


Abstract
Troglitazone glucuronidation in human liver and intestine microsomes and recombinant UDP-glucuronosyltransferases (UGTs) were thoroughly characterized. All recombinant UGT isoforms in baculovirus-infected insect cells (UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15) exhibited troglitazone glucuronosyltransferase activity. Especially UGT1A8 and UGT1A10, which are expressed in extrahepatic tissues such as stomach, intestine, and colon, showed high catalytic activity, followed by UGT1A1 and UGT1A9. The kinetics of the troglitazone glucuronidation in the recombinant UGT1A10 and UGT1A1 exhibited an atypical pattern of substrate inhibition when the substrate concentration was over 200 μM. With a Michaelis-Menten equation at 6 to 200 μM troglitazone, the Km value was 11.1 ± 5.8 μM and the Vmax value was 33.6 ± 3.7 pmol/min/mg protein in recombinant UGT1A10. In recombinant UGT1A1, theKm value was 58.3 ± 29.2 μM and theVmax value was 12.3 ± 2.5 pmol/min/mg protein. The kinetics of the troglitazone glucuronidation in human liver and jejunum microsomes also exhibited an atypical pattern. TheKm value was 13.5 ± 2.0 μM and theVmax value was 34.8 ± 1.2 pmol/min/mg for troglitazone glucuronidation in human liver microsomes, and theKm value was 8.1 ± 0.3 μM and theVmax was 700.9 ± 4.3 pmol/min/mg protein in human jejunum microsomes. When the intrinsic clearance was estimated with the in vitro kinetic parameter, microsomal protein content, and weight of tissue, troglitazone glucuronidation in human intestine was 3-fold higher than that in human livers. Interindividual differences in the troglitazone glucuronosyltransferase activity in liver microsomes from 13 humans were at most 2.2-fold. The troglitazone glucuronosyltransferase activity was significantly (r = 0.579, p < 0.05) correlated with the β-estradiol 3-glucuronosyltransferase activity, which is mainly catalyzed by UGT1A1. The troglitazone glucuronosyltransferase activity in pooled human liver microsomes was strongly inhibited by bilirubin (IC50 = 1.9 μM), a typical substrate of UGT1A1. These results suggested that the troglitazone glucuronidation in human liver would be mainly catalyzed by UGT1A1. Interindividual differences in the troglitazone glucuronosyltransferase activity in S-9 samples from five human intestines was 8.2-fold. The troglitazone glucuronosyltransferase activity in human jejunum microsomes was strongly inhibited by emodin (IC50 = 15.6 μM), a typical substrate of UGT1A8 and UGT1A10, rather than by bilirubin (IC50 = 154.0 μM). Therefore, it is suggested that the troglitazone glucuronidation in human intestine might be mainly catalyzed by UGT1A8 and UGT1A10.
Troglitazone is an antidiabetic agent that increases the insulin sensitivity of target tissues in noninsulin-dependent diabetes mellitus (Nolan et al., 1994; Fujiwara et al., 1998). Although troglitazone-associated hepatitis is thought to be rare, there are clinical reports of severe hepatic reactions associated with the use of troglitazone. About 1.9% of patients experience an increase in alanine aminotransferase levels greater than 3 times the upper limit of the normal range (Watkins and Whitcomb, 1998). Hepatic injury was reported to occur after short- and long-term troglitazone treatment in the United States (Physicians' Desk Reference, 2000) and after troglitazone treatment for a period longer than 4 weeks in Japan (Kuramoto et al., 1998). The hepatic toxicity of troglitazone was not observed in any experimental animals tested, including monkeys, which show similar metabolite profiles as humans (Summary Basis for Approvals, 1997).
Troglitazone has been reported to be metabolized to sulfate, glucuronide, and an oxidized quinone-type metabolite in human and monkeys (Physicians' Desk Reference, 2000). To clarify the mechanisms of liver injury induced by troglitazone, we previously performed several studies from the aspect of metabolism. We clarified that oxidation of troglitazone to the quinone-type metabolite is catalyzed by cytochrome P450 (P450)1 2C8 and 3A4 in human liver microsomes (Yamazaki et al., 1999) and that troglitazone and its quinone-type and sulfate conjugate metabolites, but not pioglitazone and rosiglitazone, exhibited inhibitory effects on human CYP2C and CYP3A4 activities (Yamazaki et al., 2000). Furthermore, we clarified that troglitazone induces apoptosis in human hepatoma cells (Yamamoto et al., 2001) and found a novel quinone epoxide metabolite with cytotoxicity in human hepatoma cells (Yamamoto et al., 2002). However, the mechanism by which troglitazone causes liver dysfunction in certain individuals is still unclear. Recently, it has been reported that the inhibition of troglitazone sulfation, which is recognized as a major metabolic pathway (Loi et al., 1999), may result in increased hepatotoxicity due to exposure to the parent drug or increased metabolism by alternate pathways (Kostrubsky et al., 2000).
