


Abstract
2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), formed during the cooking of foods, induces colon cancer in rodents. PhIP is metabolically activated by cytochromes P450 (P450s). To evaluate the role of hepatic P450s in the bioactivation of PhIP, we used Reductase Conditional Null (RCN) mice, in which cytochrome P450 oxidoreductase (POR), the unique electron donor to P450s, can be specifically deleted in hepatocytes by pretreatment with 3-methylcholanthrene (3-MC), resulting in the loss of essentially all hepatic P450 function. RCN mice were treated orally with 50 mg/kg b.wt. PhIP daily for 5 days, with and without 3-MC pretreatment. PhIP-DNA adducts (i.e., N-(deoxyguanosin-8-yl)-2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine [dG-C8-PhIP]), measured by liquid chromatography-tandem mass spectrometry, were highest in colon (1362 adducts/108 deoxynucleosides), whereas adduct levels in liver were ∼3.5-fold lower. Whereas no differences in PhIP-DNA adduct levels were found in livers with active POR versus inactivated POR, adduct levels were on average ∼2-fold lower in extrahepatic tissues of mice lacking hepatic POR. Hepatic microsomes from RCN mice with or without 3-MC pretreatment were also incubated with PhIP and DNA in vitro. PhIP-DNA adduct formation was ∼8-fold lower with hepatic microsomes from POR-inactivated mice than with those with active POR. Most of the hepatic microsomal activation of PhIP in vitro was attributable to CYP1A. Our results show that PhIP-DNA adduct formation in colon involves hepatic N-oxidation, circulation of activated metabolites via the bloodstream to extrahepatic tissues, and further activation, resulting in the formation of dG-C8-PhIP. Besides hepatic P450s, PhIP may be metabolically activated mainly by a non-P450 pathway in liver.
Introduction
Environmental factors play an important role in human cancer (Wild, 2009). Heterocyclic aromatic amines (HAAs) are carcinogenic compounds formed in meats, fish, and poultry prepared under common household cooking practices (Schut and Snyderwine, 1999; Turesky, 2002; Knize and Felton, 2005). 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) is one of the most abundantly formed of more than 20 HAAs that are mutagenic in both bacteria and mammalian cells (Gooderham et al., 2002). PhIP induces tumors in the colon, prostate, and mammary gland of rats. These are the organ sites that are principally associated with diet-related cancer in Western countries, implying that PhIP could be a human carcinogen (Dingley et al., 1999; Gooderham et al., 2002; Zhu et al., 2003; Tang et al., 2007). The International Agency for Research on Cancer has classified PhIP as possibly carcinogenic to humans (group 2B). The Report on Carcinogens of the National Toxicology Program concluded that PhIP is “reasonably anticipated” to be a human carcinogen.
The genotoxicity of PhIP is related to metabolic activation, resulting in an electrophilic species that is capable of forming DNA adducts (Schut and Snyderwine, 1999). Cytochromes P450 (P450s) are the most important enzymes in the initial oxidation of PhIP, forming the intermediate N-OH-PhIP. On the basis of evidence primarily from in vitro experiments, CYP1A2 has been identified to have high specificity and catalytic activity for PhIP N-hydroxylation, although CYP1A1 and CYP1B1 are also able to catalyze PhIP N-hydroxylation but generally at a lower capacity (Shimada et al., 1996). Subsequent metabolism by N-acetyltransferases or sulfotransferases converts N-OH-PhIP into esters capable of undergoing heterolytic cleavage to produce a PhIP-nitrenium ion, which is the ultimate reactive species that bonds with DNA (Schut and Snyderwine, 1999). The major covalent DNA adduct detected in vivo resulting from exposure of experimental animals and humans to PhIP is N-(deoxyguanosin-8-yl)-2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (dG-C8-PhIP) (Snyderwine et al., 2002; Zhu et al., 2003).
