


Abstract
Flutamide is used for prostate cancer therapy but occasionally induces severe liver injury. Flutamide is hydrolyzed in the body into 5-amino-2-nitrobenzotrifluoride (FLU-1) and then further oxidized. In our previous study, N-hydroxy FLU-1 (FLU-1 N-OH) was detected in the urine of patients and exhibited cytotoxicity in rat primary hepatocytes. In the present study, we have assessed the roles of FLU-1 N-oxidation and hepatic glutathione (GSH) depletion in liver injury. FLU-1 (200 mg/kg p.o.) was administered to C57BL/6 mice for 5 days together with 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) (3 mg/kg i.p.) for the first 3 days. Mice were fasted for the last 2 days to deplete hepatic GSH. Administration of FLU-1 alone did not affect serum alanine aminotransferase activities (ALT), whereas coadministration of FLU-1 and TCPOBOP significantly increased ALT in fasted mice but not in nonfasted mice. Microsomal FLU-1 N-hydroxylation was enhanced approximately 5 times by TCPOBOP treatment. Flutamide metabolite-protein adducts were detected in liver microsomes incubated with FLU-1 N-OH, but not with FLU-1 and flutamide, by immunoblotting using antiflutamide antiserum. In the presence of mouse liver cytosol, FLU-1 N-OH was reduced back into FLU-1. This enzymatic reduction required NAD(P)H as a cofactor. The reduction was enhanced by the coexistence of NAD(P)H and GSH, whereas it was markedly inhibited by allopurinol (20 μM). By using purified bovine xanthine oxidase, the reduction was observed in the presence of NAD(P)H. These results suggest that FLU-1 N-OH is involved in flutamide-induced hepatotoxicity and that cytosolic reduction of FLU-1 N-OH plays a major role in protection against flutamide-induced hepatotoxicity.
Flutamide is a nonsteroidal antiandrogen that has been used for prostate cancer therapy. Flutamide is reported to induce severe liver dysfunction in patients, although hepatotoxicity was not recognized in preclinical studies using experimental animals. Approximately 3 per 10,000 users of flutamide were estimated to develop severe liver injury (Wysowski and Fourcroy, 1996). In most cases, flutamide caused liver dysfunction after a latency period (approximately 16 weeks). This flutamide-induced liver injury is not acute but delayed (Thole et al., 2004; Manso et al., 2006). Although reactive metabolites of flutamide have been considered to be involved in hepatotoxicity induction (Berson et al., 1993; Fau et al., 1994; Matsuzaki et al., 2006; Tevell et al., 2006; Kang et al., 2007), the mechanism of flutamide-induced hepatotoxicity in humans remains obscure.