Glucuronidation is catalyzed by UGT (Miners and Mackenzie, 1991). It is well known that there are many isoforms of mammalian UGT enzymes (Tukey and Strassburg, 2000). Human UGTs are expressed in a tissue-specific manner in hepatic and extrahepatic tissues (Strassburg et al., 1997). The distribution of individual UGT isoforms is believed to determine tissue-specific metabolism and detoxification. UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B4, UGT2B7, UGT2B8, UGT2B10, UGT2B15, and UGT2B17 are expressed in the liver (Burchell et al., 2001). UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A10, UGT2B7, UGT2B10, and UGT2B15 are expressed in human intestine (Strassburg et al., 2000; Tukey and Strassburg, 2000; Burchell et al., 2001). Until now, in humans, the troglitazone glucuronosyltransferase activity has been studied only in the liver (Yoshigae et al., 2000). It has been reported that troglitazone inhibits bilirubin glucuronidation (catalyzed by UGT1A1) and 1-naphthol glucuronidation (mainly catalyzed by UGT1A6) in human liver microsomes (Yoshigae et al., 2000; Ito et al., 2001). Furthermore, it has also been reported that recombinant UGT1A1, UGT1A4, UGT1A6, and UGT1A9 in microsomes from human B-lymphoblastoid cells have a capacity for troglitazone glucuronidation (Yoshigae et al., 2000). In the present study, the troglitazone glucuronosytrasferase activities in human intestinal S-9 and jejunum microsomes were compared with those in human liver microsomes. We first demonstrated that human jejunum microsomes exhibit prominently higher troglitazone glucuronosyltransferase activity than human liver microsomes. To identify the human UGT isoform(s) responsible for troglitazone glucuronidation, the activities in commercially available recombinant UGTs in microsomes from baculovirus-infected insect cells were determined. Furthermore, an inhibition analysis and correlation analysis using human liver and jejunum microsomes were performed.
Materials and Methods
Materials.
Troglitazone and troglitazone glucuronide were kindly provided by Sankyo (Tokyo, Japan). UDP-glucuronic acid, alamethicin, β-estradiol, emodin, p-nitrophenyl β-glucuronide, and α-naphthyl β-glucuronide were purchased from Sigma-Aldrich (St. Louis, MO). Bilirubin, 4-nitrophenol, 1-naphthol, and imipramine hydrochloride were from Wako Pure Chemicals (Osaka, Japan). Morphine hydrochloride was purchased from Takeda Chemical Industries (Osaka, Japan). Morphine-3-glucuronide was kindly provided by Dr. Kazuta Oguri (Kyusyu University, Fukuoka, Japan). Pooled human liver microsomes (H161) and microsomes from 13 individual human liver (H003, H023, H042, H043, H056, H064, H066, H089, H093, H095, H112, HK23, and HK34) were purchased from BD Gentest (Worburn, MA). The glucuronosyltransferase activities of β-estradiol and bilirubin in these human liver microsomes were provided as typical activities for UGT1A1 by the manufacturer. The glucuronosyltransferase activities of trifluoperazine and propofol in these human liver microsomes were also provided as typical activities for UGT1A4 and UGT1A9, respectively. Recombinant human UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15 expressed in baculovirus-infected insect cells (Supersomes) and control microsomes from insect cells infected with wild-type baculovirus were also from BD Gentest. Recombinant UGT1A1, UGT1A3, UGT1A6, UGT1A7, and UGT1A10 expressed in baculovirus-infected insect cells (Baculosomes) were from PanVera (Madison, WI). Polyclonal rabbit anti-human UGT1A antibodies were from BD Gentest. The human intestine specimens were provided by National Disease Research Interchange (Philadelphia, PA) through HAB Discussion Group (Chiba, Japan) according to the International Partnership between both nonprofit organizations. The intestinal S-9 fractions (HIS-052, HIS-053, HIS 078, HIS 084, and HIS-111) were prepared by HAB Biomedical Research Institute (Chiba, Japan). The human jejunum microsomes (HJM0040) were obtained from KAC (Shiga, Japan). All other chemicals and solvents were of the highest grade commercially available.
Troglitazone Glucuronidation Assay.
A typical incubation mixture (200-μl total volume) contained 25 mM Tris-HCl buffer, pH 7.4, 5 mM MgCl2, 3 mM UDP-glucuronic acid, 50 μg of alamethicin/mg microsomal, 1.0 mg/ml human liver microsomes (2 mg/ml human intestinal S-9, 0.5 mg/ml human jejunum microsomes, or 1 mg/ml for recombinant UGT), and 100 μM troglitazone. The reaction was initiated by the addition of troglitazone and the reaction mixture was incubated for 20 min. The reaction was then terminated by boiling for 10 min. After removal of the protein by centrifugation at 10,000 rpm for 5 min, a 100-μl portion of the sample was subjected to HPLC. Chromatography was performed using an LC-6A pump (Shimadzu, Kyoto, Japan), an SPD-6 UV detector (Shimadzu), an SIL-6B autosampler (Shimadzu), a C-R4A integrator (Shimadzu), and a CTO-6A column oven (Shimadzu) with a YMC-pack A302 column (4.6 × 150 mm, 5 μm; YMC, Kyoto, Japan). The flow rate was 1.5 ml/min and the column temperature was 35°C. The eluate was monitored at 230 nm with a noise-base clean Uni-3 (Union, Gunma, Japan). The Uni-3 can reduce the noise by integrating the output and both increase the signal 3-fold by differentiating the output and an additional 5-fold by further amplification with an internal amplifier, resulting in a maximum 15-fold amplification of the signal. The mobile phase was 39% CH3CN/0.05% H3PO4 (v/v). The retention times of troglitazone glucuronide and troglitazone were 6.2 and 50.0 min, respectively. None of these chromatograms showed any interfering peaks with troglitazone glucuronide (data not shown). The quantification of troglitazone glucuronosyltransferase activities was performed by comparing by the HPLC peak heights to those of authentic standards.
