


Abstract
Glucuronidation via UDP-glucuronosyltransferase (UGT) is an increasingly important clearance pathway. In this study intrinsic clearance (CLint) values for buprenorphine, carvedilol, codeine, diclofenac, gemfibrozil, ketoprofen, midazolam, naloxone, raloxifene, and zidovudine were determined in pooled human liver microsomes using the substrate depletion approach. The in vitro clearance data indicated a varying contribution of glucuronidation to the clearance of the compounds studied, ranging from 6 to 79% for midazolam and gemfibrozil, respectively. The CLint was ob tained using either individual or combined cofactors for cytochrome P450 (P450) and UGT enzymes with alamethicin activation and in the presence and absence of 2% bovine serum albumin (BSA). In the presence of combined P450 and UGT cofactors, CLint ranged from 2.8 to 688 μl/min/mg for zidovudine and buprenorphine, respectively; the clearance was approximately equal to the sum of the CLint values obtained in the presence of individual cofactors. The unbound intrinsic clearance (CLint, u) was scaled to provide an in vivo predicted CLint; the data obtained in the presence of combined cofactors resulted in 5-fold underprediction on average. Addition of 2% BSA to the incubation with both P450 and UGT cofactors reduced the bias in the clearance prediction, with 8 of 10 compounds predicted within 2-fold of in vivo values with the exception of raloxifene and gemfibrozil. The current study indicates the applicability of combined cofactor conditions in the assessment of clearance for compounds with a differential contribution of P450 and UGT enzymes to their elimination. In addition, improved predictability of microsomal data is observed in the presence of BSA, in particular for UGT2B7 substrates.
Glucuronidation is an important reaction in the metabolism of drugs (Williams et al., 2004). It is catalyzed by UDP-glucuronosyltransferase (UGT), a superfamily of membrane-bound enzymes that catalyze the conjugation of d-glucuronic acid to various endo- and xenobiotics. Known human UGT enzymes are classified into two families, UGT1 and UGT2 (Mackenzie et al., 2005). Of the hepatically expressed enzymes UGT1A1, 1A4, 1A9, 2B7, and 2B15 appear to be of greatest significance in drug elimination (Miners et al., 2004; Kiang et al., 2005). UGTs are primarily involved in conjugation of metabolites from oxidation reactions. However, if a suitable electrophilic acceptor group is present, UGTs can also conjugate drugs directly without any prior oxidation step, as seen in the case of buprenorphine (Picard et al., 2005) and diclofenac (Kumar et al., 2002).
Microsomes are commonly used as an in vitro system to predict the metabolic clearance of new chemical entities (Obach, 1999; Soars et al., 2002; Rawden et al., 2005; Rostami-Hodjegan and Tucker, 2007). They offer an advantage over other in vitro systems (e.g., human cryopreserved hepatocytes) because of ease of preparation, wide availability at a low cost, and ease of transport and storage. However, incubations require cofactors for cytochrome P450 (P450) and UGT reactions to be added. The active site of UGTs is on the luminal side of the endoplasmic reticulum, resulting in an in vitro latency. To overcome this phenomenon in vitro, detergents or pore-forming agents (e.g., alamethicin) are added to allow UGT activation (Fisher et al., 2000; Boase and Miners, 2002). Assessment of glucuronidation clearance is further complicated by a lack of glucuronide standards; therefore, substrate depletion offers an alternative approach. However, this approach for the assessment of glucuronidation clearance has been applied in only a limited number of studies (Mohutsky et al., 2006).
Prediction of clearance from microsomal in vitro data has shown a general trend toward underprediction in the case of both P450 and glucuronidated drugs (Mistry and Houston, 1987; Soars et al., 2002; Ito and Houston, 2005; Riley et al., 2005; Miners et al., 2006), with studies reporting 10- to 30-fold underprediction of clearance (Mistry and Houston, 1987; Miners et al., 2006). Investigation into these studies showed that incubation conditions greatly affect the clearance prediction for the glucuronidated drugs (Boase and Miners, 2002; Soars et al., 2003). Although alamethicin has been shown to enhance UGT activity without having any detrimental effect on P450 enzymes (Fisher et al., 2000), the general utility of alamethicin-activated microsomes for the prediction of glucuronidation clearance is still arguable (Engtrakul et al., 2005).
Glucuronidation clearance has been investigated recently in the presence of bovine serum albumin (BSA) in the microsomal incubations (Rowland et al., 2007, 2008b). The authors reported that longchain fatty acids (linoleic and arachidonic acid) released during microsomal incubations competitively inhibit UGT2B7 and UGT1A9 enzymes, with no effect on UGT1A1, UGT1A6, and UGT1A4 (Rowland et al., 2007, 2008b). The addition of BSA to incubations sequesters the fatty acids, resulting in a 9- to 10-fold increase in the intrinsic clearance (CLint) of propofol (Rowland et al., 2008b) and zidovudine (Rowland et al., 2007). A comparable “albumin effect” on the CYP2C9 substrate phenytoin was also observed (Rowland et al., 2008a).
