


Abstract
CYP2C8 with a modified N-terminal sequence (2C8H) crystallizes as a dimer, but it is not known whether native CYP2C8 exists as a dimer in natural membranes. We have examined the organization of 2C8H and CYP2C8 expressed in bacterial membranes and mammalian endoplasmic reticulum membranes, respectively, by cysteine scanning and cross-linking or oxidation of sulfhydryl groups. In both forms of CYP2C8, cross-linked dimers were observed that were eliminated by mutation of Cys-24 in the linker region. Introduction of individual cysteines in the N-terminal 21-amino acid membrane-spanning signal anchor resulted in a pattern of cross-linking consistent with an α-helical structure for the signal anchor. In the linker region, cross-linking was observed for cysteine substituted at residues 22, 23, or 24, just before three Arg residues, indicating close apposition of the two linker sequences despite the neighboring positive charges. Introduction into the F-G loop region of cysteine pairs optimally located for cross-linking based on the crystal structure resulted in cross-linked dimers in the Cys-24 mutant. Deletion of the signal anchor sequence eliminated cross-linking mediated by Cys-24 or by cysteines introduced in the F-G loop regions, indicating that the signal anchor interaction is required for stable dimer formation. These results indicate that the signal anchor sequence and the F-G loop region form interfaces for CYP2C8 intermolecular interactions in natural membranes.
Introduction
Microsomal cytochromes P450 (P450s) are integral membrane proteins located in the endoplasmic reticulum (ER). The N-terminal signal anchor sequence is the single membrane-spanning helix in P450s (Sakaguchi et al., 1987; Shimozawa et al., 1993; Szczesna-Skorupa and Kemper, 1993), but the catalytic domain of the protein probably penetrates partially into the membrane (Williams et al., 2000a; Schleinkofer et al., 2005). The orientation and organization of P450s in the membrane are important for their function (Ohta et al., 1992, 1994). A second integral membrane protein, cytochrome P450 reductase donates electrons to P450s in their catalytic cycles so both proteins must be oriented properly for optimal electron transfer (Black and Coon, 1982). Cytochrome b5 also interacts with some P450s and enhances their activity (Pompon and Coon, 1984). In addition, substrates of P450s are often highly hydrophobic and concentrated in the membrane, and in some models of P450s, the substrate access channel is oriented toward the membrane so that the substrate can enter the active site from the lipid membrane environment (Williams et al., 2003; Scott et al., 2004; Wester et al., 2004). Despite its importance for their function, there is little direct evidence demonstrating the orientation and organization of P450s in natural membranes.
Crystal structures were determined initially for soluble bacterial forms and later for solubilized mammalian forms with the N-terminal signal anchor sequences deleted (reviewed in Poulos and Johnson, 2005). All of the P450s have a similar three-dimensional folding structure. From the structures or by homology modeling, the orientation of the catalytic domain bound to the membrane has been predicted for several P450s based on the distribution of hydrophobic residues on the surface of the molecules (Williams et al., 2000b). Residues predicted to insert into the membrane include 30 to 45 before helix A, 60 to 69 after helix A, 376 to 379 in β strand 2-2, and 211 to 228 in the F-G loop (numbered according to CYP2C8). Studies of the residues of CYP2C2 that were embedded in the membrane by analyzing changes in the fluorescence of Trp residues that were substituted in the protein were largely consistent with the predicted membrane interactions (Ozalp et al., 2006). An exception was a Trp in the F-G loop region, residue Trp-225, which behaved as if it were in an aqueous environment, rather than the predicted membrane environment. In the crystal structure of CYP2C8, which crystallized as a dimer, the F-G loop formed the dimer interface (Schoch et al., 2004). In the dimer, the F-G loop was predicted to be outside of the membrane (Fig. 1A), which would be consistent with the Trp fluorescence studies (Ozalp et al., 2006). The Trp studies used purified P450s and artificial membranes, and the formation of the dimer in crystals occurred with solubilized protein with the signal anchor deleted and at high concentrations. An important question is whether CYP2C8 bound to natural membranes would form dimers as predicted by the crystal structure.

A, the structure of 2C8dH (Protein Data Bank 1PQ2), which crystallizes as a dimer, is shown with atoms displayed as spheres. Charged residues are black, and hydrophobic residues are gray. The lines representing the surface of the membrane at the protein membrane interface are shown as proposed for P450s as monomers and dimers (Ozalp et al., 2006). The structure was rendered using the Chimera program of the University of California at San Francisco. B, a ribbon diagram depicts the interaction of F-G loop and F and G helices at the dimeric interface based on the 2C8dH crystal structure produced with PyMOL. Trp-212 and Cys-225 and Arg-206 and Gly-226 are the closest residue pairs based on the distance of their α carbons, which are shown in stick form. Cys-216, the other Cys in the F-G loop besides Cys-225, is also shown in stick form. C, sequences of the N-terminal regions of the CYP2C8 constructs with the Cys residues underlined. Native CYP2C8 (2C8) was expressed in human AD-293 cells. 2C8H contains a modified N-terminal signal anchor sequence, 2C8dH has the signal anchor and linker sequences deleted, and 2C8dH+C has the signal anchor sequence deleted. 2C8H, 2C8dH, and 2C8dH+C were expressed in E. coli XL-1. The MAKKTSSKG sequence at the N terminus of 2C8dH and 2C8dH+C increases their expression in bacteria and promotes solubility.