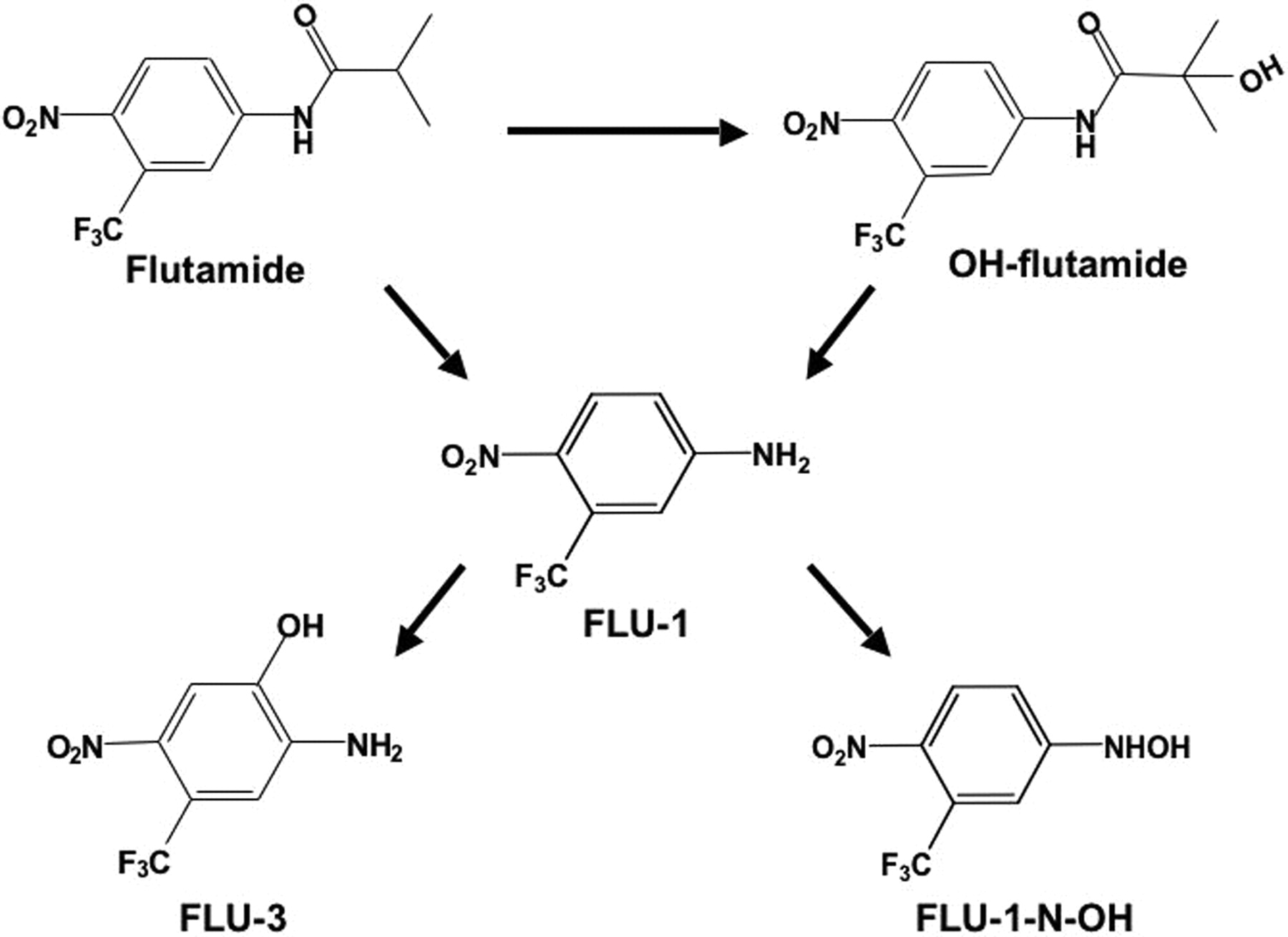
Flutamide is rapidly absorbed from the gastrointestinal tract after p.o. administration and metabolized by hepatic CYP1A2 into 2-hydroxyflutamide (OH-flutamide) (Schulz et al., 1988; Shet et al., 1997; Aizawa et al., 2003; Takashima et al., 2003) (Fig. 1). Flutamide and OH-flutamide are partially hydrolyzed into an arylamine metabolite, 5-amino-2-nitrobenzotrifluoride (FLU-1) (Schulz et al., 1988; Goda et al., 2006). Plasma concentration of FLU-1 was considerably higher in patients who had exhibited liver dysfunction, and lower CYP1A2 activity in patients was found to be associated with onset of liver injury (Ozono et al., 2002; Aizawa et al., 2003). In addition, Cyp1a2-null mice that mainly produced FLU-1 displayed hepatotoxicity by continuous administration of flutamide in contrast to the metabolites formed by wild-type mice (Matsuzaki et al., 2006). These findings suggest that FLU-1 may possibly mediate flutamide-induced liver injury. Although 5-amino-2-nitro-4-hydroxybenzotrifluoride (FLU-3) is the main metabolite in the urine of patients (Goda et al., 2006), FLU-3 did not exhibit obvious cytotoxicity. The other metabolite, N-hydroxy-5-amino-2-nitrobenzotrifluoride (FLU-1 N-OH), is considered to be involved in flutamide hepatotoxicity.

Metabolic pathways of flutamide.
Arylamines are metabolically activated by cytochrome P450 (P450)-mediated N-hydroxylation. Electrophilic N-hydroxylamine is a generally unstable and highly reactive intermediate that reacts with intracellular molecules such as proteins, lipids, and DNA to induce various types of toxicity, including hepatotoxicity (Kato and Yamazoe, 1994; Ju and Uetrecht, 2002; Turesky, 2002; Uetrecht, 2002). In our previous study, the N-hydroxy metabolite of FLU-1 (FLU-1 N-OH) was detected in urine of patients taking flutamide as FLU-1 N-OH glucuronide (Goda et al., 2006). Metabolism of FLU-1 into FLU-1 N-OH was catalyzed mainly by CYP3A4 in humans (Goda et al., 2006). FLU-1 N-OH showed a potent cytotoxicity in rat primary hepatocyte (D. Nagai, R. Goda, E. Ichimura, Y. Akiyama, C. Nishimura, K. Nishikawa, M. Miyata, and Y. Yamazoe, manuscript submitted for publication). However, FLU-1 N-OH was quite stable. Amount of FLU-1 N-OH (free plus conjugates) in urine did not differ between patients with and without hepatic dysfunction (Goda et al., 2006). The role of FLU-1 N-OH in liver injury has not been fully identified.