In this study, CLint values for 10 drugs with differential contribution of P450 and UGT pathways were determined in pooled human liver microsomes using the substrate depletion approach. The data set included buprenorphine, carvedilol, codeine, diclofenac, gemfibrozil, ketoprofen, midazolam, naloxone, raloxifene, and zidovudine. The aim of the current study was to investigate the utility of alamethicin-activated human liver microsomes to estimate the fraction metabolized via either P450 (fmCYP) or UGT (fmUGT) using individual P450 and UGT cofactors. In addition, the utility of combined cofactor conditions (P450 + UGT) for the prediction of clearance was investigated, either in the absence or presence of 2% BSA. The general implications of these findings on the suitability of alamethicin-activated microsomes for predicting the clearance for compounds with parallel P450 and UGT pathways are discussed.
Materials and Methods
Chemicals. Buprenorphine, codeine, diclofenac, gemfibrozil, ketoprofen, midazolam, naloxone, raloxifene, NADP+, UDPGA, isocitric acid, EDTA, alamethicin (from Trichoderma viride), BSA, and isocitric acid dehydrogenase were purchased from Sigma Chemical (Poole, Dorset, UK). Carvedilol and zidovudine were purchased from Sequoia Research Products (Pangbourne, West Berkshire, UK).
P450 Incubation Conditions. Pooled human liver microsomes (n = 22; BD Gentest, Woburn, MA) were diluted to a final concentration of 0.2 mg of protein/ml for raloxifene and midazolam, 1 mg of protein/ml for buprenorphine, carvedilol, diclofenac, gemfibrozil, ketoprofen, and naloxone, and 1.5 mg of protein/ml for codeine and zidovudine. Microsomes were diluted in 0.1 M phosphate buffer (pH 7.4) and added to drug solutions to give a final concentration of 5 μM (incubation volume 0.12 ml) for all of the compounds in the data set with the exception of midazolam for which a concentration of 1 μM was used. Drug solutions and microsomes were preincubated for 5 min, and the incubation was initiated by the addition of a NADPH-regenerating system containing NADP+ (1 mM), isocitric acid (7.5 mM), magnesium chloride (10 mM), and isocitric acid dehydrogenase (1.2 units/ml).
UGT Incubation Conditions. Pooled human liver microsomes (n = 22) were diluted as above in 0.1 M phosphate buffer containing magnesium chloride (3.4 mM), the chelating agent, EDTA (1.15 mM), and saccharic acid lactone (115 μM) (conditions adapted from Ogilvie et al. (2006). The microsomes were activated by the addiction of alamethicin at a final concentration of 50 μg/mg microsomal protein and left on ice for 15 min. Drug solutions and microsomes were then preincubated for 5 min, and the reaction was initiated by the addition of UGT cofactor solution containing UDPGA (5 mM).
Combined P450 and UGT Incubation Conditions. Microsomes and drug solutions were treated as described for UGT conditions. The reaction was initiated by the addition of a combined cofactor solution containing NADP+ (1 mM), isocitric acid (7.5 mM), magnesium chloride (10 mM), isocitric acid dehydrogenase (1.2 units/ml), and UDPGA (5 mM). For all incubations, the reaction was terminated at the required time points (0, 2.5, 5, 10, 20, 30, 45, and 60 min) by the addition of 120 μl of acetonitrile containing the relevant internal standard. For experiments in the presence of BSA (both individual and combined cofactors) the following additions were made to the method. Microsomes were diluted in the incubation buffer containing BSA (final 2%). Alamethicin was then added to the microsomes in buffer and left on ice for 15 min. All of the clearance data represent the mean ± S.D. of three separate experiments.
Microsomal Binding. Microsomal binding was determined using high-throughput dialysis as described previously (Gertz et al., 2008). The extent of binding in the presence of BSA was determined following a slightly modified method using a high-throughput dialysis kit (HTDialysis, LLC, Gales Ferry, CT) with membranes with a molecular weight cutoff of 12 to 14 kDa. Microsomes, diluted in buffer with 2% BSA and the drug investigated, were added to the donor side, and phosphate buffer was added to the acceptor side. After reaching equilibrium, aliquots were taken from both sides after 6 h and quenched in ice-cold acetonitrile containing the relevant internal standard.