There are few studies of the oligomerization of P450s in native membranes. Analysis of the interactions of P450 fluorescent protein hybrids by FRET and BiFC in living cells demonstrated that CYP2C2 formed homo-oligomers, whereas CYP2E1 did not and that the homo-oligomerization was dependent on the signal anchor sequence (Szczesna-Skorupa et al., 2003; Ozalp et al., 2005). These studies could not distinguish whether the oligomers were dimers or higher order oligomers. Solubilized P450s also have been shown to form homo-oligomers containing up six or eight protein molecules in some cases, which were mediated by the signal anchor sequence (Von Wachenfeldt and Johnson, 1995). Interactions among P450s may have functional significance because coexpression of a second P450 with a P450 can either inhibit or increase the activity of the first P450 (Cawley et al., 2001; Hazai and Kupfer, 2005; Subramanian et al., 2009, 2010; Reed et al., 2010). The functional significance of homo-oligomerization is not clear, but oligomerization of CYP3A4 has been shown to decrease reduction of the P450 by dithionite (Davydov et al., 2005) or the soluble flavin domain of P450BM-3 (Davydov et al., 2010).
In the present study, we examined the organization in native membranes of CYP2C8 expressed in bacterial and mammalian cells by Cys oxidation or maleimide cross-linking. These results indicate that the signal anchor sequence and the F-G loop region form interfaces for dimers of CYP2C8 bound to natural membranes.
Materials and Methods
Reagents.
Copper sulfate, 1,10-phenathroline, and N-ethylmaleimide were purchased from Sigma-Aldrich (St. Louis, MO), bis-maleimidoethane (BMOE) was from Thermo Fisher Scientific (Waltham, MA), and anti-His antibody and anti-Flag antibody were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Bacterial Strains and Plasmids.
Coding sequences for 2C8H and 2C8dH with His tags were inserted into the pCWori+ vector for expression in Escherichia coli as described previously (Richardson et al., 1995). 2C8H (Schoch et al., 2008) contains a modified signal anchor sequence and in 2C8dH (Schoch et al., 2008), the signal anchor sequence is deleted (Fig. 1C). These proteins were expressed in E. coli XL-1 Blue (Stratagene, La Jolla, CA). The coding sequence for native CYP2C8 was inserted into pCMV-5 and was expressed in AD-293 mammalian cells (Stratagene). C13S and C24S mutants were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) with the 2C8H expression vector as the template. To make F-G loop mutants C24S/W212C and C24S/R206C/G228C, the C24S expression plasmid was used as the template. 2C8H(Cys-), in which 7 Cys residues were mutated, was generated by consecutive cysteine substitutions of C13S, C24S, C51S, C216S, C225Y, C164S, and C338S beginning with the 2C8H expression plasmid as the template. Cys-225 was replaced with Tyr rather than Ser because Tyr is conserved at this position in other CYP2 proteins. To determine the proximity of the signal anchor sequences or the linker sequences to each other in neighboring 2C8H molecules, residues of the signal anchor sequence from Leu-11 to Trp-20 or residues of linker sequence from Gln-22 to Ser-24 were substituted with Cys individually. Likewise, 2C8(Cys-) expressed in AD-293 cells was made by progressive mutagenesis, following the order from C13S, C24S, C51S, C225Y, C164S, and C338S using the native CYP2C8 expression plasmid as the starting template. To construct the 2C8dH+C plasmid, 2C8dH+C cDNA was amplified by polymerase chain reaction with 2C8H as the template and a 5′ primer, which contained sequence coding for MAKKTSSKG before the coding for WRQSC in 2C8H. The polymerase chain reaction fragment was cloned into the NdeI/HindIII sites of pCWori+. All constructions were sequenced to verify that only desired mutations were present (UIUC Core Sequencing Facility, University of Illinois at Urbana-Champaign, Urbana, IL). The CO difference spectra of 2C8H(Cys-), S24C-2C8H(Cys-), and S24C-2C8H expressed in bacteria indicated that the mutant proteins were capable of folding into holoenzymes and were expressed robustly at 500, 800, and 1800 nM, respectively.
Preparation of Membrane Vesicles.
Bacterial membranes containing 2C8H or its mutants were prepared as described previously (Richardson et al., 1995). One milliliter of overnight culture in Super Broth was inoculated into 100 ml of terrific broth containing 100 μg of ampicillin/ml. Cultures were shaken at 37°C for approximately 3 h until OD600 reached approximately 0.35, at which time 0.5 ml of 100 mM δ-aminolevulinic acid was added. Twenty minutes later, isopropyl β-d-thiogalactoside was added to 1 mM to induce the expression of the 2C8 variants, and the cell cultures were shifted to 30°C and grown for 48 h. Cells were harvested by centrifugation at 8000g for 10 min at 4°C, and the cell pellet was resuspended in 15 ml of 25 mM sodium phosphate, pH 7.4, 20% glycerol, and 5 mM DTT. Lysozyme was added to the cell suspension to a final concentration of 0.2 mg/ml. After gentle stirring for 30 min at 4°C, an equal volume of ice-cold distilled water was added, and the mixture was incubated for additional 15 min at 4°C. The resulting spheroblasts were centrifuged at 9000g for 10 min, and the pellets were frozen in a dry ice/ethanol bath and stored at −80°C. The frozen pellets were thawed by incubation at 37°C and were resuspended in 12 ml of 25 mM sodium phosphate, pH 7.4, 20% glycerol, and 5 mM DTT, and phenylmethylsulfonyl fluoride was added to a final concentration of 1 mM. Cells were disrupted by sonication at a power level of 5, with four 30-s bursts with 30-s intervals between sonication with a Thermo Fisher Scientific (model 100) sonifier. The cell lysate was centrifuged at 17,000g for 10 min to pellet cell debris, and the supernatant was decanted and centrifuged at 100,000g for 60 min. After centrifugation, the membrane pellet was resuspended in 1 ml of 25 mM sodium phosphate, pH 7.4, 20% glycerol, and was stored at −20°C.