Kinetic Analyses.
The kinetics studies were performed using human liver microsomes, recombinant human UGT1A1 and UGT1A10 expressed in baculovirus-infected insect cells (Supersomes), and human jejunum microsomes. In determining the kinetic parameters, the troglitazone concentration ranged from 6 μM to 1 mM. Kinetic parameters were estimated from the fitted curves using a computer program of KaleidaGraph (Synergy Software, Reading, PA) designed for nonlinear regression analysis. The following equations were applied for Michaelis-Menten kinetics, with eq. 1 using the activities at 6 to 200 μM troglitazone or substrate inhibition kinetics, and eq. 2 using the activities at 6 μM to 1 mM troglitazone:
Metabolite Inhibition Analysis of Troglitazone Glucuronosyltransferase Activity in Human Liver Microsomes.
The inhibitory effects of troglitazone glucuronide per se on the troglitazone glucuronosyltransferase activity in the human liver microsomes were determined. Troglitazone glucuronide was added as an inhibitor to the incubation mixture at a final concentration of 0.06 to 1 μM. Troglitazone glucuronosyltransferase activity in the pooled human liver microsomes at 100 μM troglitazone was calculated with the formed metabolite by subtracting the content of the metabolite added as an inhibitor from the total content of metabolite.
Inhibition Analysis of Troglitazone Glucuronosyltransferase Activity in Human Liver and Jejunum Microsomes.
Six compounds were tested for their inhibitory effects on the troglitazone glucuronosyltransferase activity. Bilirubin is a typical substrate for UGT1A1 (Bosma et al., 1994; Senafi et al., 1994; King et al., 1996). β-Estradiol is a typical substrate for UGT1A1 (and UGT1A9 as a minor enzyme) (Bosma et al., 1994; Senafi et al., 1994; Hanioka et al., 2001a,b). 4-Nitrophenol is a substrate for UGT1A6 and UGT1A9 (Hanioka et al., 2001a,b). Imipramine is a substrate for UGT1A4 (and UGT1A3 as a minor enzyme) (Green et al., 1995, 1998). Emodin is a substrate for UGT1A8 and UGT1A10 (Cheng et al., 1999). Morphine is a substrate for UGT2B7 (Coffman et al., 1997). For the determination of the IC50 values, the concentration of troglitazone was set at 100 μM. Bilirubin and 4-nitrophenol were dissolved in dimethyl sulfoxide and ethanol, respectively. β-Estradiol and emodin were dissolved in methanol. Imipramine hydrochloride, and morphine hydrochloride were dissolved in water. The final concentration of the organic solvents in the reaction mixture was <1% (v/v). The troglitazone glucuronosyltransferase activities in pooled human liver microsomes (H161) and in individual human jejunum microsomes (HJM0040) at 100 μM troglitazone were determined as described above.
Other Glucuronidation Assays.
4-Nitrophenol glucuronosyltransferase activity in human liver microsomes was determined as described previously (Hanioka et al., 2001a). Briefly, a typical incubation mixture (200-μl total volume) contained 50 mM potassium phosphate buffer, pH 7.4, 0.2% Triton N-101, 3 mM UDP-glucuronic acid, 1.0 mg/ml human liver microsomes, and 500 μM 4-nitrophenol. The reaction was initiated by the addition of UDP-glucuronic acid and was then incubated at 37°C for 5 min. The reaction was terminated by boiling at 100°C for 2 min and adding 2.8 ml of 0.2 M glycine buffer, pH 10.4. After removal of the protein by centrifugation at 2000 rpm for 20 min, a 50-μl portion of the sample was subjected to HPLC. The HPLC instrument was the same as described above. Chromatographic separations were performed on a Mightysil RP-18 (4.6 × 150 mm; 5 μm) column (Kanto Chemical, Tokyo, Japan). The flow rate was 1.2 ml/min and the column temperature was 35°C. The eluate was monitored at 302 nm by the UV detector. The mobile phase was 5% CH3OH/0.05M KH2PO4, pH 6.5. The retention times of 4-nitrophenol glucuronide and 4-nitrophenol were 3.0 and 14.5 min, respectively. Formation of the metabolite was quantified by comparing the peak areas in the incubations to a standard curve containing known amounts of the metabolite.