LC-MS/MS. The LC-MS/MS system used consisted of a Waters 2790 with a Micromass Quattro Ultima triple quadruple mass spectrometer (Waters, Milford, MA). Samples from the microsomal incubations were centrifuged at 2500 rpm for 10 min, and an aliquot of 10 μl was injected into the LC-MS/MS system. Varying gradients of four mobile phases were used, the composition of which were as follows: A, 90% water and 0.05% formic acid with 10% acetonitrile; B, 10% water and 0.05% formic acid with 90% acetonitrile; C, 90% water and 10 mM ammonium acetate with 10% acetonitrile; and D, 10% water and 10 mM ammonium acetate with 90% acetonitrile. For buprenorphine, diclofenac, gemfibrozil, midazolam, naloxone, and raloxifene a Luna C18 column (Phenomenex, Torrance, CA) (3 μ, 50 × 4.6 mm) was used for chromatographic separation of analytes. For carvedilol a Luna phenyl-hexyl column (Phenomenex) (5 μ, 30 × 4.6 mm) was used for chromatographic separation of analytes. The flow rate was set at 1 ml/min, and this was split to 0.25 ml/min before entering the mass spectrometer. The details on the internal standards, mass transitions, and retention times have been outlined previously (Gertz et al., 2008).
Data Analysis. The CLint value determined with both P450 and UGT cofactors present in the incubation and in the presence and absence of 2% BSA was corrected for the corresponding fraction unbound in the incubation (fumic) and scaled to a whole body clearance (milligrams per minute per kilogram) using eq. 1 (Houston, 1994; Obach, 1999):  with a mean scaling factor for a 30-year-old individual of 40 mg protein/g liver (range of 13–54 mg protein/g of liver) (Barter et al., 2007) and a liver weight of 21.4 g liver/kg body weight (Ito and Houston, 2005). The observed hepatic clearance from in vivo intravenous data were converted to an in vivo CLint value using the well-stirred and parallel tube liver models, defined in the eqs. 2 and 3, respectively (Ito and Houston, 2005):
with a mean scaling factor for a 30-year-old individual of 40 mg protein/g liver (range of 13–54 mg protein/g of liver) (Barter et al., 2007) and a liver weight of 21.4 g liver/kg body weight (Ito and Houston, 2005). The observed hepatic clearance from in vivo intravenous data were converted to an in vivo CLint value using the well-stirred and parallel tube liver models, defined in the eqs. 2 and 3, respectively (Ito and Houston, 2005): 
 where fup is the fraction unbound in the plasma, CLb is hepatic blood clearance, RB is the blood to plasma concentration ratio, and QH is the hepatic blood flow (20.7 ml/min/kg) (Brown et al., 2007; Yang et al., 2007).
where fup is the fraction unbound in the plasma, CLb is hepatic blood clearance, RB is the blood to plasma concentration ratio, and QH is the hepatic blood flow (20.7 ml/min/kg) (Brown et al., 2007; Yang et al., 2007).
For buprenorphine and zidovudine, the calculated observed CLb values exceeded the QH; therefore, due to the sensitivity of the well-stirred liver model to the QH, the CLb value was set at 90% of hepatic blood flow for these two drugs. For consistency, a 90% cutoff was applied for these compounds for both liver models used. For raloxifene, no intravenous clearance data were available; therefore, the observed CLint was calculated from an oral clearance using eq. 4:  When the RB was not available a value of 1 was assumed for basic compounds (buprenorphine, codeine, and raloxifene) and a value of 1 – hematocrit (i.e., 0.55) was assumed for acidic compounds (e.g., gemfibrozil and ketoprofen).
When the RB was not available a value of 1 was assumed for basic compounds (buprenorphine, codeine, and raloxifene) and a value of 1 – hematocrit (i.e., 0.55) was assumed for acidic compounds (e.g., gemfibrozil and ketoprofen).

Intrinsic clearance for buprenorphine, raloxifene, and ketoprofen obtained in human liver microsomes using either individual (P450 and UGT) or combined (P450 + UGT) cofactor incubation conditions.
The in vitro fmUGT and fmCYP were determined from the CLint obtained in the presence of individual P450 (CLint, CYP) and UGT (CLint, UGT) cofactors using eqs. 5 and 6, respectively: 
 The predicted CLint values (from in vitro data obtained with combined cofactors and in the presence or absence of BSA) for the current data set (n = 10) were compared with the observed CLint values obtained from the literature. The bias in the predicted CLint was assessed from the geometric mean of the ratio of the predicted and the actual value [average-fold error (afe), eq. 7]. The root mean squared prediction error (rmse; eqs. 8 and 9) provided a measure of precision for the predictions of the CLint values (Sheiner and Beal, 1981; Obach et al., 1997):
The predicted CLint values (from in vitro data obtained with combined cofactors and in the presence or absence of BSA) for the current data set (n = 10) were compared with the observed CLint values obtained from the literature. The bias in the predicted CLint was assessed from the geometric mean of the ratio of the predicted and the actual value [average-fold error (afe), eq. 7]. The root mean squared prediction error (rmse; eqs. 8 and 9) provided a measure of precision for the predictions of the CLint values (Sheiner and Beal, 1981; Obach et al., 1997): 


Results
Clearance via UGT and P450 enzymes was investigated for 10 selected compounds in human liver microsomes using either individual or combined cofactors for these enzymes. The depletion plots for all of the compounds showed a linear time profile, with the exception of diclofenac for which depletion was best described by a biphasic profile; in this case the initial linear phase of depletion plots was used to calculate the CLint. Figure 1 shows the unbound CLint obtained for three compounds, buprenorphine (CLint, CYP > CLint, UGT), raloxifene (CLint, UGT > CLint, CYP), and ketoprofen (CLint, CYP = CLint, UGT) as representative examples of clearance data obtained with either individual P450 or UGT cofactors or in the presence of combined cofactors (P450 + UGT). For buprenorphine, carvedilol, zidovudine, and midazolam the clearance by P450 enzymes was greater than the clearance by UGT enzymes, whereas the opposite trend was observed for the remaining six compounds. For all the drugs in the data set, the CLint with combined cofactors present was comparable with the sum of the individual CLint, CYP and CLint, UGT (Tables 1 and 2).