To prepare membrane vesicles from mammalian cells, 10 μg of pCMV plasmid expressing CYP2C8 or its mutants were transfected into AD-293 cells in 100 mm plates using standard methods. Forty-eight hours after transfection, cells were harvested by centrifugation at 500g for 5 min and were washed once with ice-cold phosphate-buffered saline. The cells were homogenized in 3 ml of 1 M sucrose and 25 mM KOH-HEPES, pH 7.4, with 30 strokes with a B pestle in a Dounce homogenizer. The homogenized cells were centrifuged at 1000g for 10 min to pellet cell debris. The supernatant was centrifuged again at 100,000g for 60 min to pellet the microsomal membranes, which were resuspended in 300 μl of 25 mM sodium phosphate, pH 7.4, and 20% glycerol.
Cross-Linking Experiments.
Oxidative cross-linking of Cys residues using copper-phenanthroline was performed as described previously (Lee et al., 1994; Lee et al., 1995). Bacterial or mammalian membranes were adjusted to a concentration of 1 μg of protein/μl by adding 25 mM sodium phosphate, pH 7.4, and 20% glycerol. The membrane samples were incubated with 50 μl of 2 mM CuSO4, 6 mM phenanthroline, 25 mM sodium phosphate, pH 7.4, and 20% glycerol at room temperature or 0°C. The reaction was stopped by adding 20 μl of 875 mM N-ethylmaleimide. For the reactions at 0°C, the membrane sample and copper-phenanthroline solution were cooled on ice before the reaction was started. For cross-linking experiments with BMOE, 200 μl of membrane samples adjusted to 1 μg of protein/μl were mixed with BMOE at a final concentration of 0.4 mM at 37°C or room temperature. At different time points, 20-μl aliquots were withdrawn, and the reaction was quenched by addition of DTT to a final concentration of 50 mM and further incubation for 15 min (Giron-Monzon et al., 2004). Samples were analyzed by SDS-PAGE under nonreducing conditions. CYP2C8 was detected by Western analysis with anti-His or anti-flag antibody for experiments with bacterial or mammalian membranes, respectively. Cross-linking experiments were performed at least twice, and representative results are shown.
Results
2C8H, but Not 2C8dH, Forms Cross-Linked Dimers in Bacterial Membranes.
To initially examine the organization of CYP2C8 in membranes, we expressed 2C8H and 2C8dH in bacteria and examined cross-linking patterns of these proteins by oxidation of Cys with copper-phenanthroline or with the sulfhydryl cross-linker BMOE. 2C8H contains a functional, N-terminal signal anchor (Fig. 1C) that is modified to increase expression levels in bacteria (Barnes et al., 1991; Richardson et al., 1995) so that the protein is inserted normally into the membrane via the signal anchor sequence. In 2C8dH, the source of the protein for crystallization and structure determination, the first 27 amino acids in the signal anchor sequence were replaced with a short hydrophilic sequence, MAKKTSSKG, so although it is associated with the membrane, it is not an integral membrane protein (Schoch et al., 2004).
Oxidation of 2C8H bound to bacterial membranes at room temperature resulted in rapid formation within 30 s of a complex with the molecular weight expected for a dimer, the amount of which progressively decreased with time as higher order complexes progressively increased (Fig. 2A). At 0°C, the kinetics of complex formation was slower, and the dimer was the predominant cross-linked complex. Likewise, treatment of the membranes with BMOE at 0°C or room temperature (Fig. 2B) or 37°C (Fig. 2C) resulted in rapid formation of apparent dimers, the amount of which then decreased as the amount of higher order complexes increased. The rapid formation of apparent cross-linked dimer followed by formation of higher complexes is consistent with the presence of a preexisting dimer bound to the membrane that is rapidly cross-linked, followed by less specific cross-linking resulting from random interactions to form the larger complexes.

2C8H, but not 2C8dH, is cross-linked in membranes. Bacterial membranes were treated with Cu2+-phenanthroline (A) to oxidize the Cys or with BMOE (B) to cross-link Cys residues at the indicated temperatures. The proteins were separated by SDS-PAGE and the 2C8 proteins were detected by Western analysis with anti-His antibodies. The positions of molecular weight (M.W.) markers are indicated on the left and asterisks denote the expected mobility of the 2C8H dimer.
It is possible that the apparent dimer of 2C8H is a heterodimer of 2C8H with another bacterial membrane protein. To exclude this possibility, after cross-linking with BMOE, the proteins were affinity-purified by binding to Ni2+-nitrilotriacetic acid and separated by SDS-PAGE (Supplemental Fig. S1). The gel band corresponding to the size of a 2C8H dimer was excised, and the proteins present were identified by mass spectrometry (data not shown). Only peptides contained within 2C8H were detected, confirming that the band contained a 2C8H dimer.
In contrast to 2C8H, no formation of dimers was observed for 2C8dH by oxidation of Cys (Fig. 2A) or by cross-linking with BMOE (Fig. 2C). Some higher order oligomers were observed at later times, presumably as a result of random interactions. 2C8H has Cys at positions 13 and 24, whereas these Cys are deleted in the modified N terminus of 2C8dH. It is possible, therefore, that these Cys mediate the cross-linking so that dimers of CYP2C8dH exist, but cannot be cross-linked or, alternatively, that the hydrophobic signal anchor sequence is required for formation of membrane-bound dimers.
Cross-Linking of CYP2C8H Is Mediated by Cys-24 in the Linker Sequence.