1-Naphthol glucuronosyltransferase activity in human liver microsomes was determined as described previously (Mackenzie and Hanninen, 1980). Briefly, a typical incubation mixture (200-μl total volume) contained 50 mM potassium phosphate buffer, pH 7.4, 0.2% Triton N-101, 3 mM UDP-glucuronic acid, 0.5 mg/ml human liver microsomes, and 50 μM 1-naphthol. The reaction was initiated by the addition of UDP-glucuronic acid followed by incubation at 37°C for 2.5 min. The reaction was terminated by boiling for 2 min and adding 1.8 ml of phosphate-buffered saline. After removal of the protein by centrifugation at 2000 rpm for 20 min, the fluorescence of the supernatant was measured at excitation 290 nm and emission 343 nm by an RF5000 fluorescence detector (Shimadzu). Formation of the metabolite was quantified by comparing the fluorescence in the incubations to a standard curve containing known amounts of the metabolite.
Morphine glucuronosyltransferase activity in human liver microsomes was determined as described previously (Milne et al., 1991). Briefly, a typical incubation mixture (250-μl total volume) contained 50 mM potassium phosphate buffer, pH 7.4, 0.2% Triton N-101, 3 mM UDP-glucuronic acid, 1.0 mg/ml human liver microsomes, and 1 mM morphine. The reaction was initiated by the addition of UDP-glucuronic acid and was then incubated at 37°C for 30 min. The reaction was terminated by adding 250 μl of ice-cold acetonitrile. After removal of the protein by centrifugation at 2000 rpm for 20 min, a 50-μl portion of the sample was subjected to HPLC. The HPLC instrument was the same as described above. Chromatographic separations were performed on a Mightysil RP-18 (4.6 × 150 mm; 5 μm) column (Kanto Chemical). The flow rate was 1.2 ml/min and the column temperature was 35°C. The mobile phase was 20% CH3CN/0.8 mM sodium dodecyl sulfate/0.05 M KH2PO4, pH 1.9. The eluate was monitored at excitation 210 nm and emission 350 nm by an 821-FPS fluorescence detector (Jasco, Tokyo, Japan). The retention times of morphine 3-glucuronide and morphine were 3.1 and 8.0 min, respectively. Formation of the metabolite was quantified by comparing the peak areas in the incubations to a standard curve containing known amounts of the metabolite.
Immunoblot Analysis of Recombinant UGT 1A Isoforms.
SDS-polyacrylamide gel electrophoresis and immunoblot analysis of recombinant UGT1A isoforms were performed according to Laemmli (1970). The microsomes from baculovirus-infected insect cells (2 μg) were separated on 7.5% polyacrylamide gel and transferred electrophoretically to a nitrocellulose membrane. Polyclonal anti-human UGT1A antibodies have been reported to detect human UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A9, and UGT1A10 by the manufacturer. Biotinylated anti-rabbit IgG and VECTASTAIN ABC kit (Vector Laboratories, Burlingame, CA) were used for diaminobenzidine staining.
Correlation Analyses.
Correlation analyses between troglitazone glucuronidation and the other glucuronidation activities were determined by Pearson's product moment method. A p value of less than 0.05 was considered statistically significant.
Results
Troglitazone Glucuronidation in Recombinant UGT Isoforms.
Ten recombinant UGT isoforms expressed in baculovirus-infected insect cells were used to determine their troglitazone glucuronosyltansferase activities (Fig. 1). In Supersomes, UGT1A10 (31.0 ± 0.2 pmol/min/mg protein) and UGT1A8 (17.6 ± 3.3 pmol/min/mg protein) exhibited high troglitazone glucuronosyltransferase activities (Fig. 1A). All other isoforms (UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, GT2B7, and UGT2B15) exhibited low troglitazone glucuronosyltransferase activities. In Baculosomes, UGT1A10 (56.4 ± 3.5 pmol/min/mg protein) exhibited the highest troglitazone glucuronosyltransferase activities (Fig. 1B). Other UGTs also exhibited weak glucuronosyltransferase activities.

Troglitazone glucuronosyltransferase activity in recombinant human UGTs in baculovirus-infected insect cells and immunoblot analysis of the recombinant UGT1A isoforms.
Troglitazone glucuronosyltransferase activities in the recombinant UGT in baculovirus-infected insect cells of Supersomes from BD Gentest (A) or Baculosomes from PanVera (B) were determined at 100 μM troglitazone. Each column represents the mean ± S.D. of triplicate determinations. Immunoblot analysis of the recombinant UGT1A isoforms was performed using anti-human UGT1A antibodies (C). Each lane contains 2 μg of microsomal protein.
Immunoblot Analysis of Recombinant UGT 1A Isoforms.
In Supersomes, band density with all UGT1A isoforms was almost similar except for UGT1A3. In Baculosomes, UGT1A3, UGT1A6, and UGT1A10 exhibited weak detectable band, although UGT1A1 and UGT1A7 exhibited a clear band.
Kinetics of Troglitazone Glucuronidation in Recombinant UGT1A1 and UGT1A10 (Supersomes).