Clearance obtained for 10 drugs in the presence of individual P450 and UGT cofactors
Data are mean ± S.D. References for major UGT enzymes are available at http://www.pharmacy.manchester.ac.uk/capkr/.
Clearance obtained for 10 drugs in the presence and absence of BSA using combined P450 and UGT cofactors and scaled to in vivo
Table 1 shows the unbound in vitro CLint, obtained in the incubations with individual cofactors and the estimated in vitro fmUGT and fmCYP values for the 10 compounds studied. The unbound CLint, CYP ranged from 2.4 to 472 μl/min/mg for codeine and buprenorphine, respectively. Zidovudine had the lowest clearance by UGT enzymes (2.2 μl/min/mg), whereas raloxifene had the highest CLint, UGT (444 μl/min/mg). The CLint calculated with the individual cofactors was used to estimate an in vitro contribution of UGT and P450 enzymes, as described in eqs. 5 and 6, respectively. The in vitro clearance data indicated a varying contribution of glucuronidation to the clearance of the compounds studied, ranging from 6 to 79% for midazolam and gemfibrozil, respectively. The fmCYP ranged from 0.21 to 0.94 for gemfibrozil and midazolam, respectively (Table 1).
In the presence of both cofactors in the incubation, the CLint ranged from 2.8 to 688 μl/min/mg for zidovudine and buprenorphine, respectively (Table 2). The unbound intrinsic clearance (CLint, u) obtained under these incubation conditions were scaled using the mean human microsomal scaling factor of 40 mg protein/g liver (Ito and Houston, 2005; Barter et al., 2007) (Fig. 2A). The observed CLint covered a 500-fold range with codeine and raloxifene at the lower and upper end of the clearance (Table 3). With use of the well-stirred liver model, the predicted CLint obtained from data in the presence of both cofactors gave a bias of 8.8 and poor precision (rmse of 4566). However, use of the parallel tube model reduced bias by approximately 50% with no significant effect on the precision (Fig. 2A). Independent of the model used, zidovudine and codeine were poorly predicted, with an 18-fold underprediction of clearance observed for these compounds when the parallel tube model was used. The range of scaling factors (13–54 mg protein/g liver) had a marginal effect on the prediction of clearance; however, they influence the extent of underprediction observed.
In vivo clearance values for 10 drugs investigated and the main parameters used in in vitro-in vivo extrapolation
References for plasma clearance, RB, and fup are available at http://www.pharmacy.manchester.ac.uk/capkr/.
When the well-stirred liver model was used, variability in the QH had a pronounced effect on highly cleared compounds such as buprenorphine. An exponential increase in the estimated in vivo CLint was observed when the hepatic clearance approached hepatic blood flow (>95% of QH), resulting in a significant underprediction of clearance from in vitro data. For example, in the case of buprenorphine, a 52-fold underprediction of buprenorphine clearance was observed when the hepatic clearance was set at 99% of QH, whereas setting the hepatic clearance at 90% of the QH resulted in only 5-fold underprediction. Therefore, for drugs for which the observed clearance approached hepatic blood flow, the CLB was limited to 90% of the QH. For consistency, an analogous approach was also applied when the parallel tube liver model was used.
The experiments with alamethicin-activated human liver microsomes using individual and combined P450 and UGT cofactors were also performed in the presence of 2% BSA. Table 2 shows the clearance values (combined cofactors) obtained in the presence and absence of BSA corrected for the extent of nonspecific binding. In the absence of BSA, the fumic ranged from 0.1 to 0.99 for buprenorphine and codeine, respectively. In the presence of BSA, the fumic ranged from 0.008 to 0.99 for diclofenac and codeine, respectively, with the largest decrease in fumic observed for diclofenac (approximately 100-fold). The increase in the individual P450 and UGT CLint estimates in the presence of BSA is shown in Table 4. The -fold increase in CLint, uGT in the presence of BSA ranged from 0.9 to 12.1 for buprenorphine and gemfibrozil, respectively. On average, a 50% increase in fmUGT was observed in the presence of BSA for UGT2B7 substrates. Where available an estimate of the fmUGT was also obtained from renal excretion data and compared with the experimental values (Fig. 3). For naloxone, in vivo and in vitro fmUGT estimates were comparable in the absence of BSA. However, for buprenorphine, codeine, zidovudine, and ketoprofen the experimental fmUGT was lower than the value estimated in vivo. In contrast, the fmUGT obtained in the presence of 2% BSA was more comparable with the in vivo estimates for these drugs (Fig. 3). In the case of gemfibrozil, the in vitro fmUGT was higher than the extent of glucuronidation estimated in vivo (0.40), independent of the addition of BSA to the microsomal incubation.
