8 but not in recombinant CYP3A4. Additionally, gemfibrozil inhibited paclitaxel 6α-hydroxylation [Ki(IC50) = 75 μM (91 μM)], a CYP2C8 marker reaction, but did not inhibit testosterone 6β-hydroxylation (CYP3A4). The present in vitro findings suggest that inhibition of CYP2C8 activity by gemfibrozil at least partially explains the interaction between gemfibrozil and cerivastatin. The formation of M-23 acid from cerivastatin is mediated mainly by CYP2C8 and thus may be a suitable CYP2C8 probe reaction. Inhibition of CYP2C8-mediated metabolism by gemfibrozil warrants further in vivo exploration.
Cerivastatin, a cholesterol lowering drug, was recently withdrawn from the market due to its high risk of the life-threatening adverse effect, rhabdomyolysis (Charatan, 2001). Fatal rhabdomyolysis after cerivastatin treatment was reported most frequently when the drug was combined with the lipid lowering fibrate gemfibrozil (Pogson et al., 1999; Farmer, 2001; Staffa et al., 2002). Gemfibrozil increases the risk of myopathy associated with simvastatin and lovastatin, too. It was recently found to increase plasma concentrations of the active acid forms of simvastatin and lovastatin, suggesting a pharmacokinetic basis as a mechanism of gemfibrozil-statin interactions (Backman et al., 2000; Kyrklund et al., 2001).
Cerivastatin is administered as the pharmacologically active hydroxy acid form. It is cleared exclusively via biotransformation and subsequent biliary (70%) or renal (30%) excretion of the formed metabolites; no unchanged drug is excreted in humans (Mück, 1998a). According to the manufacturer of cerivastatin, in vitro studies in human liver microsomes have indicated that the biotransformation of cerivastatin is catalyzed by cytochrome P450 (P4501) CYP2C8 and 3A4, forming two major metabolites, desmethylcerivastatin (M-1) and the hydroxylated metabolite (M-23) (Mück, 1998a). The same P450 isoforms also mediate the biotransformation of M-1 and M-23 to a tertiary minor metabolite M-24. The in vitro study of Boberg et al. (1997) suggested that CYP3A4 plays a major role in the metabolism of cerivastatin. However, in vivo investigations have revealed that potent CYP3A4-inhibitors, such as erythromycin and itraconazole, increase only slightly plasma concentrations of cerivastatin (Mück et al., 1998b; Kantola et al., 1999). In contrast, gemfibrozil markedly increases the plasma concentrations of cerivastatin and M-1 acid and reduces the concentrations of M-23 acid (Backman et al., 2002).
In a recent in vitro study, gemfibrozil strongly inhibited CYP2C9 activity (Ki = 5.8 μM) and moderately inhibited CYP2C19 (Ki = 24 μM) and CYP1A2 (Ki = 82 μM) activity, with no appreciable effect on CYP2A6, CYP2D6, CYP2E1, and CYP3A4 activities (Wen et al., 2002). However, there seem to be no published studies on the effect of gemfibrozil on CYP2C8 activity in vitro. Therefore, we have studied the effects of gemfibrozil on the oxidative metabolism of cerivastatin in vitro, to clarify the mechanism of the gemfibrozil-cerivastatin interaction.
Experimental Procedures
Materials.
Ketoconazole was obtained from Orion Pharma (Espoo, Finland). Sulfamethoxazole, trimethoprim, tranylcypromine, sulfaphenazole, quinidine, pyridine, paclitaxel, testosterone, and NADPH were purchased from Sigma-Aldrich (St. Louis, MO). S-mephenytoin, 6β-hydroxytestosterone, and 6α-hydroxypaclitaxel were purchased from Ultrafine Chemicals Ltd. (Manchester, UK). Erythromycin and cerivastatin were obtained from Serva (Heidelberg, Germany) and Bayer AG (Leverkusen, Germany), respectively. Pooled human liver microsomes (prepared from five male, and five female human liver microsomal samples) were obtained from BD Gentest (Woburn, MA). Microsomes from baculovirus-infected cells engineered to express the cDNA encoding human CYP2C8 and CYP3A4 were also purchased from BD Gentest. Other chemicals and reagents were obtained from Merck (Darmstadt, Germany).