UGT1A1 is a major isoform in human liver (Bock et al., 1999). UGT1A8 and UGT1A10 are both expressed in the intestine but not in the liver, and the substrate specificity of these isoforms overlaps (Radominska-Pandya et al., 1999). Therefore, kinetic analyses of troglitazone glucuronidation in recombinant UGT1A1 and UGT1A10 (Supersomes) were performed. As shown in Fig.2A, the kinetics of troglitazone glucuronidation in recombinant UGT1A1 at 6 to 200 μM troglitazone fitted to the Michaelis-Menten kinetics. However, when the troglitazone concentration exceeded 200 μM, the troglitazone glucuronosyltransferase activity gradually decreased. A possible explanation for this phenomenon is substrate inhibition or metabolite inhibition. When the apparent enzyme kinetic parameters were estimated by fitting to the Michaelis-Menten eq. 1 with the initial velocity values at 6 to 200 μM troglitazone, theKm value was 58.3 ± 29.2 μM and Vmax was 12.3 ± 2.5 pmol/min/mg protein (Table 1). When the apparent enzyme kinetic parameters were estimated by fitting to the substrate inhibition eq. 2 with the velocity values at 6 μM to 1 mM troglitazone, the Km value was 141.1 ± 85.8 μM and Vmax value was 23.8 ± 9.9 pmol/min/mg protein. As shown in Fig. 2B, the kinetics of troglitazone glucuronidation in recombinant UGT1A10 at 6 to 200 μM troglitazone fitted to the Michaelis-Menten kinetics. However, when the troglitazone concentration exceeded 100 μM, the troglitazone glucuronosyltransferase activity decreased. When the apparent enzyme kinetic parameters were estimated by fitting to the Michaelis-Menten eq. 1 with the initial velocity values at 6 to 200 μM troglitazone, the Km value was 11.1 ± 5.8 μM and Vmax value was 33.6 ± 3.7 pmol/min/mg protein. When the apparent enzyme kinetic parameters were estimated by fitting to the substrate inhibition eq. 2 with the velocity values at 6 μM to 1 mM troglitazone, theKm value was 497.1 ± 2793.3 μM and Vmax value was 532.5 ± 2828.1 pmol/min/mg protein.

Kinetics of troglitazone glucuronidation in recombinant UGT1A1 (A) or UGT1A10 (B) in microsomes from baculovirus-infected insect cells (Supersomes).
The solid line represents the fitting curve to the Michaelis-Menten eq.1 (6–200 μM, closed circles). The dotted line represents the fitting curve to the substrate inhibition eq. 2 (6 μM–1 mM, closed and open circles). The troglitazone glucuronosyltransferase activity was determined as described under Materials and Methods. Each data point represents the mean of duplicate determinations.
Kinetic parameters of troglitazone glucuronidation in recombinant UGT isoforms and in human liver and jejunum microsomes
Kinetics of Troglitazone Glucuronidation in Human Liver or Jejunum Microsomes.
Kinetic analyses of troglitazone glucuronidation in human liver (H161) or jejunum (HJM0040) microsomes were performed. As shown in Fig.3, the kinetics at 6 to 200 μM troglitazone fitted to the Michaelis-Menten kinetics. However, when the troglitazone concentration exceeded 200 μM, the troglitazone glucuronosyltransferase activity gradually decreased. In human jejunum microsomes, troglitazone glucuronide was not detected at 750 μM and 1 mM troglitazone (Fig. 3B). When the apparent enzyme kinetic parameters were estimated by fitting to the Michaelis-Menten eq. 1 with the initial velocity values at 6 to 200 μM troglitazone, theKm values in human liver and jejunum microsomes were 13.5 ± 2.0 and 8.1 ± 0.3 μM andVmax values were 34.8 ± 1.2 and 700.9 ± 4.3 pmol/min/mg protein, respectively (Table 1). When the apparent enzyme kinetic parameters were estimated by fitting to the substrate inhibition eq. 2 with the velocity values at 6 μM to 1 mM troglitazone, the Km values in human liver and jejunum microsomes were 49.4 ± 26.3 and 62.7 ± 122.8 μM and Vmax values were 70.2 ± 22.2 and 2138.5 ± 2990.4 pmol/min/mg protein, respectively.
Kinetics of troglitazone glucuronidation in human liver (H161) (A) or jejunum (HJM0040) (B) microsomes.
The solid line represents the fitting curve to the Michaelis-Menten eq.1 (6 μM-200 μM, closed circles). The dotted line represents the fitting curve to the substrate inhibition eq. 2 (6 μM–1 mM, closed and open circles). The troglitazone glucuronosyltransferase activity was determined as described under Materials and Methods. Each data point represents the mean of duplicate determinations.
Metabolite Inhibition of Troglitazone Glucuronosyltransferase Activity in Human Liver Microsomes.