Fold difference and fmUGT estimates obtained for 10 drugs obtained in the presence of BSA using individual P450 and UGT cofactors
In the presence of BSA, the unbound CLint obtained with combined P450 and UGT cofactors ranged from 13.2 to 2143 μl/min/mg protein for zidovudine and diclofenac, respectively. Addition of 2% BSA resulted in 0.84- to 13-fold increases in CLint for buprenorphine and gemfibrozil, respectively, as illustrated in Fig. 4. When 2% BSA was added, there was an increase in CLint by more than 2-fold for all UGT2B7 substrates in the data set (6 of 10 drugs), whereas a negligible effect was observed for drugs glucuronidated via UGT1A1, with the exception of raloxifene for which a 3.3-fold increase in CLint was observed. The effect was minimal on all drugs with predominant P450 pathways that are not CYP2C9-mediated. For example, a 1.5-fold increase was observed for midazolam, for which UGT1A4 and CYP3A4 are involved in the metabolism.
The impact of 2% BSA on the prediction of CLint was also assessed, as shown in Fig. 2B. Prediction of clearance from the in vitro data obtained in the presence of BSA reduced the bias and extent of underprediction, resulting in 8 of 10 compounds within 2-fold of in vivo values when the parallel tube liver model was used (Fig. 2B). However, incorporation of BSA in the incubation resulted in significant overprediction (18-fold) of gemfibrozil clearance, in contrast to data without BSA, for which predicted and observed clearances were in very good agreement (Table 2). For raloxifene, clearance was underpredicted even after the addition of BSA.
Discussion
In recent years an increasing number of studies have been performed to ascertain the suitability of microsomes to accurately assess the glucuronidation of drugs in vitro (Boase and Miners, 2002; Soars et al., 2002; Miners et al., 2004; Mohutsky et al., 2006). Incubations with microsomes are often carried out to investigate P450 and UGT metabolism individually; however, both pathways of metabolism are not commonly evaluated for a single compound (Engtrakul et al., 2005; Mohutsky et al., 2006). Methods for studying glucuronidation in microsomes have varied considerably, which has led to questions about the suitability of this in vitro system to accurately predict the glucuronidation clearance (Engtrakul et al., 2005). The current study assesses the use of microsomes in the prediction of clearance for compounds with parallel P450 and UGT elimination pathways and the utility of this system to obtain estimates of fmCYP and fmUGT in vitro. The impact of the addition of 2% BSA to microsomal incubations on the clearance prediction was also investigated.

Prediction of clearance from in vitro data obtained in the presence of combined P450 and UGT cofactors and in the absence (A) and presence (B) of 2% BSA.  , buprenorphine; ▾, carvedilol; ▴, codeine; □, diclofenac;
, buprenorphine; ▾, carvedilol; ▴, codeine; □, diclofenac;  , gemfibrozil; ▪, ketoprofen;
, gemfibrozil; ▪, ketoprofen;  , midazolam;
, midazolam;  , naloxone; •, raloxifene; ○, zidovudine. Error bars indicate range of scaling factors on the y-axis from 13 to 54 mg/g liver (Barter et al., 2007) and a range of QH on the x-axis from 17 to 25.5 ml/min/kg (Kato et al., 2003).
, naloxone; •, raloxifene; ○, zidovudine. Error bars indicate range of scaling factors on the y-axis from 13 to 54 mg/g liver (Barter et al., 2007) and a range of QH on the x-axis from 17 to 25.5 ml/min/kg (Kato et al., 2003).
Depletion of parent compound is a common method to determine CLint, and it has been shown to be comparable with a metabolite formation approach (Obach, 2001; Jones and Houston, 2004). Due to a lack of glucuronide standards available, a depletion approach was used to obtain the CLint for this data set. For most of the compounds, the protein concentration and time course used were greater than the proposed optimal values of 0.5 mg/ml and an incubation time of 30 min (Jones and Houston, 2004). These conditions ensured that greater than 20% metabolism was obtained during the incubations to distinguish from any baseline variability in the analytical methodology. For low clearance compounds codeine and zidovudine (zidovudine has high hepatic clearance in vivo), microsomal protein concentrations were >1 mg/ml. However, because these two compounds are not highly bound to the microsomal incubation matrix (Gertz et al., 2008), binding did not restrict clearance estimation. CLint, UGT obtained for low clearance compounds were comparable with values obtained by metabolite formation (Boase and Miners, 2002; Soars et al., 2002), indicating the suitability of depletion data.