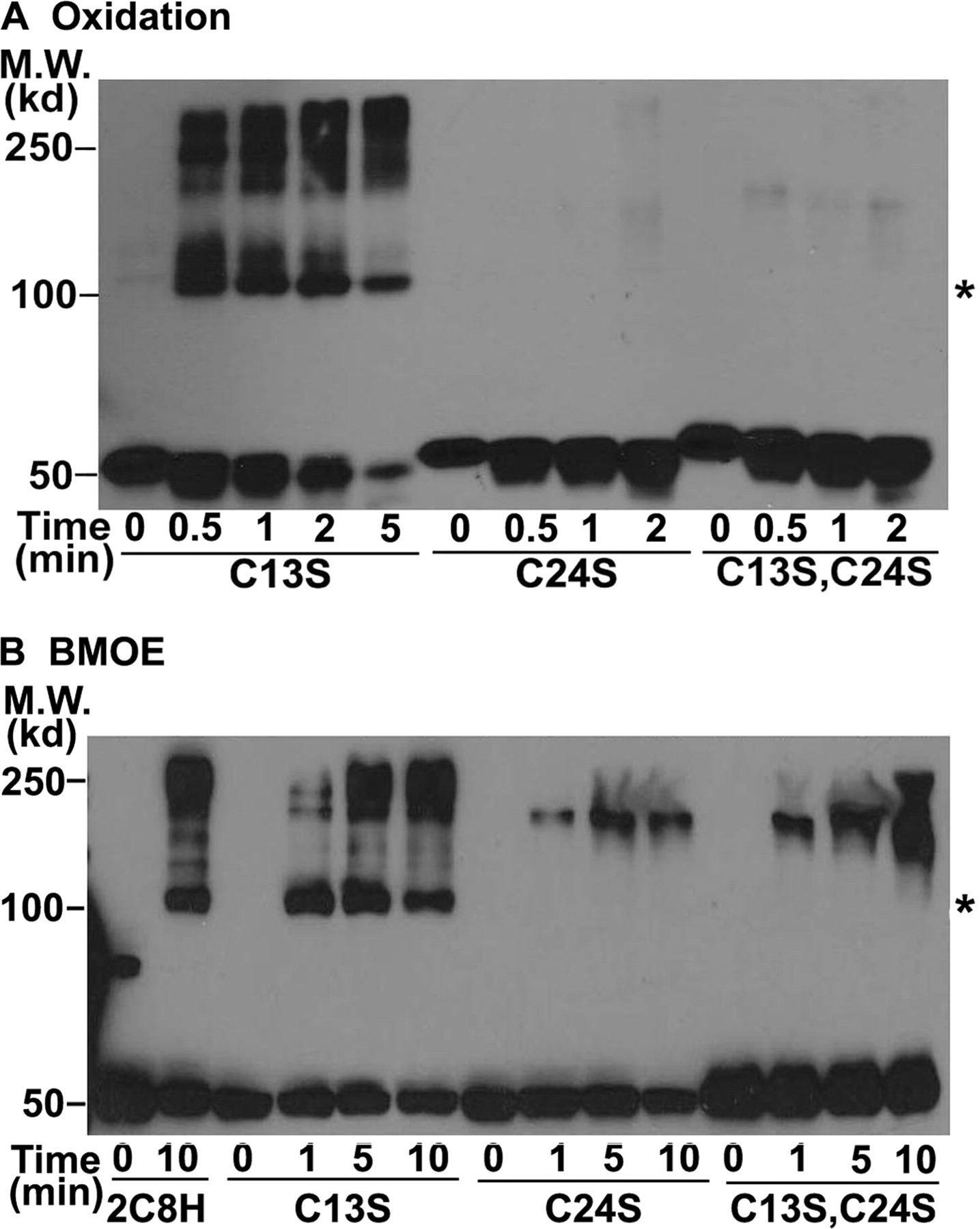
To determine which Cys was responsible for formation of the 2C8H cross-linked dimers, we substituted Ser for Cys-13 in the signal anchor or Cys-24 in the linker region because these Cys were missing in 2C8dH. In addition, the signal anchor sequence has been shown to mediate aggregation of solubilized P450s so that Cys-13 was a likely candidate to mediate cross-linking. We were surprised to find that the kinetics and formation of cross-linked dimers by oxidation of Cys (Fig. 3A) or with BMOE (Fig. 3B) of 2C8H with Ser substituted for Cys-13 (Fig. 3, A and B) were similar to those of wild-type 2C8H (Fig. 2), indicating that Cys-13 was not involved in the cross-linking. In contrast, substitution of Ser for Cys-24 or for both Cys-24 and Cys-13 completely eliminated the formation of cross-linked dimers with higher order oligomers forming at later times, suggesting that Cys-24 in the linker sequence is responsible for the formation of cross-linked dimers of 2C8H.