In rodents, P450-mediated N-oxidation of PhIP occurs primarily in the liver. In the present study we used Reductase Conditional Null (RCN) mice (Finn et al., 2007) to determine the effect of diminished hepatic P450 activity on PhIP-DNA adduct formation in liver and other tissues. In RCN mice, NADPH:cytochrome P450 oxidoreductase (POR) can be deleted conditionally in the liver by 3-methylcholanthrene (3-MC) because a rat CYP1A1 promoter drives Cre recombinase expression, resulting in loss of hepatic POR-mediated P450 activity within 2 weeks after administration. To detect and quantify dG-C8-PhIP adducts in DNA, we used a recently validated online column-switching liquid chromatography (LC)-electrospray ionization (ESI)-tandem mass spectrometry (MS/MS) method (Singh et al., 2010).
Materials and Methods
Chemicals.
The synthesis of PhIP was performed at the Biochemical Institute for Environmental Carcinogens (Grosshansdorf, Germany) according to a method described previously (Lindström, 1995): m.p. >300°C (sublimation); UV-visible (methanol) λmax (ε, square centimeters per millimole) 214 (30,500), 275 (10,000). No impurities were detectable by reverse-phase high-performance liquid chromatography and NMR. 1H-NMR data matched the literature values (Felton and Knize, 1986).
Animal Treatment and Microsome Isolation.
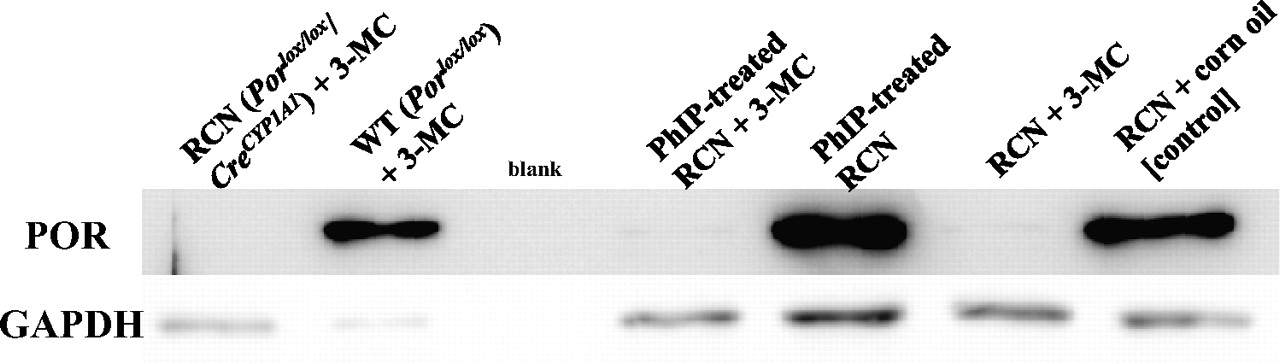
RCN (Porlox/lox/CreCYP1A1) mice (Finn et al., 2007) on a C57BL/6 background were bred in-house at the Biomedical Research Institute (Dundee, UK). In brief, Por floxed mice (Porlox/lox) (Henderson et al., 2003) were crossed with a transgenic line expressing Cre recombinase under the control of the rat CYP1A1 promoter (CreCYP1A1) (Ireland et al., 2004) to generate the mouse line Porlox/lox/CreCYP1A1. Pretreatment of RCN mice with 3-MC (40 mg/kg b.wt. i.p. in corn oil) 2 weeks before PhIP administration resulted in hepatic POR loss (Fig. 1). PhIP was dissolved in corn oil at a concentration of 10 mg/ml. Groups of adult female RCN mice (3 months old, 25–30 g) were dosed by oral gavage daily for 5 days with 50 mg/kg b.wt. PhIP (n = 3). Control mice (n = 3) received corn oil only. Animals were sacrificed 24 h after the last dose, and several organs (liver, lung, forestomach, glandular stomach, small intestine, colon, spleen, kidney, and bladder) were removed, snap-frozen, and stored at −80°C until analysis. All animal experiments were performed under license in accordance with the law and with local ethics approval. Hepatic microsomes from RCN mice (with and without 3-MC pretreatment) were isolated as described previously (Stiborová et al., 2003). Pooled microsomal fractions were used for further analysis.