To ensure that substrates have access to the active site of the UGT enzyme on the luminal side of the endoplasmic reticulum, alamethicin is commonly used as an alternative to detergents (Fisher et al., 2000; Boase and Miners, 2002). In this study alamethicin was found not to affect P450 activity, which is particularly apparent when individual and combined cofactor studies are compared, as the sum of the individual CLint is approximately equal to the combined cofactor CLint for all of the drugs investigated (Tables 1 and 2). This result is supported by Fisher et al. (2000), who showed that alamethicin had minimal effects on P450 activity when the CYP3A substrate testosterone was studied. Determination of CLint in the presence of either P450 or UGT cofactors allowed the calculation of the in vitro fraction metabolized by the corresponding pathways. In this study we determined the fmCYP and fmUGT for 10 compounds with varying success, compared with renal excretion data (Tables 1 and 4). Improved correlation was observed when BSA was included in the incubation for 4 of 6 drugs for which the in vivo fmUGT data were available. Discrepancy between in vitro and in vivo data (Fig. 3) may have arisen because of the methods for determining the in vivo fmUGT: the estimates were obtained from the amount of glucuronide excreted in the urine, which does not take into account the glucuronide metabolites excreted in the bile/feces. The discrepancy in gemfibrozil estimates may be a result of the stability issues affecting the quantification of the acyl-glucuronide metabolites in vivo (Spahn-Langguth and Benet, 1992), which may lead to an underestimation of the fraction glucuronidated. The fmUGT in vitro may represent a useful initial estimate of the importance of metabolism via glucuronidation and can be incorporated in the prediction of clearance or drug-drug interactions. However, in the absence of information on the potential contribution of renal and biliary clearance to drug elimination, caution is needed in interpretation of these in vitro estimates.
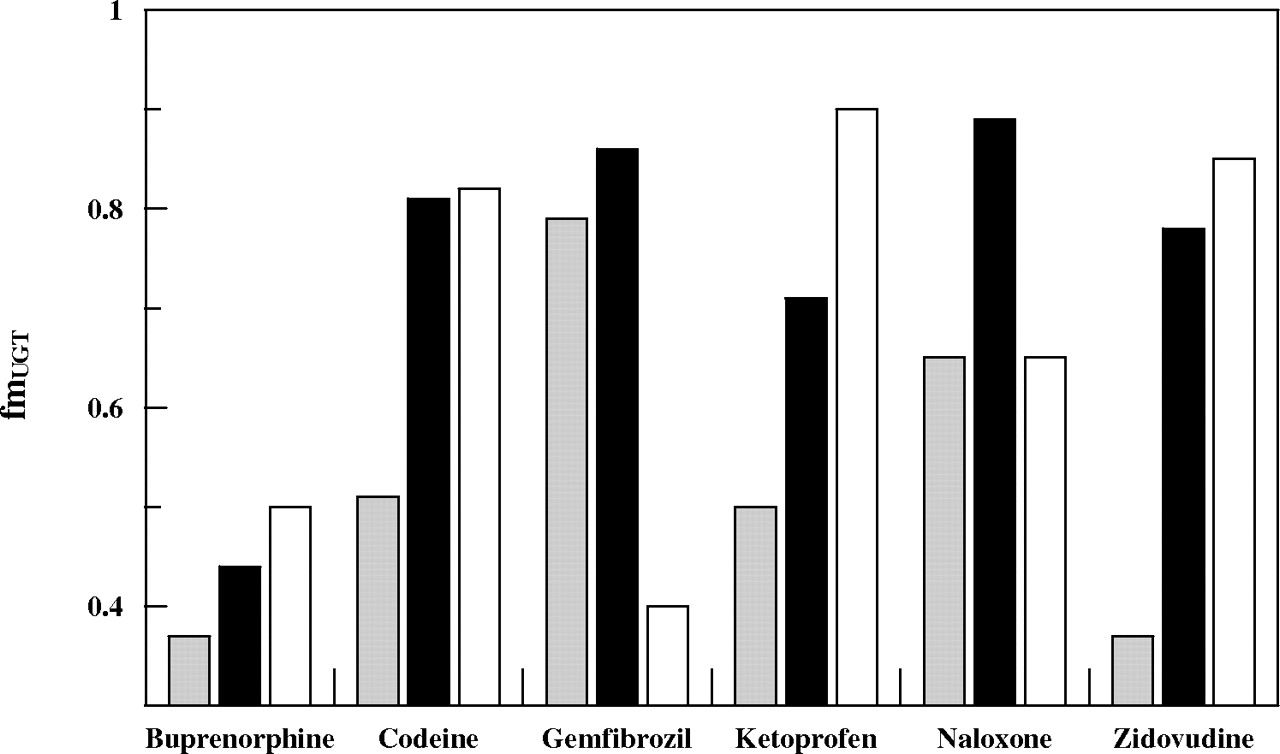
Comparison of the in vitro fmUGT – BSA ( ) and fmUGT + BSA (▪) with the in vivo fmUGT (□) obtained from renal excretion data for six compounds investigated.