Assays for the Oxidative Metabolism of Cerivastatin.
The incubation mixture, in a final volume of 200 μl, contained 1 mg/ml microsomal protein, 0.1 M sodium phosphate buffer (pH 7.4), 1.0 mM NADPH, 5 mM MgCl2, and various concentrations of cerivastatin (substrate stock solution dissolved in water and subsequently diluted using the 0.1 M sodium phosphate buffer) in the presence or absence of one of the putative inhibitors. After incubation in a shaking water bath (37°C) for 5 to 30 min, the reactions were terminated by adding 200 μl ice-cold acetonitrile, and the samples were cooled on ice for 15 min. The samples were then removed to glass tubes containing 1 ml sodium acetate buffer (0.1 M, pH 4.5). Lovastatin acid (50 μl of 1 μg/ml) was subsequently added as an internal standard (ISTD). The samples were then extracted with 6 ml of methyl-tert-butyl ether for 20 min. After centrifugation, the supernatant was evaporated to dryness. The residue was then reconstituted with 75 μl of a solution (50:50, v/v) containing acetonitrile and 10 mM ammonium acetate buffer (pH = 5). Thereafter 5 to 20 μl of the sample was subjected to LC-MS-MS analysis. The formation rates of M-1 and M-23 were confirmed to be linear with respect to the microsomal protein concentration (up to 1 mg/ml) and incubation time (up to 30 min) used. All human liver microsomal incubations were performed in duplicate.
Incubations with the recombinant P450 isoforms were performed using the same conditions as described for human liver microsomes, except that the mixture contained 5 pmol of P450 (CYP2C8 or CYP3A4) and was incubated for 15 min (CYP3A4) or 20 min (CYP2C8). The formation rates of M-1 and M-23 were linear over this period of time. The incubations in the expressed enzymes were performed in duplicate.
Inhibition Studies.
The effects of 11 isoform-selective P450 inhibitors as well as gemfibrozil (1–250 μM) on the P450-mediated oxidative metabolism of cerivastatin were studied. Furafylline (20 μM) was used as a selective inhibitor of CYP1A2, tranylcypromine (1 μM) for CYP2A6, trimethoprim (100 μM) for CYP2C8, sulfaphenazole (10 μM), and sulfamethoxazole (500 μM) for CYP2C9, S-mephenytoin (300 μM) for CYP2C19, quinidine (10 μM) for CYP2D6, pyridine (100 μM) for CYP2E1, ketoconazole (1 μM), troleandomycin (100 μM), and erythromycin (100 μM) for CYP3A4 (Hargreaves et al., 1994; Newton et al., 1995; Eagling et al., 1998; Hickman et al., 1998; Taavitsainen et al., 2001; Wen et al., 2002). The stock solutions of the inhibitors were prepared in the 0.1 M sodium phosphate buffer (pH 7.4) with, in some cases, minimal use of methanol (final concentration less than 1%). Gemfibrozil was dissolved in acetonitrile. After evaporation of acetonitrile to dryness, gemfibrozil was reconstituted in the incubation medium. All the inhibitors and their matched controls (containing an equal volume of methanol in the case that it was used) were preincubated with the incubation medium at 37°C for 15 min, either in the presence or absence of 1.0 mM NADPH. After the preincubation, cerivastatin (final concentration 0.01–1 μM) was added either with or without 1.0 mM NADPH.
Effects of Gemfibrozil on P450 Activities.
Activities of CYP3A4 (testosterone 6β-hydroxylation) and CYP2C8 (paclitaxel 6α-hydroxylation) were determined as described elsewhere (Wen et al., 2002). The marker substrate concentrations used, 25 μM testosterone and 1 to 5 μM paclitaxel, were comparable with or around the Km values of the reactions. Gemfibrozil was either coincubated at 37°C for 15 min with the marker substrate and NADPH (1 mM) before the reaction was initiated with human liver microsomes (0.1 mg/ml) or preincubated with human liver microsomes and NADPH for 15 min before adding the marker substrate.