Expression of POR in livers of RCN mice treated orally with 50 mg/kg b.wt. PhIP for 5 days without and with 3-MC pretreatment (single dose of 40 mg/kg b.wt. 3-MC 14 days before PhIP treatment), as determined by Western blotting. WT, wild type.
Expression of POR by Western Blotting.
Western blot analysis was performed as described previously (Forrester et al., 1992) using 5 μg of hepatic microsomal protein/lane and polyclonal antisera raised against human POR (Smith et al., 1994).
Microsomal Incubations.
The incubation mixtures in a final volume of 750 μl consisted of 50 mM potassium phosphate buffer (pH 7.4), 1 mM NADPH, 1 mg of mouse hepatic microsomal protein, 0.5 mg of calf thymus DNA, and 0.5 mM PhIP (dissolved in dimethyl sulfoxide). The reaction was initiated by addition of NADPH. Incubations with microsomes were performed at 37°C for 60 min. α-Naphthoflavone (α-NF) was used to inhibit CYP1A-mediated activation of PhIP in microsomes (Stiborová et al., 2001, 2005). α-NF was dissolved in 7.5 μl of methanol to yield a final concentration of 0.1 mM in the incubation mixtures. Mixtures containing α-NF were incubated at 37°C for 10 min with NADPH before addition of PhIP and then were incubated for a further 60 min at 37°C. Control incubations were performed without NADPH. After extraction with ethyl acetate, DNA was isolated from the residual water phase by standard phenol/chloroform extraction.
PhIP-DNA Adduct Detection by LC-ESI-MS/MS Analysis.
Genomic DNA from whole tissue was isolated by a standard phenol-chloroform extraction method, and DNA adducts were measured by LC-ESI-MS/MS analysis. The development and validation of the LC-ESI-MS/MS method to detect and quantify dG-C8-PhIP adducts in DNA is described in detail elsewhere (Singh et al., 2010). For analysis, a 15-μl aliquot, equivalent to 50 μg of hydrolyzed DNA containing 1000 fmol of dG-C8-[13C10]PhIP internal standard, was injected and analyzed using an online column-switching valve system (Singh et al., 2010). The hydrolyzed DNA samples were analyzed in positive ESI-MS/MS selected reaction monitoring mode for the [M + H]+ ion to base [B + H2]+ transitions of m/z 490 to 374 for dG-C8-PhIP and m/z 500 to 379 for dG-C8-[13C10]PhIP. The level of the adduct in the DNA sample was determined from the ratio of the peak area of the dG-C8-[13C10]PhIP internal standard and is expressed as adducts per 108 deoxynucleosides (Singh et al., 2010).
Enzyme Activity.
CYP1A activity was measured with 3-cyano-7-ethoxycoumarin as substrate in a 96-well format as described previously (Martin et al., 2010).
Results and Discussion
We have previously used Hepatic P450 Reductase Null (HRN) mice to investigate hepatic versus extrahepatic P450-mediated carcinogen metabolism (Arlt et al., 2005, 2006, 2008; Stiborová et al., 2008; Levová et al., 2011). In HRN mice, hepatic POR expression has been deleted, resulting in almost complete ablation of hepatic P450 function (Henderson et al., 2003). For example, we showed that hepatic P450s seem to be more important for detoxication of benzo[a]pyrene in vivo despite being important for its bioactivation in vitro (Arlt et al., 2008). In HRN mice, the deletion of the Por gene occurs neonatally and although HRN mice develop normally, they exhibit a number of phenotypic changes associated with the loss of P450 function, including hepatic lipid accumulation, reduced bile acid production, increased constitutive P450 expression, and decreased plasma cholesterol and triglyceride levels, which may have an impact on the pharmacokinetics of xenobiotics studied. In RCN mice, hepatic POR can be deleted conditionally using a rat CYP1A1 promoter to drive Cre recombinase expression (Finn et al., 2007). Thus, the use of this promoter provides a tightly regulated method for controlling expression of the transgene in vivo shortly before the animal experiment by the administration of inducers (i.e., 3-MC) that act through the aryl hydrocarbon receptor. In addition, RCN mice can be used as their own control. As shown in Fig. 1, administration of 40 mg/kg b.wt. 3-MC led to a complete and specific deletion of the hepatic Por gene within 14 days; no expression of POR was observed by Western blotting in hepatic microsomes isolated from RCN mice pretreated with 3-MC used in the present study. It is noteworthy that 14 days after a single intraperitoneal dose of 40 mg/kg b.wt. 3-MC, there is no hepatic CYP1A protein evident (Finn et al., 2007), although some other hepatic P450s are induced, consistent with the elevated hepatic P450 expression seen in the HRN model and driven by lipid accumulation (Henderson et al., 2003; Finn et al., 2007).