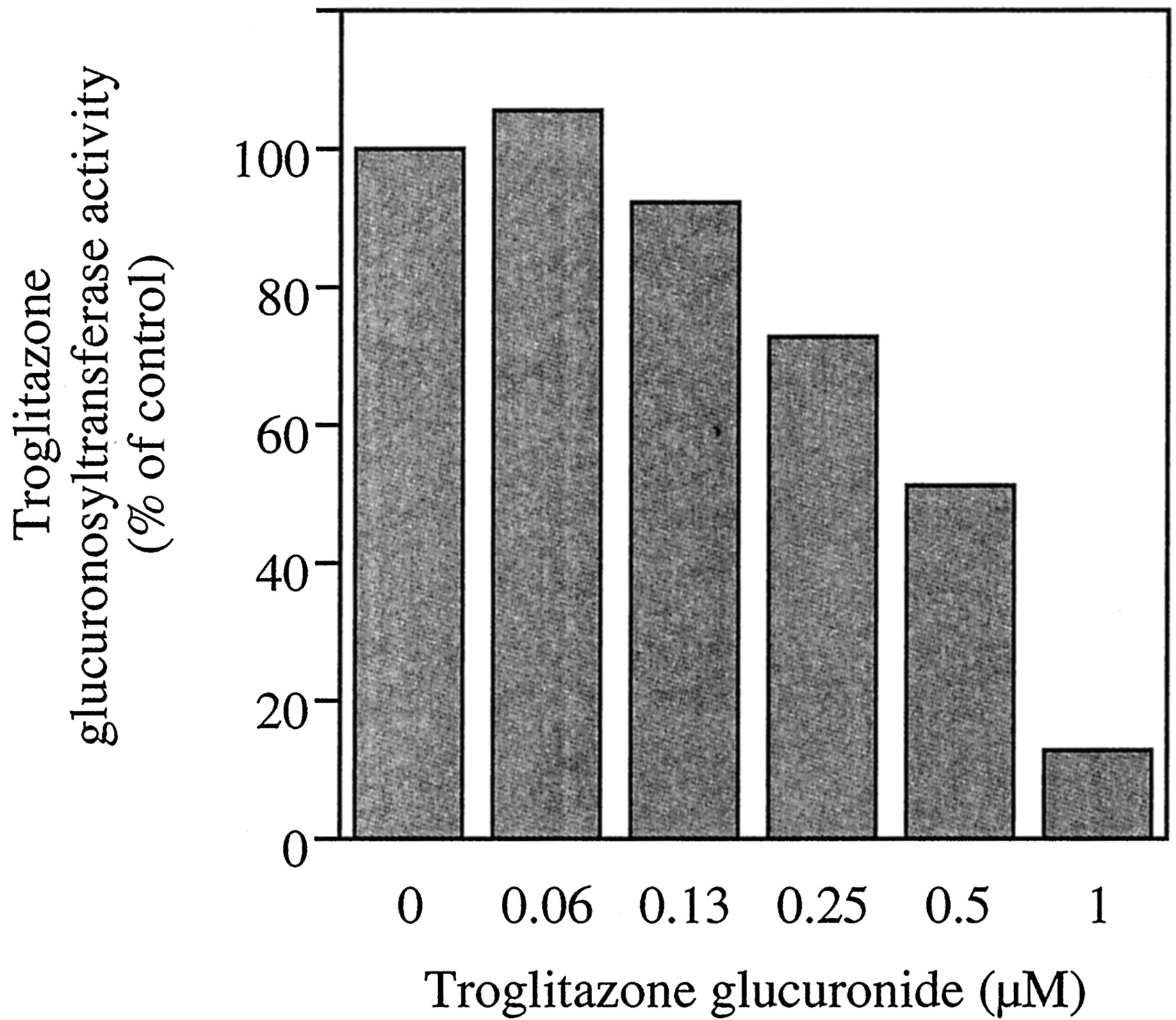
The inhibitory effects of troglitazone glucuronide per se on the troglitazone glucuronosyltransferase activity in the pooled human liver microsomes were determined. Troglitazone glucuronide strongly inhibited the troglitazone glucuronosyltransferase activity in human liver microsomes (Fig. 4), giving the IC50 value of 0.5 μM. We confirmed that the incubation of troglitazone glucuronide with the human liver microsomes and UDP-glucuronic acid, but without troglitazone did not affect the concentration of the added troglitazone glucuronide under the present experimental condition, suggesting that there was no further metabolism of troglitazone glucuronide.

Metabolite (troglitazone glucuronide) inhibition of troglitazone glucuronosyltransferase activity in human liver microsomes.
Troglitazone glucuronide was added to the incubation mixture as an inhibitor. Troglitazone glucuronosyltransferase activity in pooled human liver microsomes (H161) was determined at 100 μM as described under Materials and Methods. Each data point represents the mean of duplicate determinations. The troglitazone glucuronosyltransferase activity in the pooled human liver microsomes in the absence of the inhibitor was 34.2 pmol/min/mg protein.
Interindividual Variability of Troglitazone Glucuronosyltransferase Activity from 13 Human Livers and Correlation Analyses.
Troglitazone glucuronosyltransferase activities in microsomes from 13 human livers were determined at 100 μM troglitazone (Fig.5A). The interindividual difference in troglitazone glucuronosyltransferase activity was at most 2.2-fold (12.0 ± 1.7–27.1 ± 2.3 pmol/min/mg protein). Correlation analyses were performed between the troglitazone glucuronosyltransferase activity and β-estradiol (UGT1A1), bilirubin (UGT1A1), trifluoperazine (UGT1A4), or propofol glucuronosyltransferase activities provided by the manufacturer, or 4-nitrophenol (UGT1A6), 1-naphthol (UGT1A1, UGT1A6, UGT1A8, and UGT1A9), or morphine (UGT2B7) glucuronosyltransferase activities determined in the present study. The troglitazone glucuronosyltransferase activities in the 13 human liver microsomes were significantly (r = 0.579,p < 0.05) correlated with the β-estradiol 3-glucuronosyltransferase activities (Fig. 5B). However, the troglitazone glucuronosyltransferase did not correlated with the bilirubin glucuronosyltransferase activities (r = 0.487, p = 0.154), whereas the bilirubin glucuronosyltransferase activities were highly correlated with the β-estradiol 3-glucuronosyltransferase activities (r = 0.927, p < 0.001). In addition, the troglitazone glucuronosyltransferase activities did not correlate with the trifluoperazine (r = 0.310, p = 0.302), propofol (r = 0.453, p = 0.120), 4-nitrophenol (r = 0.306, p = 0.361), 1-naphthol (r = 0.172, p = 0.613), or morphine (r = 0.427, p = 0.160) glucuronosyltransferase activities (data not shown).