Analytical Procedures.
The concentrations of cerivastatin and its major metabolites in incubations were measured by use of liquid chromatography-ionspray tandem mass spectrometry with use of the PE SCIEX API 3000 LC-MS-MS system (MDS Sciex, Concord, ON, Canada) as described previously, with minor modifications (Jemal et al., 1999). The ion transitions monitored were m/z 460 to 356 for cerivastatin (acid), m/z 442 to 354 for cerivastatin lactone,m/z 446 to 342 for M-1 acid, a sum ofm/z 476 to 280 and m/z 476 to 278 for M-23 acid, m/z 462 to 340 for M-24 acid, and m/z 423 to 285 for lovastatin acid. The limit of quantification for cerivastatin was 0.001 μM, and the day-to-day coefficient of variation was below 15% at relevant concentrations (n = 5). The cerivastatin metabolites were produced by incubating a high concentration of cerivastatin with human liver microsomes in standard incubation conditions, and the metabolites formed were used for optimization of the assay. A signal/noise ratio of 10:1 was used as the limit of detection for cerivastatin metabolites. Assays for gemfibrozil, 6α-paclitaxel, and 6β-testosterone were carried out using high-performance liquid chromatography as described previously (Hsu et al., 1991; Wen et al., 2002). The day-to-day coefficients of variation (CV) were <7% at relevant concentrations (n = 6).
Binding Studies.
Various concentrations of cerivastatin (six concentrations, 0.01–10 μM) and gemfibrozil (six concentrations, 10–250 μM) in incubation buffers containing microsomal protein (1 mg/ml) were prepared in duplicate. Unbound fractions of cerivastatin and gemfibrozil were separated by ultrafiltration (Sallustio et al., 1997; Wang et al., 2002). The unbound concentrations of gemfibrozil in the filtrates were measured by high-performance liquid chromatography (Hsu et al., 1991), and those of cerivastatin by LC-MS-MS as described above.
Data Analysis.
In the determination of the IC50 (concentration of inhibitor to cause 50% inhibition of original enzyme activity) values, the analyte/ISTD peak area ratios were converted to percentage of control, using the control peak area ratio value as 100%. The apparent inhibitory constant (Ki) values of gemfibrozil for M-1 and M-23 were calculated using secondary plots of slopes taken from double-reciprocal plots of cerivastatin concentrations versus the analyte/ISTD peak area ratios.
Results
Studies with Human Liver Microsomes.
Two major metabolites, M-23 acid and M-1 acid, were found after incubation of clinically relevant concentrations of cerivastatin (0.01–0.04 μM) in human liver microsomes. The metabolite M-24 acid was also detectable, but its formation was much lower compared with the formation of M-1 and M-23, and its concentrations were around the detection limit of the LC-MS-MS assay. The formation of lactones of parent cerivastatin, M-1, M-23, and M-24 acid was negligible.
Trimethoprim and gemfibrozil considerably inhibited both M-23 and M-1 formation, whereas ketoconazole, troleandomycin, and erythromycin inhibited only the formation of M-1 (Fig.1). Gemfibrozil competitively inhibited the formation of M-23 and M-1, with Ki(IC50) values of 69 μM (95 μM) and 273 μM (>250 μM), respectively (Fig. 2). Preincubation of gemfibrozil with NADPH for 15 min did not enhance the inhibitory effects of gemfibrozil (data not shown).

Percent of inhibition of the formation of M-23 (left panel) and M-1 (right panel) at 0.04 μM cerivastatin by selective inhibitors of eight P450 isoforms in human liver microsomes (data represent the mean of duplicate incubations).

Inhibitory effects of gemfibrozil (0–250 μM) on the metabolism of cerivastatin in human liver microsomes.