Glutathione (GSH) is a ubiquitous tripeptide and biosynthesized from cysteine, glycine, and glutamate in mammalian tissues (Meister and Anderson, 1983). GSH is particularly present at a high concentration in liver and participates in cellular protection by scavenging radicals and reacting with electrophiles (Wu et al., 2004). Depletion of GSH is known to increase sensitivity to hepatotoxic compounds (Yang et al., 2002; Watanabe et al., 2003; Akai et al., 2007; McConnachie et al., 2007). Nucleophilic attack of GSH to electrophiles is catalyzed by glutathione S-transferases (GSTs) (Hayes et al., 2005). The role of GSH in flutamide-induced liver injury has not been sufficiently identified. In the present study, we have shown the involvement of N-hydroxy metabolite and protective role of cytosolic reduction of the metabolite in flutamide-induced liver injury using a mouse model.
Materials and Methods
Materials. FLU-1 N-OH (>95%) and antiflutamide antiserum were provided by Nippon Kayaku (Tokyo, Japan). FLU-1, GSH (reduced form), GSH (oxidized form), and 1-chloro-2,4-dinitrobenzene (CDNB) were purchased from Wako Pure Chemicals (Osaka, Japan). Flutamide, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP), β-naphthoflavone (β-NF), pregnenolone-16α-carbonitrile (PCN), 2,4-dinitrofluorobenzene, allopurinol, pyrazole, dicumarol [3,3′-methylene-bis(4-hydroxycoumarin)], human GST (G8642), equine GST (G6511), rabbit GST (G8261), xanthine oxidase from bovine milk (X4500), and bovine serum albumin (BSA) were purchased from Sigma-Aldrich Japan K.K. (Tokyo, Japan). Umbelliferone was purchased from Nacalai Tesque (Kyoto, Japan). Cellulose membrane was purchased from Viskase Companies, Inc. (Darien, IL).
Animals and Treatments. C57BL/6 Cr Slc male mice were purchased from Japan SLC (Hamamatsu, Shizuoka, Japan) and housed under standard 12-h light/12-h dark cycle with access to a standard rodent diet CE-2 (Clea, Tokyo, Japan) and water ad libitum. Age-matched groups of 9- to 10-week-old mice were used in all the experiments.
Flutamide (200 mg/kg) or FLU-1 (200 mg/kg) was suspended in 0.5% carboxymethylcellulose solution and administered p.o. to mice for 5 days. These are toxicological doses. TCPOBOP (3 mg/kg) was suspended in corn oil and administered i.p. for the first 3 days. Mice were fasted for the last 2 days to deplete hepatic GSH and were euthanized 4 h after the last administration. The experimental protocol was shown as Fig. 2. Animal experiments were performed under the instruction of Tohoku University Animal Care and Use Committee.

Experimental protocol to develop mouse model for flutamide-induced liver injury. C57BL/6 male mice (10 weeks old) were used. Flutamide or FLU-1 (200 mg/kg) was given p.o. for 5 days, and TCPOBOP (3 mg/kg weight) was given i.p. for the first 3 days. Mice were fasted for the last 2 days to deplete hepatic GSH and were euthanized 4 h after the last treatment, and blood and livers were collected.
Alanine Aminotransferase Activities in Serum. Blood was obtained from inferior vena cava of mice. Serum was prepared from blood by centrifugation at 20,400g for 20 min. Serum alanine aminotransferase (ALT) activities were measured using a commercial kit, Transaminase CII-B-test Wako (Wako Pure Chemicals).
Preparation of Mouse Liver Microsomes and Cytosols. Mouse livers were homogenized in 75 mM potassium-phosphate buffer, pH 7.4, containing 75 mM KCl and centrifuged at 9000g for 20 min at 4°C. The supernatant was centrifuged at 105,000g for 60 min at 4°C. For microsome preparation, the final pellet was washed twice with 75 mM potassium-phosphate buffer, pH 7.4, containing 75 mM KCl and suspended in 75 mM potassium-phosphate buffer, pH 7.4, containing 20% glycerol. Cytosolic fraction was obtained from 105,000g centrifugation during microsome preparation. Protein concentration was determined by the method of Lowry et al. (1951) with BSA as the standard.