After treatment of RCN mice with PhIP, dG-C8-PhIP-DNA adducts were determined in various organs by LC-MS/MS. Representative LC-MS/MS ion chromatograms for liver DNA are shown in Fig. 2, inset. The highest DNA adduct levels were observed in colon (1362 adducts/108 deoxynucleosides or 100%), lower levels were observed in kidney (76%), lung (73%), glandular stomach (57%), and spleen (56%), and the lowest levels were observed in small intestine (37%), forestomach (33%), liver (29%), and bladder (13%) (Fig. 2). No dG-C8-PhIP adducts were observed in control (untreated) tissues (data not shown). These results are consistent with colon as a tumor target organ of PhIP in rodents (Sugimura et al., 2004). Previous studies have identified PhIP-DNA adducts in rat and mouse colon (Kaderlik et al., 1994; Snyderwine et al., 2002; Metry et al., 2009), and they have also been identified in human colon (Malfatti et al., 2006).

Quantitative LC-ESI-MS/MS analysis of dG-C8-PhIP in organs of RCN mice treated orally with 50 mg/kg b.wt. PhIP for 5 days without and with 3-MC pretreatment (single dose of 40 mg/kg b.wt. 3-MC 14 days before PhIP treatment). F, fold difference in DNA binding in RCN mice treated with PhIP compared with RCN mice treated with PhIP and inducer (i.e., 3-MC). Values are given as mean ± S.D. (n = 3); each DNA sample was determined by two independent measurements. Comparison was performed by t test analysis: *, P < 0.01, different from RCN mice treated with PhIP but without inducer. Inset, typical LC-MS/MS selected reaction monitoring ion chromatograms for hydrolyzed liver DNA depicting the transition m/z 490 to 374 and m/z 500 to 379 for dG-C8-PhIP and dG-C8-[13C10]PhIP, respectively.
Although hepatic deletion of POR had no significant effect on dG-C8-PhIP-DNA adduct levels in the liver, significantly lower levels of adducts (up to 2.8-fold) were formed in all extrahepatic tissues (P < 0.01) of RCN mice that lack hepatic POR (Fig. 2). We also determined DNA adduct formation by PhIP in incubations with calf thymus DNA in the presence of microsomes isolated from livers of RCN mice with and without 3-MC pretreatment (Fig. 3A). NADPH, a cofactor for P450-dependent oxidation of PhIP, stimulated the formation of dG-C8-PhIP-DNA adducts; DNA adduct levels were ∼8-fold higher in hepatic microsomes with active POR compared with microsomes isolated from livers of RCN mice that lack hepatic POR. Because hepatic P450 enzyme activity should be essentially obliterated by the conditional deletion of POR in hepatocytes of RCN mice pretreated with 3-MC, the level of PhIP activation to DNA adducts in hepatic microsomes is difficult to rationalize. The low levels of DNA adducts in microsomes lacking POR could be explained as suggested previously (Arlt et al., 2008): endoplasmic reticulum membranes from nonparenchymal cells that still contain POR are mixed with those from hepatocytes containing P450s in the process of microsome isolation. However, no clear POR band was detectable by Western blotting. The levels of PhIP-DNA adducts in microsomal incubations corresponded to the activities of CYP1A1/2 (3-cyano-7-ethoxycoumarin activity) in these microsomes (Fig. 3B). Under the experimental conditions used, α-NF, an inhibitor for CYP1A1 and CYP1A2, reduced PhIP-DNA adduct formation by 60% in microsomal incubations with active POR in vitro (Fig. 3A).