Interindividual variability in troglitazone glucuronosyltransferase activity in microsomes from 13 human livers.
A, troglitazone glucuronosyltransferase activities in microsomes from 13 human livers were determined at 100 μM troglitazone. Each column represents the mean ± S.D. of triplicate determinations. B, correlation analysis between troglitazone glucuronosyltransferase activity and β-estradiol 3-glucuronosyltransferase activity in microsomes from 13 human livers.
Troglitazone Glucuronosyltransferase Activity in Human Intestinal S-9 Sample.
Troglitazone glucuronosyltransferase activities in S-9 samples from five human intestines were determined (Fig.6). The interindividual difference in the troglitazone glucuronosyltransferase activity was about 8.2-fold (0.5–4.1 pmol/min/mg protein).

Troglitazone glucuronosyltransferase activity in human intestinal S-9.
The troglitazone glucuronosyltransferase activity in S-9 samples from five human intestines was determined at 100 μM troglitazone. Each column represents the mean of duplicate determinations.
Inhibition Analyses of Troglitazone Glucuronidation in Human Liver or Jejunum Microsomes.
The inhibitory effects of bilirubin (UGT1A1), β-estradiol (UGT1A1 and UGT1A9), 4-nitrophenol (UGT1A6 and UGT1A9), imipramine (UGT1A3 and UGT1A4), emodin (UGT1A8 and UGT1A10), and morphine (UGT2B7) on the troglitazone glucuronosyltransferase activity in human liver or jejunum microsomes were determined. As shown in Fig.7A, the troglitazone glucuronosyltransferase activity in the pooled human liver microsomes was prominently inhibited by bilirubin (IC50 = 19.0 μM). In contrast, the inhibitory effects of emodin (IC50 = 134.0 μM), β-estradiol (IC50 = 261.2 μM), 4-nitrophenol (IC50 = 379.2 μM), imipramine (IC50 > 500 μM), and morphine (IC50 > 500 μM) were weak. As shown in Fig.7B, the activity in human jejunum microsomes was prominently inhibited by emodin (IC50 = 13.7 μM). The inhibitory effects of bilirubin (IC50 = 154.0 μM), β-estradiol (IC50 = 361.8 μM), 4-nitrophenol (IC50 = 486.5 μM), imipramine (IC50 > 500 μM), and morphine (IC50 > 500 μM) were weak.
Inhibitory effects of typical substrates for UGT isoforms on troglitazone glucuronosyltransferase activity in human liver (A) or jejunum (B) microsomes.
Troglitazone glucuronosyltransferase activities at 100 μM troglitazone in pooled human liver microsomes (H161) and human jejunum microsomes (HJM0040) were determined as described under Material and Methods. Bilirubin (UGT1A1), β-estradiol (UGT1A1 and UGT1A9), 4-nitrophenol (UGT1A6 and UGT1A9), imipramine (UGT1A3 and UGT1A4), emodin (UGT1A8 and UGT1A10), and morphine (UGT2B7) were used as inhibitors. The troglitazone glucuronosyltransferase activities in the pooled human liver and jejunum microsomes in the absence of inhibitor were 32.5 and 650.6 pmol/min/mg protein, respectively.
Discussion
All UGT isoforms expressed in human liver, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7, and UGT2B15, exhibited troglitazone glucuronosyltransferase activity. With immunoblot analysis, relative expression levels of recombinant UGT1A isoforms were determined. Microsomes from UGT1A3 Supersomes, UGT1A3, UGT1A6, and UGT1A10 Baculosomes showed weak band in comparison with the other UGT1A isoforms. It might be due to low expression level or low reactivity of the antibodies to the expression systems. In Supersomes, high troglitazone glucuronosyltransferase activities in UGT1A8 and UGT1A10 were not due to the differences in the expression levels. In Baculosomes, UGT1A10 exhibited high troglitazone glucuronosyltransferase activity in spite of weak immunoreactive band. The troglitazone glucuronosyltransferase activity in human liver microsomes was significantly correlated only with the β-estradiol 3-glucuronosyltransferase activity that was mainly catalyzed by UGT1A1. Furthermore, the activity in human liver microsomes was strongly inhibited by bilirubin, a specific substrate for UGT1A1. These results suggest that the troglitazone glucuronidation in human liver microsomes might be mainly catalyzed by UGT1A1. It has been reported that in Gunn rats, which are hereditarily deficient in the UGT1 family, the metabolic profile of troglitazone was much the same as that of Wistar rats (Watanabe et al., 2000), suggesting no contribution of UGT1 family to troglitazone glucuronidation. Concerning the contradiction with our results, the possibility is considered that other UGT isoforms might compensate for UGT1 family in Gunn rats or that the UGT isoforms in rats which are responsible for troglitazone glucuronidation might differ from those in humans as Yoshigae et al. (2000) have also reported.