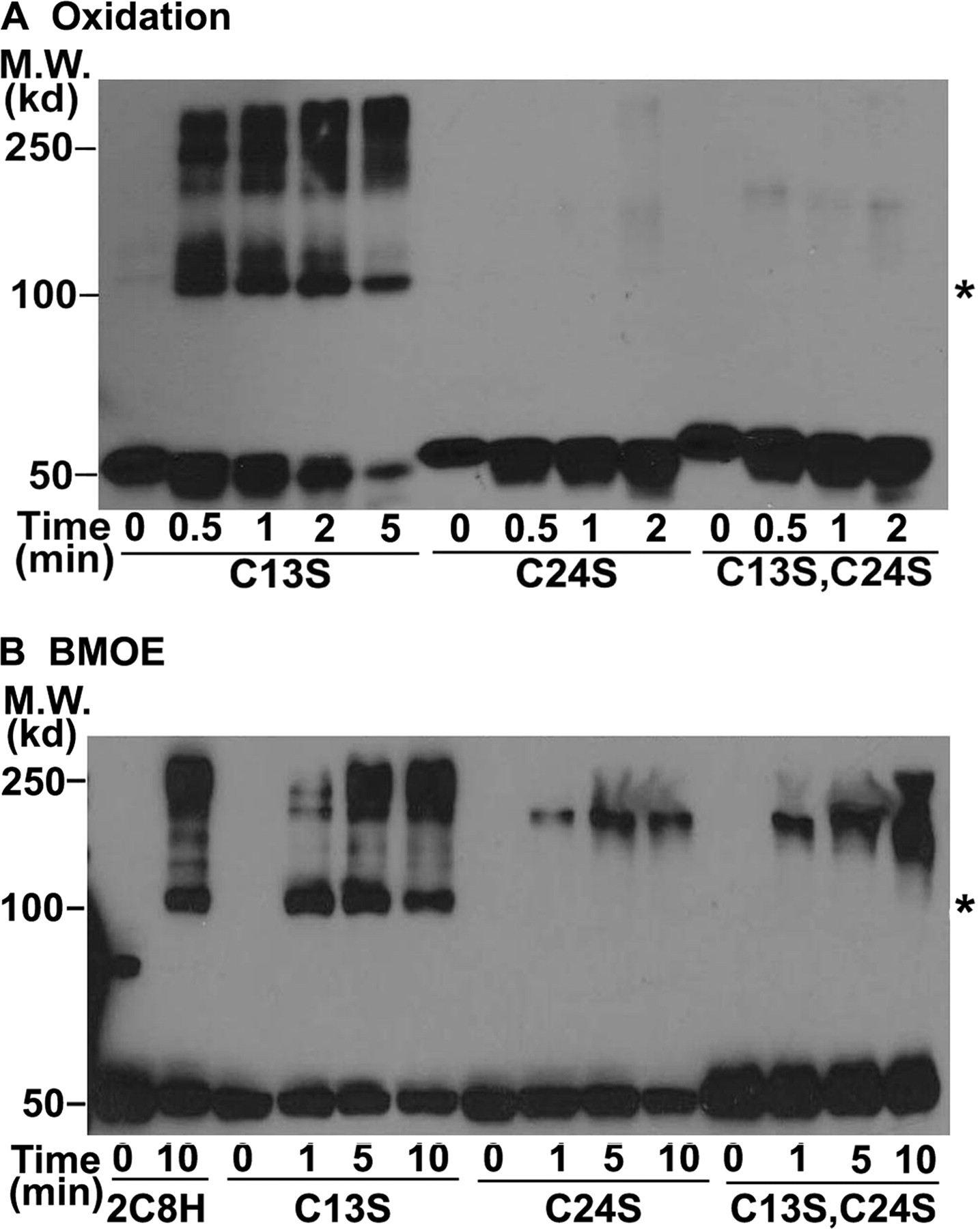
Cross-linking of 2C8H is mediated by Cys-24 in the linker sequence, not by Cys-13 in the signal anchor. 2C8H and its C13S and C24S mutants were expressed in bacteria, and the proteins were cross-linked by oxidation of Cys (A) or BMOE treatment (B) and analyzed as described in the legend to Fig. 1. Asterisks denote the expected mobility of the 2C8H dimer. M.W., molecular weight.
2C8H contains 14 Cys so that Cys-24 could either cross-link with Cys-24 in the second 2C8H molecule or with another Cys. To reduce nonspecific dimer formation and determine whether cross-linking between the two Cys-24 residues in the two 2C8H molecules was responsible for the formation of dimers, seven Cys residues, including Cys-13 and Cys-24, were mutated to Ser, except for Cys-225, which was replaced by Tyr. The mutated Cys included Cys-216 and Cys-225 in the F-G loop region, which is part of the dimerization interface in the crystallized 2C8H, Cys-51, which, based on the crystal structure, would be closest to Cys-24 and other surface Cys, Cys-164, and Cys-338. As expected, no cross-linked dimers were observed with the mutant protein, termed 2C8H(Cys-) in which Cys-24 had been mutated (Fig. 4). Interestingly, reintroduction of Cys at position 24 resulted in formation of the cross-linked dimers indicating that Cys-24 in one 2C8H molecule was forming an oxidized cystine with Cys-24 on the second 2C8H molecule in the dimer. Furthermore, substitution of Cys at either position 22 or 23 also resulted in dimer formation. The amounts of higher order complexes were greatly reduced in the mutant with multiple substitutions for Cys, which is consistent with the formation of these complexes by cross-linking of surface Cys due to random collisions in the membrane. These results indicate that the linker sequences from residues 22 to 24 in the two subunits of the dimer are in close apposition to each other so that cross-links can occur at these positions. This occurs despite the multiple positive charges in this region of the linker sequence, which would be expected to prevent the close apposition.

2C8H with Cys present at positions 22, 23, and 24 in the linker sequence of CYP2C8 can form cross-linked dimers. To reduce the formation of higher order complexes, the 2C8H(Cys-) mutant in which 7 Cys were mutated was used, and Cys was substituted at either residue 22, 23, or 24. The proteins were expressed in bacteria, cross-linked by oxidation of Cys (A) or BMOE treatment (B), and analyzed as described in the legend to Fig. 2. Asterisks denote the expected mobility of the 2C8H dimer. M.W., molecular weight.
Cross-Linking of CYP2C8 in Mammalian Membranes.