A, effect of the CYP1A inhibitor α-NF on DNA adduct formation by PhIP activated with hepatic microsomes isolated from RCN mice, without or with 3-MC pretreatment (single dose of 40 mg/kg b.wt. 3-MC and 14 days recovery). F, fold difference in DNA binding in RCN mice pretreated with 3-MC compared with RCN mice without inducer (i.e., 3-MC). Values are given as means ± range of four determinations (duplicate analyses of two independent in vitro incubations). Control, without NADPH cofactor. ND, not detected. B, CYP1A enzymatic activity in hepatic microsomes isolated from RCN mice with or without 3-MC pretreatment. Values are given as means ± S.D.; rates were determined by linear regression of fluorescence values (RFU) of four parallel incubations with seven time-points between 0 and 12 min. Pooled hepatic microsomal samples were used for analyses.
CYP1A2 is a hepatic P450 enzyme that shows strong interindividual variation in expression in humans (Nakajima et al., 1994), and it has been hypothesized that greater CYP1A2 activity produces higher levels of DNA adducts by HAA such as PhIP. Case-control studies have supported the notion that rapid CYP1A2 activity in conjunction with rapid N-acetyltransferase acetylation activity is a risk factor for colorectal cancer in individuals eating well done cooked meat (Lang et al., 1994; Le Marchand et al., 2001). It was shown that PhIP-DNA adduct levels were significantly lower in Cyp1a2-null mice relative to those in wild-type animals, corroborating the importance of CYP1A2 in the bioactivation of PhIP in vivo (Snyderwine et al., 2002). In contrast, PhIP carcinogenesis in Cyp1a2-null mice using the neonatal bioassay, known to cause liver tumors, was independent of CYP1A2 expression (Kimura et al., 2003). These results suggest that although the metabolic activation of PhIP is performed primarily by CYP1A2, another pathway unrelated to CYP1A2 appears to be responsible for PhIP carcinogenesis in neonatal mice. Other hepatic P450s, such as the CYP3A4 ortholog, potentially contribute to N-oxidation of PhIP in Cyp1a2-null mice (Snyderwine et al., 2002; Kimura et al., 2003). It is also noteworthy that in wild-type mice, PhIP 4′-hydroxylation (detoxication) is the predominant pathway in liver, whereas in mice humanized for CYP1A2, PhIP is preferentially metabolized by N2-hydroxylation (activation) (Cheung et al., 2005). Indeed, CYP1A-humanized mice developed PhIP-induced colon tumors, whereas no tumors were found in the similarly treated wild-type mice (Cheung et al., 2011).
In the present study, we showed a predominant role of hepatic CYP1A enzymes in the activating pathways of PhIP in vitro but only found a nonsignificant decrease in PhIP-DNA adduct formation in livers of RCN mice lacking POR in vivo. In contrast, a previous study showed that DNA adduct formation by the aromatic amine 3-aminobenzanthrone, which is metabolically activated by P450s (i.e., CYP1A1 and CYP1A2), was reduced (by up to 80%) in the livers of HRN mice (Arlt et al., 2006). If hepatic CYP1A in particular, or hepatic P450s in general, are involved in the activation of PhIP in vivo, it would be predicted that there would be elevated levels of PhIP-DNA adducts in extrahepatic tissues and reduced levels in the liver of RCN mice pretreated with 3-MC, relative to those in RCN mice without 3-MC pretreatment. However, although adduct levels in liver of RCN mice lacking POR were only marginally lower, surprisingly, we actually found significantly lower adduct levels in extrahepatic tissues.