) and fmUGT + BSA (▪) with the in vivo fmUGT (□) obtained from renal excretion data for six compounds investigated.
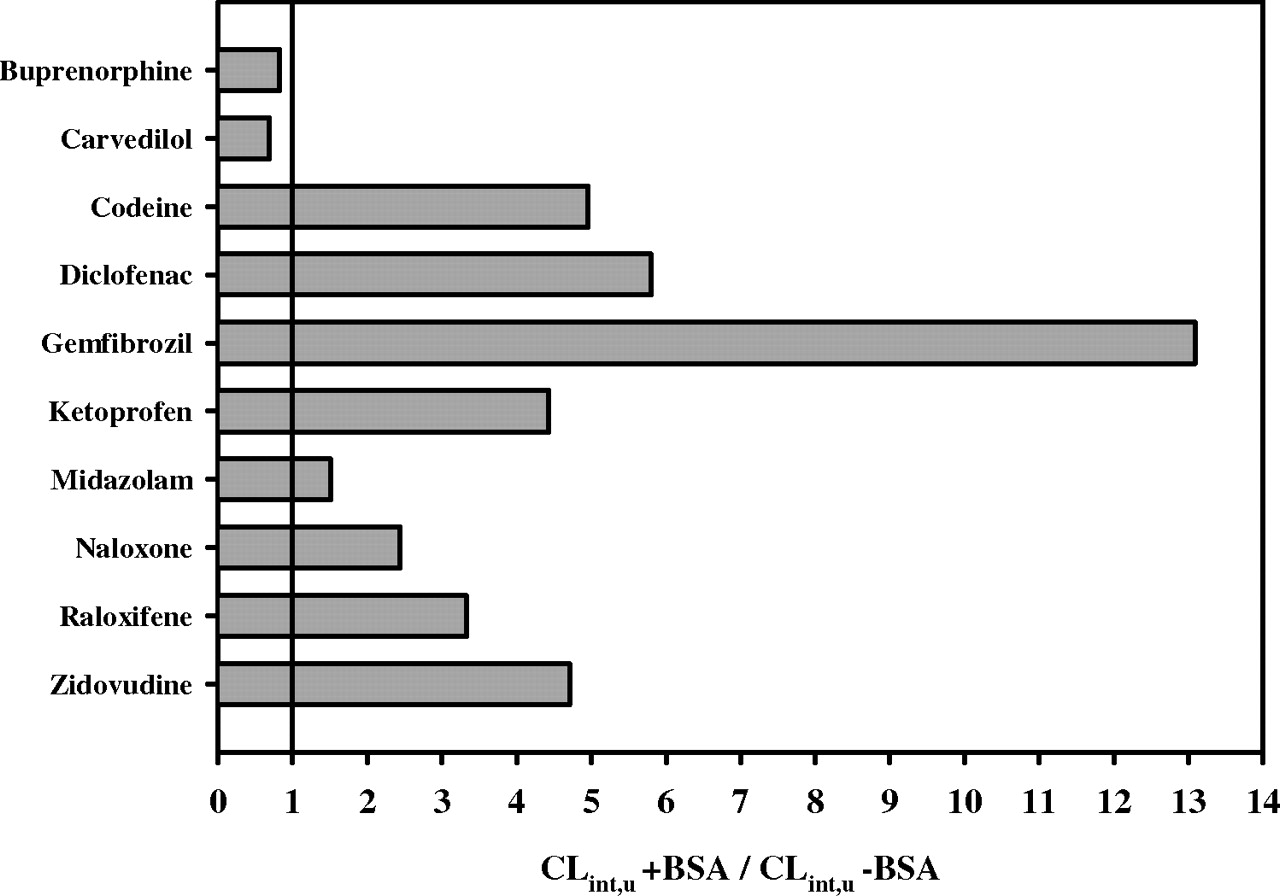
-Fold difference in CLint, u for 10 compounds obtained in the presence and absence of 2% BSA. The CLint was obtained in the presence of P450 and UGT cofactors in both cases. The solid line indicates the CLint, u + BSA/CLint – BSA ratio of 1.
For three compounds investigated metabolism was observed via both direct P450 and UGT pathways although the literature indicates only glucuronidation. In the case of naloxone and raloxifene, a P450 component accounting for approximately 30% of the total clearance was identified. In the case of zidovudine, a significant depletion was also observed in the presence of P450 cofactor (Table 1). However, when BSA is added to the incubation, this trend is reversed, with glucuronidation accounting for 78% of zidovudine metabolism (Table 4), in agreement with the previously reported UGT contribution (Blum et al., 1988). In the case of midazolam, direct glucuronidation accounted for a small proportion of the total clearance (6%), consistent with recent reports on N-glucuronidation via UGT1A4 (Klieber et al., 2008). Therefore, cautious interpretation of the clearance data obtained in the presence of individual cofactors is required, especially if the pathways of metabolism are unknown.