The composition of the membrane of E. coli differs from that of the ER membrane of mammalian cells, which could affect the conformation and organization of 2C8H in the membrane. In addition, 2C8H is expressed at relatively high levels in the bacteria, and 2C8H has a modified N-terminal signal anchor sequence. To determine whether dimer formation of native CYP2C8 bound to the mammalian membrane was similar to that observed for 2C8H in bacterial membranes, native CYP2C8 was expressed in mammalian AD-293 cells. ER membranes were isolated and proteins were cross-linked as described for the bacterial membranes above. In the mammalian cells, the pattern of cross-linked CYP2C8 complexes after Cys oxidation (Fig. 5A) or cross-linking with BMOE (Fig. 5A) was very similar to that observed for 2C8H in bacterial membranes (Fig. 2). There was rapid formation of the dimer with additional higher order forms present. In addition, just as with 2C8H in bacteria, substitution of Ser for Cys-24 eliminated the cross-linked dimers but not the higher order complexes (Fig. 5B). Furthermore, cross-linked dimers were not observed for CYP2C8(Cys-) by oxidation or with BMOE (Fig. 5C) in mammalian ER membranes, but reintroduction of a Cys at position 24 restored dimer formation just as was observed for 2C8H in bacterial membranes (Fig. 5C). These results provide strong evidence that the same CYP2C8 dimer is formed in mammalian and bacterial membranes and confirmed that the bacterial membranes were a good model for the mammalian membranes.

Cross-linking of CYP2C8 and its mutants in mammalian membranes. Native CYP2C8 (A), the C24S mutant of CYP2C8 (B), or CYP2C8 with the 7 Cys mutated (Cys-) or the S24C mutant of the (Cys-) mutant (C) was expressed in AD-293 cells. The cells were lysed, and the microsomal membranes were isolated by centrifugation. The membranes were treated with Cu2+-phenanthroline (oxidation) or BMOE as indicated, and the proteins were analyzed as described for bacterial membranes in the legend to Fig. 2. Asterisks denote the expected mobility of the 2C8H dimer. M.W., molecular weight.
N-Terminal Anchor Sequence Forms a Dimer Interface.
Structures of all microsomal P450s were determined for modified P450s in which the signal anchor sequence had been deleted to prevent aggregation of the purified protein, except for aromatase for which the signal anchor sequence was disordered in the structure (Ghosh et al., 2010). The structures, therefore, provide no insight into the role of the signal anchor in dimerization. However, the signal anchor sequence of CYP2C2 has been shown to be involved in homo-oligomerization in living mammalian cells by FRET and BiFC analysis (Szczesna-Skorupa et al., 2003; Ozalp et al., 2005), and the signal anchor is known to mediate aggregation of solubilized P450 (von Wachenfeldt et al., 1997; Cosme and Johnson, 2000). The signal anchor is, therefore, a reasonable candidate for forming a dimerization interface. To examine the role of the signal anchor in more detail, Cys was introduced at each position from 11 to 20 in the 2C8H(Cys-) mutant. The 2C8H(Cys-) mutant was used to reduce the formation of the higher order complexes. Cys substituted at most positions resulted in formation of cross-linked dimers by oxidation of Cys (Fig. 6). Although Cys at most positions resulted in cross-linking, the amount of cross-linked dimer formed relative to remaining monomer was relatively low or absent for positions 12, 15, and 18 compared with their neighboring residues. These minima of reaction with three-residue spacing would be consistent with a helical structure for the signal sequence and a mirror image interaction between helices so that residues on the back side from the interface would have a reduced probability of cross-linking. The mutation of the 7 Cys residues in 2C8H(Cys-) may also subtly alter the signal sequence interaction because Cys at position 13 resulted in cross-linking in the Cys- mutant, but not in 2C8H(C24S) (Fig. 3). These results demonstrate that the signal anchor sequence of 2C8H formed a dimerization interface in the membrane.

The N-terminal anchor sequence is part of a dimer interface. A–D, in the (Cys-) mutant of 2C8H, with mutations of 7 Cys, Cys was substituted at each position from 11 to 20 in the signal anchor sequence, and the bacterial membranes expressing these mutants of 2C8H(Cys-) were analyzed as described in the legend to Fig. 2. Asterisks denote the expected mobility of the 2C8H dimer. M.W., molecular weight.
The F-G Loop Forms a Dimer Interface.
In the 2C8dH crystal structure, the F-G loop formed a dimerization interface. Because mutation of Cys-24 in 2C8H eliminated cross-linked dimers, we introduced pairs of Cys in the F-G loop region of the Cys-24 mutant that would be close to each other at the dimer interface based on the crystal structure (Fig. 1B). Cys-225 and Trp-212 form the major van der Waals interactions between the two 2C8H molecules in the dimer so Cys was substituted for Trp-212 in one mutant. Arg-206 and Gly-228 have the shortest distance between their α-carbons of the residues in the F-G interface region (Fig. 1B) so that Cys was substituted for these residues in a second mutant. Cross-linked dimers formed after oxidation of the Cys for each of these mutant proteins (Fig. 7A), but cross-linking with BMOE was not observed (Fig. 7B). The lack of cross-linking with BMOE may be related to the dynamics of the proteins (see Discussion). These results indicate that the F-G loop region forms a dimer interface in the membrane-bound dimer and suggest that the interface interactions are the same as those observed in the crystal structure.

Cross-linking of 2C8H and its F-G loop mutants in bacterial membranes. Based on the 2C8dH crystal structure, as shown in Fig. 1B, Trp-212 and Cys-225 and Arg-206 and Gly-228 in the F-G loop are the closest two pairs of residues. To determine whether the F-G loop forms a dimer interface, Cys was substituted for Trp-212 or for Arg-206 and Gly-228 in the C24S mutant of 2C8H, and the mutants were expressed in bacteria, and membranes were treated with (A) Cu2+-phenanthroline or (B) BMOE cross-linker. The proteins in the membranes were analyzed as described in the legend to Fig. 2. Asterisks denote the expected mobility of the 2C8H dimer. M.W., molecular weight.