A, effects of gemfibrozil on the formation of M-1 (open circles) and M-23 (solid circles) from cerivastatin (0.01 μM) in human liver microsomes. B and C, representative secondary plots for the inhibition of the formation of M-1 (B) and M-23 (C) by gemfibrozil in human liver microsomes. Data represent the mean of duplicate determinations.
Studies with Recombinant CYP2C8 and CYP3A4.
Both recombinant CYP2C8 and CYP3A4 catalyzed the formation of M-23 and M-1. However, when 0.01 μM cerivastatin was used, the formation rates of M-23 were about 14-fold lower in recombinant CYP3A4 than in recombinant CYP2C8, whereas recombinant CYP2C8 and CYP3A4 catalyzed equally the formation of M-1. Gemfibrozil dose dependently inhibited the formation of M-23 (IC50 = 68 μM) and M-1 (IC50 = 78 μM) in recombinant CYP2C8 but did not show appreciable effect on the formation of these metabolites in recombinant CYP3A4 (Fig. 3).

The effect of gemfibrozil on the metabolism of cerivastatin (0.01 μM) to M-23 (left panel) and M-1 (right panel) in recombinant CYP2C8 (open circles) and CYP3A4 (solid circles).
Data represent the mean of duplicate determinations.
Effects of Gemfibrozil on CYP3A4 and CYP2C8 Activities.
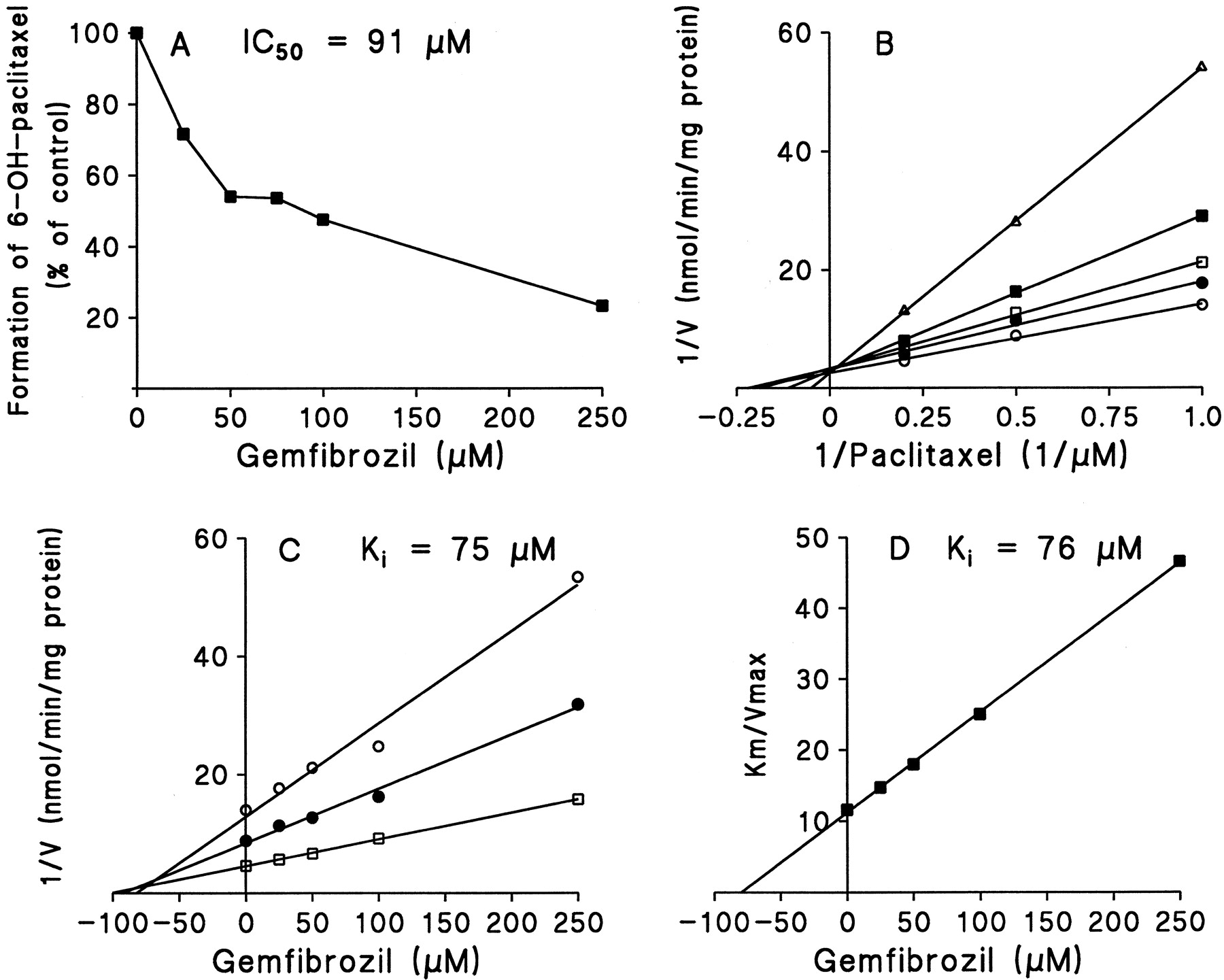
With concentrations up to 250 μM, gemfibrozil showed practically no inhibitory effect on the formation of 6β-hydroxytestosterone in human liver microsomes (data not shown). However, gemfibrozil competitively inhibited the biotransformation of paclitaxel to 6α-hydroxypaclitaxel, with an estimatedKi (IC50) value of 75 μM (91 μM) (Fig. 4).

Inhibitory effects of gemfibrozil (0–250 μM) on paclitaxel 6α-hydroxylation in human liver microsomes.
A, dose-dependent inhibitory effects of gemfibrozil on the formation of 6α-hydroxypaclitaxel from paclitaxel (1 μM) in human liver microsomes. B, a double-reciprocal plot obtained from a 20-min incubation of human liver microsomes with NADPH and paclitaxel (a CYP2C8 marker, 1–5 μM) in the absence (○) or presence of 25 μM (●), 50 μM (■), 100 μM (▪) or 250 μM (▵) gemfibrozil. C, a Dixon plot obtained from a 20-min incubation of human liver microsomes with 1 μM (○), 2 μM (●), and 5 μM (■) of paclitaxel in the absence or presence of gemfibrozil (25–250 μM). D, a secondary plot of slopes taken from double-reciprocal plots versus gemfibrozil concentration. Each data point represents the average of duplicate determinations.