The mechanism by which DNA-binding species were generated by PhIP in the liver of RCN mice lacking POR in the present study is not known, but it is clear that the process did not involve generation of a reactive species different from that formed in wild-type mice. PhIP is a good substrate not only for CYP1A2 but also for prostaglandin H synthase (PTGS), suggesting that PTGS may be of importance to explain the PhIP-DNA adduct levels found in livers of mice lacking POR-mediated P450-catalyzed bioactivation. However, in a previous study using HRN mice, no protein expression of PTGS1 and PTGS2 was detectable in hepatic microsomes (Arlt et al., 2008). Therefore, the role of other, as yet unidentified, PhIP-activating enzymes awaits further investigation. Taken together, our study and those of other investigators (Kimura et al., 2003) suggest that PhIP may be metabolically activated mainly by a non-P450 pathway in liver. PhIP itself is an inducer of CYP1A1 (Hirata et al., 2008), and in our experiments mice were treated for 5 days; therefore, other PhIP-activating enzymes might have been induced as well.
It has been proposed that the formation of PhIP-DNA adducts in colon and other extrahepatic tissues could involve the transport of N-OH-PhIP and/or N-acetoxy-PhIP from the liver. Indeed, intravenous administration of both metabolites via the jugular vein of rats resulted in the formation of PhIP-DNA adducts in every tissue examined, with the lowest DNA binding observed in liver (Kaderlik et al., 1994). Thus, highly reactive PhIP metabolites can circulate through the bloodstream, contributing to the formation of PhIP-DNA adducts in extrahepatic tissues. We found fewer PhIP-DNA adducts in extrahepatic tissues of mice lacking POR-mediated P450-catalyzed metabolism in the liver, indicating that the liver is the major P450-dependent PhIP-activating organ and that, as a consequence, fewer bioactivated PhIP metabolites (i.e., N-OH-PhIP and N-acetoxy-PhIP) are transported to extrahepatic organs.
In summary, our results show that the formation of PhIP-DNA adducts in target tissues such as colon of mice administered oral doses of PhIP involves hepatic N-oxidation, possible further hepatic O-acetylation to N-acetoxy-PhIP, which can be circulated via the bloodstream to extrahepatic tissues where it can result in the formation of dG-C8-PhIP. Furthermore, our data suggest that, besides hepatic P450s, PhIP may be metabolically activated mainly by a non-P450 pathway in the liver, which warrants further investigation.
Authorship Contributions
Participated in research design: Arlt, Singh, Stiborová, and Gamboa da Costa.
Conducted experiments: Arlt, Singh, Stiborová, Frei, Evans, and Henderson.
Contributed new reagents or analytic tools: Gamboa da Costa, Farmer, Wolf, and Henderson.
Performed data analysis: Arlt, Singh, Stiborová, and Frei.
Wrote or contributed to the writing of the manuscript: Arlt, Singh, Stiborová, Frei, Henderson, and Phillips.
Acknowledgments
We thank Catherine Meakin and Susanne van Schelven at the Biomedical Research Institute for skilled technical assistance.
Footnotes
This work was supported by Environmental Cancer Risk, Nutrition and Individual Susceptibility as part of the European Union 6th Framework, Priority 5: “Food Quality and Safety” [Contract 513943]; Cancer Research UK; and the Grant Agency of the Czech Republic [Grant P301/10/0356].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.111.041343.
-
ABBREVIATIONS:
- HAA
- heterocyclic aromatic amine
- PhIP
- 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- P450
- cytochrome P450
- dG-C8-PhIP
- N-(deoxyguanosin-8-yl)-2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
- RCN
- Reductase Conditional Null
- POR
- cytochrome P450 oxidoreductase
- 3-MC
- 3-methylcholanthrene
- ESI
- electrospray ionization
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- α-NF
- α-naphthoflavone
- HRN
- Hepatic P450 Reductase Null
- PTGS
- prostaglandin H synthase.
- Received June 23, 2011.
- Accepted September 22, 2011.
- U.S. Government work not protected by U.S. copyright




{kind=link}
{kind=link}
{kind=link}