Metabolism of FLU-1 in Mouse Liver Microsomes. The reaction mixture consisted of 20 μg of microsomal protein, 200 μM FLU-1, 0.1 M potassium-phosphate buffer, pH 7.4, 4.8 mM MgCl2, 0.32 mM NADP+, 2.4 mM glucose 6-phosphate, and 0.26 units/ml glucose-6-phosphate dehydrogenase in a final volume of 0.2 ml. The mixture was incubated for 10 min at 37°C. The reaction was terminated by addition of 0.4 ml of ice-cold methanol containing 100 μM ascorbic acid, and 2 nmol of umbelliferone was added to the mixture as an internal standard. The mixture was centrifuged at 3000g for 5 min, and the supernatant was analyzed by high-performance liquid chromatography (HPLC) as described below.
HPLC Conditions. HPLC analysis was performed with Jasco (Tokyo, Japan) intelligent model PU-980 pump and UV-970 detector. Metabolites were separated by ODS-80 TM column (4.6 × 150 mm, 5 μm; Tosoh, Tokyo, Japan) and monitored by UV detection at 366 nm at room temperature. Flow rate was 0.75 ml/min with mobile phase of ammonium-acetate buffer (25 mM, pH 6.5) and methanol (68:32).
Detection of Cyp3a or Cyp1a in Mouse Liver Microsomes by Immunoblot Analysis. Microsomal proteins boiled for 2 min were separated by SDS-polyacrylamide gel electrophoresis (PAGE) with 8% acrylamide gel and transferred to nitrocellulose membrane. The membrane was incubated for 2 h in blocking solution consisting of phosphate-buffered saline (PBS), Tween 20 [0.05% (v/v)], and fetal calf serum [20% (v/v)]. The membrane was then treated with anti-rat CYP3A2 antibody at 1:1000 dilution or anti-rat CYP1A1 antibody at 1:10,000 dilution in PBS-Tween for 2 h at room temperature. Subsequently, the membrane was treated with alkaline phosphatase-conjugated goat anti-rabbit IgG at 1:3000 dilution in PBS-Tween for 2 h at room temperature. Thereafter, the membrane was washed five times in PBS-Tween, and antibody reactivity was visualized using 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium staining. The stained membranes were scanned with an Epson America (Torrance, CA) GT-8700 scanner, and the band intensities were measured using National Institutes of Health image (version 1.59) software (Bethesda, MD).
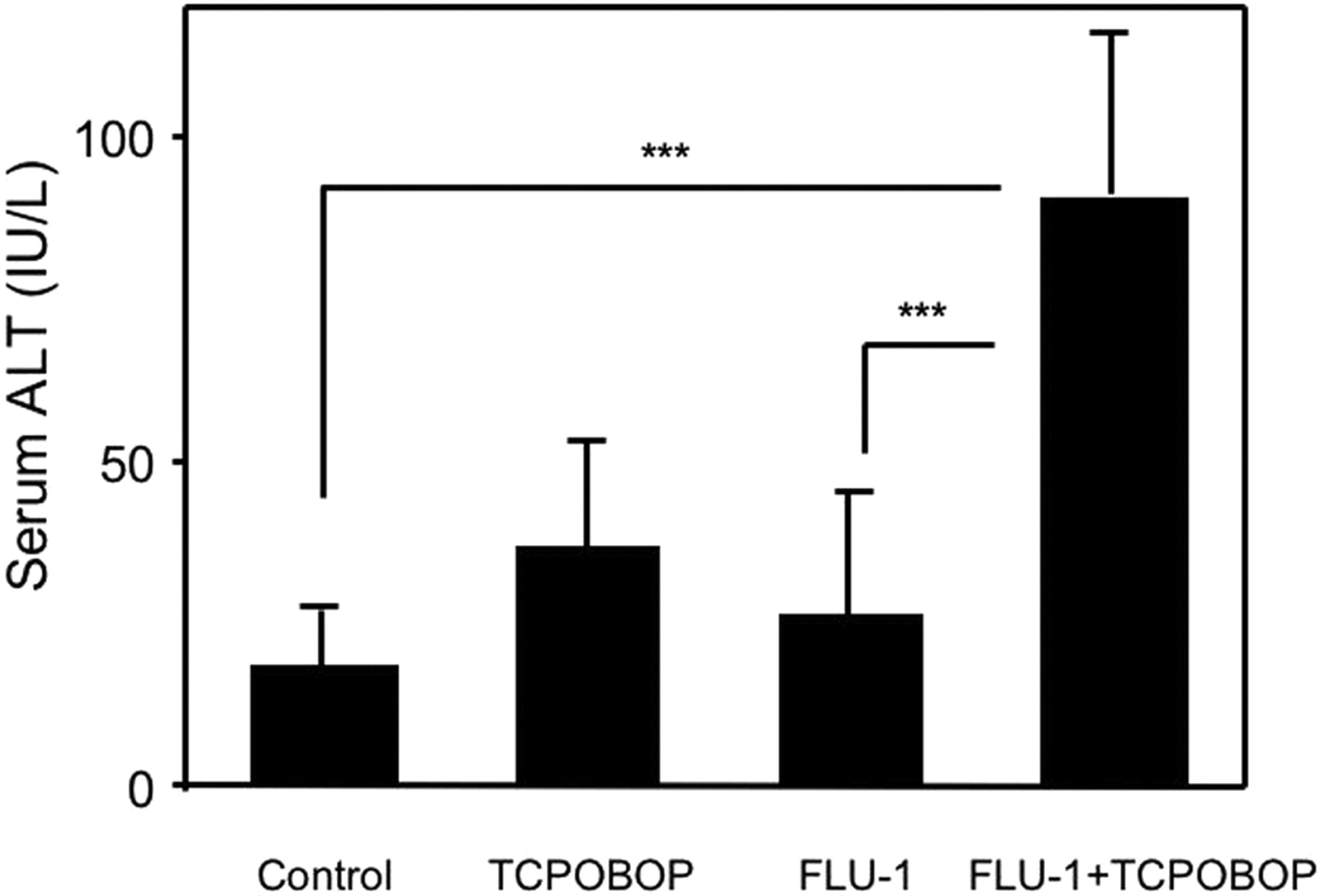
Influence of TCPOBOP administration on FLU-1-induced liver injury in fasted mice. FLU-1 and TCPOBOP were coadministered to mice as shown in Fig. 2. Mice were fasted for the last 2 days. Blood was collected 4 h after last administration. Data are shown as mean ± S.D. (n = 5–6) and analyzed by Tukey's test. ***, P < 0.001. (Significant difference between the indicated groups.)
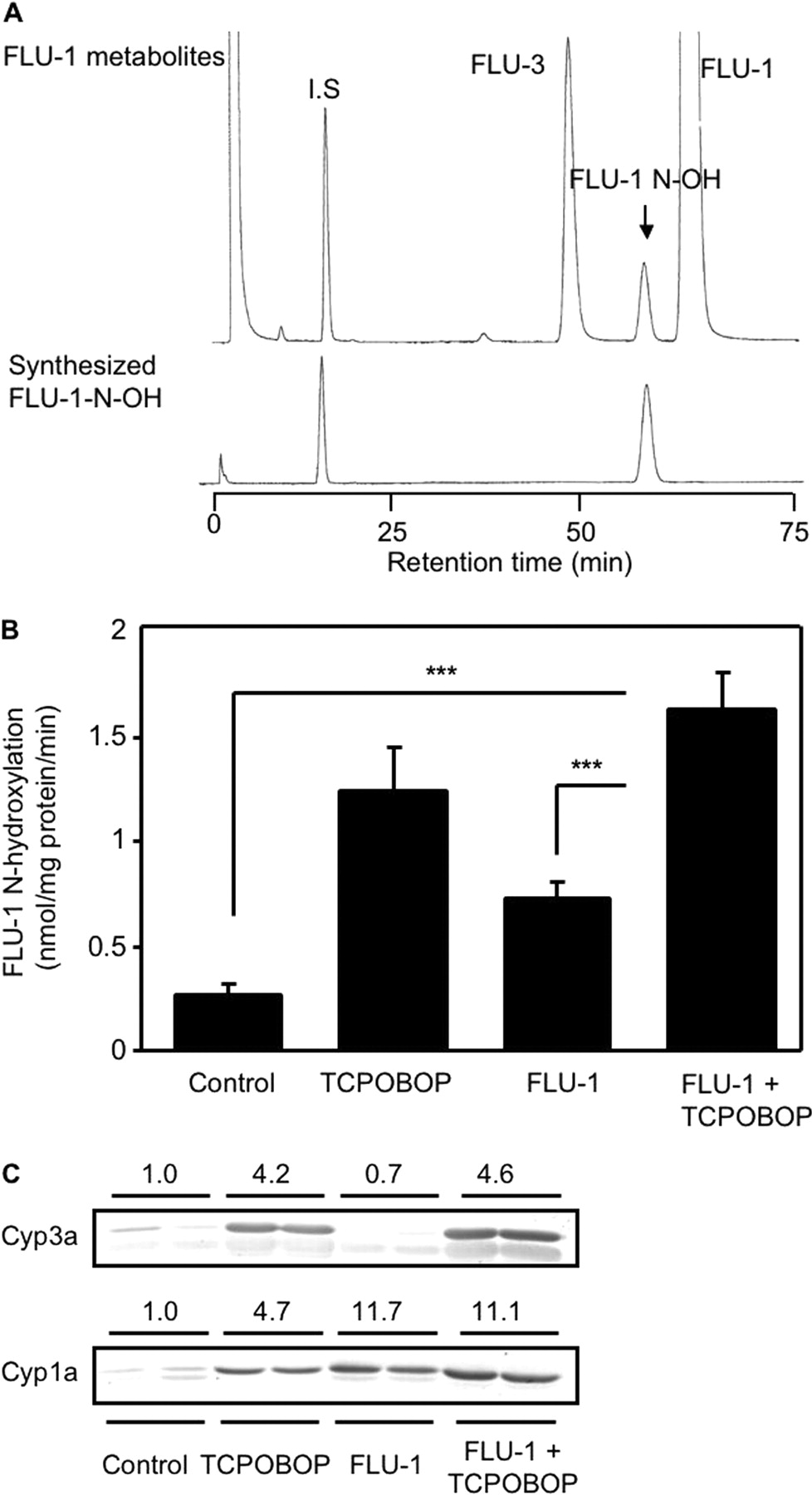