To our knowledge, this is the first study to show that UGT1A8 and UGT1A10 have high troglitazone glucuronosyltransferase activity. Because UGT1A8 and UGT1A10 are not expressed in liver, these isoforms could be responsible for the extrahepatic metabolism of troglitazone particularly in the gastrointestinal tract. As shown in Fig. 7, the troglitazone glucuronosyltransferase activity in human jejunum microsomes was prominently inhibited by emodin, a typical substrate of UGT1A8 and UGT1A10. Therefore, the involvement of UGT1A8 and UGT1A10 in the troglitazone glucuronidation in human intestine was also indicated.Strassburg et al. (2000) reported that the expressions of UGT1A1, UGT1A3, UGT1A4, and UGT1A6 were polymorphic in intestine. Therefore, the polymorphic expression might be one of the factors of the interindividual variation in the troglitazone glucuronosyltransferase activity in human intestines.
In humans, troglitazone glucuronosyltransferase activity has been studied in the liver (Yoshigae et al., 2000). However, the activity in extrahepatic tissues has not been determined. In the present study, we compared the troglitazone glucuronosyltransferase activity in human jejunum microsomes with that in human liver microsomes. We first demonstrated that the kinetics of troglitazone glucuronidation in human liver and jejunum microsomes is an atypical type of substrate inhibition at higher substrate concentrations (>200 μM). Although the solubility of troglitazone decreased at concentrations >200 μM, the phenomenon could not explain the atypical kinetics. The kinetic curves successfully fitted to the substrate inhibition equation. Furthermore, it was also indicated that the troglitazone glucuronosyltransferase activity is inhibited by troglitazone glucuronide per se (Fig. 3). The concentrations of troglitazone glucuronide added as an inhibitor in the metabolite inhibition analysis corresponded to the amounts of troglitazone glucuronide formed in usual assay conditions. Therefore, the metabolite inhibition as well as the substrate inhibition may be responsible for the atypical kinetics.Yoshigae et al. (2000) reported that the troglitazone glucuronidation in human liver microsomes did not become saturated at concentrations up to 200 μM troglitazone. Nevertheless the activity in human liver microsomes at 200 μM troglitazone was 32.9 ± 1.9 pmol/min/mg protein, which was similar to our results (Fig. 2). The reason for the contradiction in the kinetic pattern with our present data is unknown.
The maximum plasma concentrations of troglitazone have been reported to be 3.6 and 6.3 μM in humans taking troglitazone at therapeutic doses of 400 and 600 mg/day, respectively (Loi et al., 1999). In clinical use, the concentration of troglitazone is unlikely to reach 200 μM. Therefore, we adopted the kinetic parameter fitted to the Michaelis-Menten equation with <200 μM troglitazone. When the substrate concentration is sufficiently low, the in vitro clearance is expressed asVmax/Km. The clearance of troglitazone glucuronidation in human liver and intestine was 2.58 and 86.53 μl/min/mg protein, respectively (Table1). To estimate the in vitro intrinsic clearance (CLint), the following formula is used (Obach et al., 1997; Soars et al., 2002):
Troglitazone sulfate and glucuronide are excreted into bile. Both metabolites are converted back to the parent compound, troglitazone, in the small intestine by arylsulfotransferase and β-glucuronidase. In the enterohepatic circulation troglitazone would be converted to glucuronide mainly by UGT1A1 in human livers. Furthermore, in enterocytes, it might also be converted to glucuronide by UGTs such as UGT1A8 and UGT1A10. Using knockout rats of multidrug-resistant associated protein-2 (Mrp-2), it has been demonstrated that troglitazone glucuronide is a substrate of Mrp2 (Kostrubsky et al., 2001). Therefore, the troglitazone glucuronide formed in enterocytes might be excreted to the intestinal lumen via transporters such as Mrp2 expressed in the brush-border membrane. Then, the glucuronide would again be converted to troglitazone by β-glucuronidase and the troglitazone might be reabsorbed.
In conclusion, it was demonstrated that the clearance of troglitazone glucuronidation in human intestines is higher than that in human livers. UGT1A8 and UGT1A10 as well as UGT1A1 would significantly contribute to the troglitazone glucuronidation in human intestines.
Acknowledgments
We thank Dr. Tetsuo Satoh (HAB Biomedical Research Institute, Chiba, Japan) for providing human intestinal S-9 samples and Dr. Kazuta Oguri (Kyusyu University, Fukuoka, Japan) for providing morphine-3-glucuronide. We acknowledge Brent Bell for reviewing the manuscript.
Footnotes
- Abbreviations used are::
- P450
- cytochrome P450
- UGT
- UDP-glucuronosyltransferase
- HPLC
- high-performance liquid chromatography
- Mrp-2
- multidrug-resistant associated protein-2
- Received July 9, 2002.
- Accepted September 5, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}