Introduction of Cys at 212 and 225 or at 206 and 228 in the F-G loop of 2C8H(Cys-) did not result in cross-linked dimers (data not shown). It is possible that the Cys residues introduced in the F-G loop form cross-links with Cys 216 also in the F-G loop region, which is mutated in 2C8H(Cys-). However, reintroduction of Cys-216 with Cys at 225 or at 228 did not result in cross-linking (data not shown). Most likely the multiple mutations in 2C8H(Cys-) altered the conformation of the protein enough so that the structure of the dimerization interface was disrupted or altered such that the Cys pairs were not close enough for oxidation of the Cys to cystine to occur.
Signal Anchor Is Required for Dimer Formation.
Cross-linking was not observed with 2C8dH (Fig. 2, A and C), in which the signal anchor is deleted, but Cys-24, which mediates the cross-linking of 2C8H, is also not present in 2C8dH. To determine whether the lack of cross-linking was due to the absence of the signal anchor or Cys-24, the hydrophobic anchor of 2C8H was replaced with the hydrophilic N-terminal sequence in 2C8dH, MAKKTSSKG, to produce 2C8dH+C (Fig. 1C). This protein has a Cys in a position equivalent to that in Cys-24 in 2C8H. Although higher molecular weight complexes were observed, no cross-linked dimer of 2C8dH+C was observed by either oxidation of Cys or with BMOE (Fig. 8C). These results indicate that the signal anchor sequence is required for the formation of Cys-24 mediated cross-linking of 2C8H bound to membranes. We also introduced Cys into the F-G loop in 2C8dH, equivalent to the Cys substitutions, which result in cross-linked dimers in 2C8H. However, no cross-linked dimer was observed in these 2C8dH mutants (Fig. 8, A and B). These results show that in the absence of the hydrophobic anchor, neither Cys in the linker sequence nor in the F-G loop region can mediate cross-linking, which strongly suggests that formation of stable dimers bound to the membrane requires the signal sequence.

The signal anchor is required for dimer formation. A and B, 2C8dH and W212C and R206C,G228C F-G loop mutants of 2C8dH were expressed in bacteria, and membranes were treated with Cu2+-phenanthroline (A) or BMOE cross-linker (B). The F-G loop substitutions mediate cross-linking of 2C8H. In C, 2C8dH+C, which contains a Cys in a position equivalent to that of Cys-24 in 2C8H, was expressed in bacteria. A–C, the proteins in bacterial membranes were analyzed as described in the legend to Fig. 2. Asterisks denote the expected mobility of the 2C8H dimer. M.W., molecular weight.
Discussion
Although 2C8dH crystallizes as a dimer, high concentrations of a protein modified by deletion of the signal anchor are required for crystallization, which could result in artifactual dimerization (Schoch et al., 2004). In solution, 2C8dH typically elutes as a dimer from size exclusion columns during purification, and dimerization is concentration-dependent and influenced by fatty acids bound to the dimer interface (Schoch et al., 2004). In this study, we present evidence that 2C8H and native CYP2C8 exist as dimers when bound to natural bacterial and mammalian membranes, respectively. Cross-linking of Cys in either the signal anchor/linker or F-G loop regions resulted in covalently linked dimers of the 2C8 proteins. Considering the reported efficiency of cross-linking, which ranged from 25 to 80% for different Cys pairs in the human erythrocyte anion exchange protein (Taylor et al., 2001), a substantial fraction of CYP2C8 is likely to exist as a dimer.
The evidence indicates that cross-linking occurs between the Cys-24 residues in the two 2C8 molecules. Mutation of Cys-24 eliminated the cross-linked dimers. Furthermore, mutation of other Cys that potentially could cross-link to Cys-24 did not eliminate the cross-linked dimers, which indicates that cross-linked Cys-24 residues were being formed. Cys introduced at either 22 or 23 also resulted in cross-linking. Interestingly, residues 22, 23, and 24 in the linker region are surrounded by basic amino acids with Arg residues at 21, 25, 26, and 27. It is unlikely that such positively charged regions would contribute to the forces driving dimerization. The positive charges may be neutralized by negative charges in phospholipids at the membrane surface, and the interactions of the preceding N-terminal transmembrane helices of the signal anchor bring the linker regions together.
The results are consistent with a dimer structure of 2C8 bound to the membrane that is the same as that in the crystal structure. Introduction of Cys pairs into the F-G loop region of 2C8H at sites that were predicted be in close proximity based on the crystal structure resulted in formation of cross-linked dimers bound to the membrane. Therefore, the F-G loop forms a dimerization interface in the membrane-bound dimer. In addition, Cys present in the signal anchor sequence also formed cross-links. The signal anchor sequence is deleted in the crystallized protein so that the structure is not informative about the role of the signal anchor in dimerization. The present cross-linking studies indicate that both the F-G loop regions and the signal anchor/linker sequences form dimerization interfaces.