Influence of TCPOBOP administration on FLU-1 N-hydroxylation and P450s expression in mouse liver microsomes. A, HPLC chromatogram of FLU-1 metabolites in mice. FLU-1 (200 μM) was incubated for 10 min at 37°C with microsomes (0.1 mg/ml) prepared from TCPOBOP-administered mouse livers. Reaction mixture was analyzed by HPLC (λ = 366 nm). I.S., internal standard (umbelliferone). B, rate of microsomal FLU-1 N-hydroxylation was determined by HPLC (λ = 366 nm). FLU-1 (200 μM) was incubated with mouse liver microsomes (0.1 mg/ml) prepared from mice for 10 min at 37°C. Data are shown as mean ± S.D. (n = 5–6) and analyzed by Tukey's test. ***, P < 0.001. (Significant difference between the indicated groups.) C, hepatic Cyp3a and Cyp1a expression levels were determined by immunoblot analysis. Microsomal proteins (5 μg for Cyp3a and 20 μg for Cyp1a) were separated by SDS-PAGE with 8% acrylamide gel. Anti-rat CYP3A2 antibody and anti-rat CYP1A1 antibody were used for determination of mouse Cyp3a and Cyp1a, respectively. Data are shown as two representative samples. Relative expression levels are indicated.
Interaction between Flutamide and Its Metabolites with BSA. The reaction mixture consisted of 10 μM FLU-1 or FLU-1 N-OH, 0 to 1 mg/ml BSA, and 0.1 M potassium-phosphate buffer, pH 7.4, in a final volume of 0.1 ml. Incubation was carried out for the indicated period at 37°C. To remove the metabolite binding to BSA, 200 μl of ice-cold methanol was added, and the reaction mixture was centrifuged at 3000g for 5 min. Unbound form of metabolite in supernatant was measured by HPLC.