Binding Studies.
At spiked concentrations of 0.01 to 10 μM cerivastatin, and 10 to 250 μM of gemfibrozil, both compounds showed essentially no binding with the incubation medium containing 1 mg/ml microsomal protein (data not shown).
Discussion
The present in vitro studies showed that the formation of the major metabolite of cerivastatin, M-23, was markedly inhibited by gemfibrozil at clinically relevant concentrations (150 μM approximates the peak plasma concentrations of gemfibrozil after normal therapeutic doses; Backman et al., 2000). The formation of metabolite M-1 was also inhibited by gemfibrozil but to a much smaller extent. The formation of M-23 was predominantly mediated by CYP2C8, whereas both CYP2C8 and CYP3A4 catalyzed the formation of M-1. The inhibition of both metabolites formation by gemfibrozil was due to inhibition of CYP2C8 activity rather than CYP3A4.
Gemfibrozil is a nonselective inhibitor of several P450 isoforms (Ki values for CYP2C9, 2C19, 2C8, and 1A2 are 5.8, 24, 69, and 82 μM, respectively) (Wen et al., 2001) and uridine diphosphate glucuronosyltransferases (Prueksaritanont et al., 2002b). The formation of M-23 was susceptible to inhibition by trimethoprim, which is a selective inhibitor of CYP2C8 (Wen et al., 2002), and the formation rate of M-23 was 14-fold higher in CYP2C8 than in CYP3A4. Moreover, the extent of inhibition of the formation of M-23 by gemfibrozil (Ki = 69 μM) was comparable with that of the formation of 6α-hydroxypaclitaxel (a known probe reaction for CYP2C8, Ki = 75 μM). Thus, the present results indicate that the formation of M-23 is mediated mainly via CYP2C8 and that the formation of M-23 may be a suitable probe for CYP2C8 activity. However, the limited availability of cerivastatin and its metabolites may limit the usefulness of this assay.