Dimerization interfaces by both the F-G loop and the linker are not consistent with the structure of the truncated protein. The first resolvable residue in the structure is Lys-28 and the two Lys-28 residues in the dimer are approximately 60 Å apart so that the two Cys-24 residues could not be within 8 Å of each other as needed for a cross-link to form. There are two possible explanations that are consistent with the cross-linking results. First, it is possible that that there are two populations of dimers such that F-G loop dimerization occurs in one population and linker region dimerization occurs in the other. Second, binding to the membrane may result in a 2C8H conformation different from that in the crystallized protein. Lys-28 is projected into the membrane based on the crystal structure so its position in the membrane-bound protein is likely to be different. Lys-28 is followed by a hydrophobic proline-rich region, which is predicted to interact with the membrane. A membrane-dependent change in the conformation of the proline-rich regions could alter the positions of the preceding linker regions so that cross-linking could occur at Cys-24. The lack of cross-linking of Cys in the F-G loop if the signal anchor is deleted further supports the idea that both F-G and the signal anchor form dimerization interfaces in a single dimer.
In two cases, the signal anchor (data not shown) and the F-G loop region (Fig. 7), cross-linking was observed by oxidation of Cys, but not by BOME cross-linking. Similar discrepancies have been observed for other proteins. In human erythrocyte anion exchange protein (Taylor et al., 2001), for example, only oxidation cross-linking or maleimide cross-linking was observed for some Cys pairs. The differences were interpreted in terms of the dynamics of the protein. Regions in which maleimide cross-linking was not observed were considered inflexible so that the distance between Cys was not optimal for the spacer arms of the cross-linkers. In the case of 2C8H, this would predict that the signal anchor and F-G loop dimeric regions are relatively inflexible, whereas the linker region is flexible. Given the positive charges in the linker regions, it would be reasonable that the interaction might allow considerable breathing to accommodate the BOME molecule. Alternatively, the residues in the signal anchor and F-G loop regions may not be accessible to the BMOE, which was not tested.
The signal anchor sequence appears to be required for the stable formation of CYP2C8 dimers bound to the membrane because deletion of the signal anchor eliminates cross-linking of the molecule. This result is consistent with previous studies in which homo-oligomerization of the closely related CYP2C2 was examined in living mammalian cells by either FRET or BiFC. By either method, homo-oligomerization of intact CYP2C2 or only the N-terminal signal sequence of CYP2C2 fused to fluorescent proteins was observed. In contrast, for proteins in which the CYP2C2 signal anchor was replaced with the transmembrane sequence of epidermal growth factor receptor, no homo-oligomerization was observed (Szczesna-Skorupa et al., 2003). The importance of the signal anchor in homo-oligomerization is also supported by its requirement for aggregation of several P450s after solubilization and purification (von Wachenfeldt et al., 1997; Cosme and Johnson, 2000). The signal anchor sequence requirement for stable dimer formation suggests that the interaction in the F-G loop regions is relatively weak and not sufficient alone to maintain dimers of CYP2C8 bound to the membrane. In the crystallization of 2C8dH, which lacks a signal anchor sequence, the high concentrations of protein may be sufficient for the interaction between the F-G loops to mediate dimer formation even in the absence of the signal sequence. Although less likely, it remains possible that binding of 2C8dH, without a signal anchor, to membranes differs from that of CYP2C8 and is inhibitory for dimer formation. To conclusively eliminate this possibility, the structure at the atomic level of the two 2C8 forms bound to membranes would need to be determined, which is not presently feasible.
An outstanding question is whether the formation of the homodimer of CYP2C8 is functionally significant. There are theoretical considerations suggesting that dimerization might affect CYP2C8 function. Most models of the interaction of the catalytic domain of P450 monomers place the hydrophobic F-G loop within the membrane (Williams et al., 2000a). In a dimer, the monomers must be rotated so that the F-G loop is part of the dimerization interface and is no longer within the membrane (Fig. 1A). Trp scanning analysis is consistent with a position of the F-G loop outside of the membrane for a closely related P450, CYP2C2 (Ozalp et al., 2006). Such rotation might affect the interaction of P450 with its membrane-bound redox partner, cytochrome P450 reductase, or with the substrate if the substrate access channel faces the membrane. Experimentally, both hetero- and homodimerization have been shown to affect P450 function. Enzyme kinetic analysis of pairs of P450s, including 2B4/1A4, 2D6/2C9, 2C9/3A4, and 2C9/2C19 has provided evidence that hetero-interactions between the P450s affect the activity of one or both of the P450s in the dimer (Cawley et al., 2001; Hazai and Kupfer, 2005; Subramanian et al., 2009, 2010; Reed et al., 2010). Homo-oligomerization also has functional consequences. Reduction of CYP3A4 by either the small molecule dithionite or by the soluble flavin domain of P450BM-3 is inhibited by homo-oligomerization (Davydov et al., 2005, 2010). Interestingly, oligomerization of another ER membrane protein, heme oxygenase-1, mediated by its single transmembrane helix stabilized and increased the activity of this enzyme (Hwang et al., 2009). Based on these results with other P450s and heme oxygenase-1, it is likely that the homo-oligomerization of CYP2C8 has functional significance.
In conclusion, these studies have demonstrated that the dimeric structure observed in the crystal structure of 2C8dH is also present in membrane-bound native CYP2C8. In addition to the dimerization interface involving the F-G loop that is present in the crystallized modified enzyme, the signal anchor/linker regions form a dimerization interface in the native enzyme. The signal anchor interaction is required for the formation of the dimer in the native protein. Although direct evidence for the functional significance of the dimerization is lacking, such interactions have been shown to affect function of other P450s and ER membrane proteins.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants GM035897, GM031001].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.034942.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- P450
- cytochrome P450
- ER
- endoplasmic reticulum
- FRET
- fluorescence resonance energy transfer
- BiFC
- bimolecular fluorescence complementation
- BMOE
- bis-maleimidoethane
- DTT
- dithiothreitol
- PAGE
- polyacrylamide gel electrophoresis.

- Received June 11, 2010.
- Accepted August 9, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}