Detection of Flutamide-Protein Adducts by Immunoblot Analysis. Detection of flutamide-protein adducts was carried out as described previously with some modification (D. Nagai, R. Goda, E. Ichimura, Y. Akiyama, C. Nishimura, K. Nishikawa, M. Miyata, and Y. Yamazoe, manuscript submitted for publication). In brief, FLU-1 N-OH, FLU-1, flutamide, or OH-flutamide (500 μM) was incubated with microsomal protein (2 mg/ml) for 2 h at 37°C. Microsomal proteins (50 μg) boiled for 2 min were separated by SDS-PAGE with 10% acrylamide gel and transferred to nitrocellulose membrane. The membrane was incubated for 2 h in blocking solution as described above. The membrane was treated with antiflutamide antiserum at 1:3000 dilution in 0.1% BSA/PBS-Tween for 90 min at room temperature. Subsequently, the membrane was treated with alkaline phosphatase-conjugated goat anti-rabbit IgG for 60 min. Thereafter, the membrane was washed five times in PBS-Tween, and antibody reactivity was visualized using 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium staining.
Metabolism of FLU-1N-OH. The reaction mixture consisted of 0.1 M potassium-phosphate buffer, pH 7.4, 4.8 mM MgCl2,50 μM FLU-1 N-OH, and 20 μg of cytosolic protein or 1.1 μg (1.5 mU) of purified xanthine oxidase protein with and without 10 mM NAD(P)H or 10 mM GSH in a final volume of 0.2 ml. The mixture was incubated for the indicated period at 37°C. For inhibition study, CDNB (50 μM and 1.5 mM), allopurinol (20 μM), pyrazole (5 mM), and dicumarol (15 μM) were added 3 min before the start of incubation. The reaction was terminated by addition of 0.4 ml of ice-cold methanol containing 100 μM ascorbic acid, and 2 nmol of umbelliferone was added to the mixture as an internal standard. The mixture was then centrifuged at 3000g for 5 min, and the supernatant was analyzed by HPLC. For the preparation of dialyzed cytosolic protein, cytosolic fraction was dialyzed three times using cellulose membrane (UC20-32) with 0.1 M potassium-phosphate buffer, pH 7.4, containing 4.8 mM MgCl2 for 2 h at 4°C.
Measurement of Hepatic GSH Level and GSH/GSSG Level in Reaction Mixture. Liver homogenates were diluted with 10% trichloroacetic acid (1:4) and centrifuged at 9000g for 5 min to obtain nonprotein supernatants. GSH levels were determined by the method of Tietze et al. (1969). GSH/GSSG levels in reaction mixture were measured by HPLC after the amino groups of GSH/GSSG were derivatized with 2,4-dinitrofluorobenzene (Reed et al., 1980).