The presence of both cofactors for P450 and UGT in the incubation allowed the assessment of direct oxidation and glucuronidation simultaneously in alamethicin-activated microsomes. This approach has previously been reported for 7-hydroxycoumarin (Fisher et al., 2000) and buprenorphine (Mohutsky et al., 2006) and may provide a useful alternative to the individual cofactor assays for new chemical entities. Predicted CLint obtained from data in the presence of both cofactors correlated well with the observed total CLint; however, a 5.0-fold underprediction was observed. This was mainly driven by the significant underprediction seen with zidovudine, codeine, and raloxifene (predicted CLint represents only 3–6% of observed CLint). The poor prediction observed for low clearance compounds (e.g., codeine) may be confounded by the use of the depletion approach to determine CLint. Over the time course used in this study, 20% depletion of the parent compound was only just reached for these two drugs, adding a potential error in the CLint estimates. For this data set, the parallel tube liver model gave an improved prediction of clearance compared with the well-stirred liver model, reducing the bias by 50%. The improved prediction accuracy with the use of the parallel tube liver model was in good agreement with the observations by Ito and Houston (2005).
It has recently been reported that the addition of BSA to microsomal incubation decreases the Km and consequently increases the clearance estimates for UGT2B7 and UGT1A9 substrates (Rowland et al., 2007, 2008b); an analogous effect was observed on CYP2C9 (Carlile et al., 1999; Rowland et al., 2008a). The rationale is that BSA sequesters the inhibitory effect of unsaturated long-chain fatty acids released during the incubation on certain UGTs. Therefore, zidovudine was included as a control substrate in this study, and formation studies (data not shown) showed a decrease in the Km in the presence of 2% BSA from 1357 to 204 μM, in agreement with findings by Rowland et al. (2007). Within the current data set there was no substantial decrease in clearance observed in the presence of BSA. An improvement in the prediction of clearance was observed for 8 of 10 compounds when 2% BSA was added to the incubation, resulting in a bias of 1.7 (Fig. 2B). This improvement in the clearance prediction in comparison with the data obtained in the absence of BSA is predominantly driven by the increased clearance for the 6 compounds that are metabolized by UGT2B7 (Table 2; Fig. 4). In contrast to the increase in CLint observed for UGT2B7 substrates in the data set, a negligible effect was seen for most drugs glucuronidated by UGT1A1 with the exception of raloxifene for which a 3.3-fold increase in CLint was observed. This increase could be attributed to the effect on the P450-mediated pathway (exact P450 not defined) that contributes 31% to the total clearance of this drug. Raloxifene is also metabolized by UGT1A9 (Kiang et al., 2005), and the 3-fold increase in CLint could be caused by a decrease in the Km for the UGT1A9 enzyme in the presence of BSA, as reported in the case of propofol (Rowland et al., 2008b). The increase in CLint values for ketoprofen and diclofenac is most likely due to the combined effect of BSA on UGT2B7 and CYP2C9-mediated pathways (Table 4). For diclofenac, this assumption is confirmed by the comparable affinities of this drug for both CYP2C9 and UGT2B7 (Carlile et al., 1999; Kiang et al., 2005). The -fold change in CLint is in good agreement with a 5-fold increase in phenytoin CLint observed in the presence of 2% BSA (Rowland et al., 2008b). Diclofenac and phenytoin are reported to be metabolized at the same CYP2C9 binding site (Kumar et al., 2006); therefore, the effects on diclofenac are probably caused by a decrease in the CYP2C9 Km value in a manner similar to that of phenytoin (Rowland et al., 2008a), resulting in an increased CLint.
In conclusion, the current study indicates the applicability of combined cofactor conditions in the assessment of clearance for compounds with a differential contribution of P450 and UGT enzymes to their elimination. Addition of 2% BSA improved the clearance predictability of alamethicin-activated microsomal data, in particular for UGT2B7 substrates. General application of this approach in in vitro-in vivo extrapolation is promising, although the BSA effect is enzyme-specific. Underprediction observed for certain compounds (e.g., raloxifene and naloxone) regardless of the incubation conditions or models used may be attributed to potential metabolism by cytosolic enzymes or contribution of extrahepatic glucuronidation.
Acknowledgments
We thank Prof. John Miners (Flinders University, Adelaide, Australia) for useful discussions and Sue Murby and Dr. David Hallifax (University of Manchester) for assistance with analytical assays.
Footnotes
-
The work was funded by a consortium of pharmaceutical companies (GlaxoSmithKline, Lilly, Novartis, Pfizer, and Servier) within the Centre for Applied Pharmacokinetic Research at the University of Manchester.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.023853.
-
ABBREVIATIONS: UGT, UDP-glucuronosyltransferase; P450, cytochrome P450; BSA, bovine serum albumin; UDPGA, UDP-glucuronic acid; afe, average-fold error; rmse, root mean squared prediction error.
- Received August 12, 2008.
- Accepted October 1, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}