CYP2C8 is expressed at relatively high levels (6–7% of total P450 content) in human liver (Romkes et al., 1991), and its importance in drug metabolism has recently been recognized (Ong et al., 2000). It is primarily responsible for the metabolism of e.g., taxol, cerivastatin, rosiglitazone, and troglitazone and also involved in the metabolism of zopiclone, carbamazepine, verapamil, and amiodarone (Ohyama et al., 2000; Ong et al., 2000). The current results suggest that the metabolic activity of CYP2C8 in the liver can be inhibited markedly by administration of therapeutic doses of gemfibrozil. Thus, significant drug-drug interactions can be expected when gemfibrozil is coadministered with substrates of CYP2C8, in particular, if the substrates have narrow therapeutic ranges. Similarly, a significant inhibition of cerivastatin metabolism may also be expected when other CYP2C8 inhibitors [e.g., trimethoprim (Wen et al., 2002)] are coadministered with cerivastatin.
Lactonization is another important metabolic pathway of cerivastatin in humans, as evidenced by the approximate plasma levels of cerivastatin lactone and those of the M-1 and M-23 acid in vivo in humans (Kantola et al., 1999). However, lactonization of parent cerivastatin acid, M-1, and M-23 was negligible in the current in vitro conditions. Quite recently, during the final preparation of this manuscript, formation of the acyl glucuronide of cerivastatin was reported to be a significant metabolic pathway for cerivastatin in vitro (Prueksaritanont et al., 2002a,b). It was therefore assumed that cerivastatin glucuronide, and some other metabolic intermediates of cerivastatin [e.g., acyl-CoA thioester intermediates (Boberg et al., 1998)] may contribute to the formation of cerivastatin lactone in vivo (Prueksaritanont et al., 2002a). This would explain the different levels of cerivastatin lactonization observed in different in vitro conditions and in vivo.
Prueksaritanont et al. (2002b) also reported that gemfibrozil inhibited the formation of M-23 with an IC50 value of 87 μM. Although the exact mechanism of inhibition was not explored in their study, this result is in good agreement with our findings. In addition, they reported that the potency of inhibition of the formation of cerivastatin glucuronide (IC50 = 82 μM) was comparable with inhibition of the formation of M-23 (Prueksaritanont et al., 2002b). Coadministration of gemfibrozil and cerivastatin acid in vivo may inhibit not only the oxidative metabolism of cerivastatin acid but also the formation of cerivastatin glucuronide or the further metabolism of cerivastatin lactone. This may result in a more prominent inhibition of cerivastatin acid metabolism in vivo than that seen in the in vitro incubations.
To conclude, the present findings indicate that the inhibition of CYP2C8-mediated cerivastatin metabolism may contribute to the interaction between gemfibrozil and cerivastatin. Formation of M-23 acid from cerivastatin may be a useful tool for probing CYP2C8 activity both in vivo and in vitro. The in vivo interaction potential of gemfibrozil with other substrates of CYP2C8 warrants further exploration.
Footnotes
-
This study was supported by grants from the Helsinki University Central Hospital Research Fund and the National Technology Agency of Finland (Tekes).
- Abbreviations used are::
- P450
- cytochrome P450
- ISTD
- internal standard
- LC-MS-MS
- liquid chromatography-tandem mass spectrometry
- M-1
- desmethylcerivastatin
- M-23
- hydroxylated metabolite of cerivastatin
- M-24
- hydroxylated desmethylcerivastatin
- Received June 17, 2002.
- Accepted August 27, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}