Statistical Analysis. Data are shown as the mean ± S.D. Statistical differences were determined by either an unpaired Student's t test or one-way analysis of variance followed by Tukey's multiple comparison with PRISM 4.0 software (GraphPad Software Inc., San Diego, CA). P < 0.05 was considered to be statistically significant.
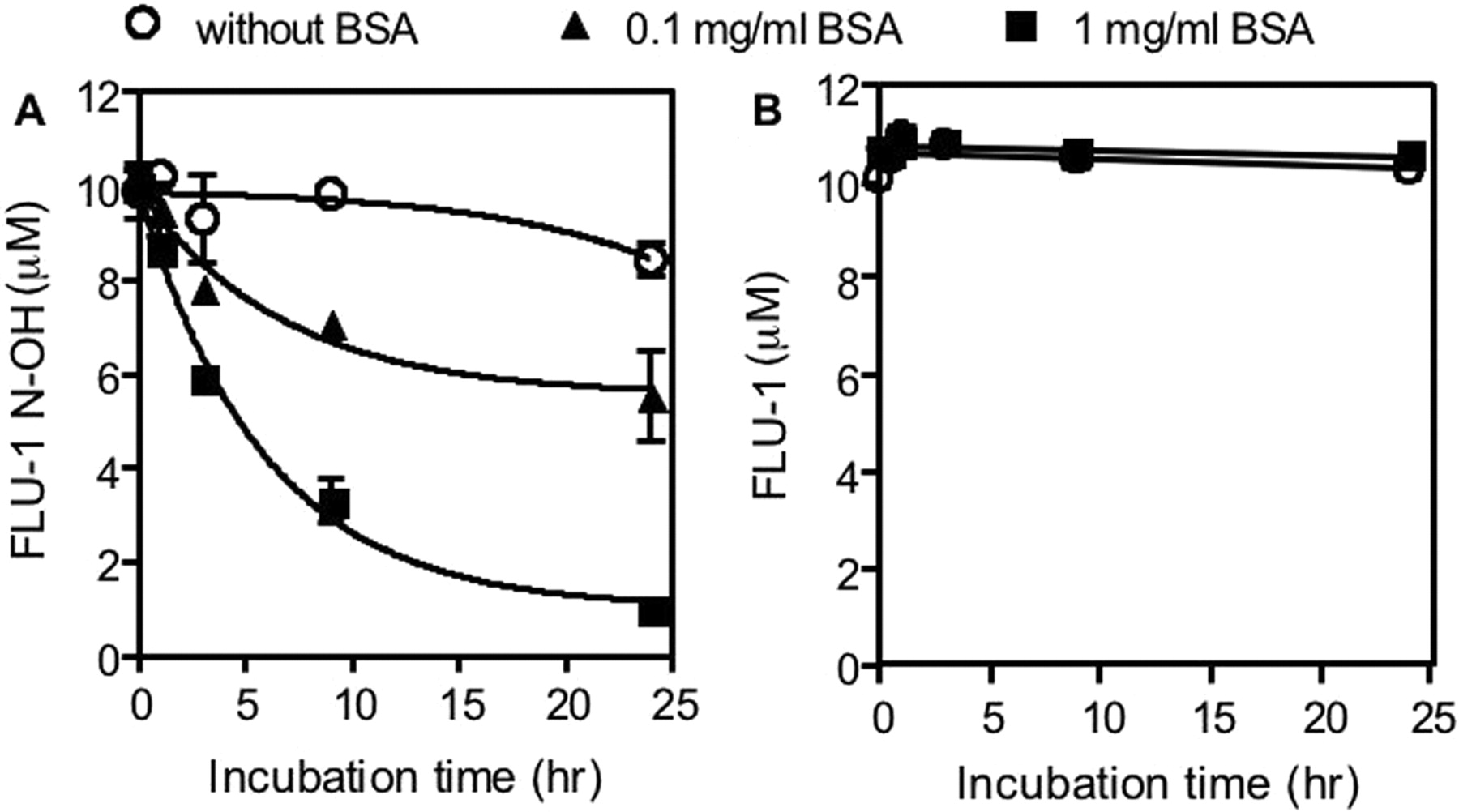
Interaction between flutamide metabolites and BSA. FLU-1 N-OH (A) or FLU-1 (10 μM) (B) was incubated with BSA at 37°C for indicated period. To remove BSA, reaction mixture was centrifuged after methanol was added. FLU-1 N-OH or FLU 1 in supernatant was determined by HPLC. Data are shown as the mean ± S.D. (n = 3).
Results
Animal Experiment with Flutamide and FLU-1. Fasting decreased hepatic GSH levels approximately 60% in C57BL/6 male mice but did not affect ALT (data not shown). TCPOBOP, a drug-metabolizing enzyme inducer, was reported to aggravate liver injury induced by acetaminophen or cocaine through P450-mediated bioactivation (Wei et al., 2000; Zhang et al., 2002). Administration of TCPOBOP or FLU-1 alone did not increase ALT, but coadministration of FLU-1 and TCPOBOP significantly increased ALT compared with vehicle control (Fig. 3). Coadministration of flutamide and TCPOBOP was also attempted but did not increase ALT compared with vehicle control (data not shown).
Influence of TCPOBOP on Microsomal FLU-1N-Hydroxylation. Formation of FLU-1 N-OH was tested in mouse liver microsomes by HPLC. FLU-1 N-OH and FLU-3 were detected as microsomal metabolites of FLU-1 (Fig. 4A). Administration of TCPOBOP (3 mg/kg for 3 days) to mice increased approximately 5-fold microsomal FLU-1 N-hydroxylation compared with vehicle treatment (Fig. 4B). FLU-1 administration (200 mg/kg for 5 days) also increased FLU-1 N-hydroxylation but less than the TCPOBOP administration. Administration of TCPOBOP significantly increased hepatic expression of Cyp1a and Cyp3a in liver microsomes (Fig. 4C). FLU-1 also increased Cyp1a expression. Treatment with rodent Cyp1a and Cyp3a inducer, β-NF and PCN, respectively, also increased hepatic FLU-1 N-hydroxylation (data not shown).
Covalent Binding of FLU-1N-OH to Mouse Liver Microsomes. To investigate the ability of FLU-1 N-OH to interact with protein, FLU-1 N-OH was incubated with BSA. Amount of FLU-1 N-OH in supernatant was decreased with incubation time and BSA concentration (Fig. 5A). Although FLU-1 N-OH in supernatant was not altered within 24-h incubation in the absence of BSA, only approximately 10% of FLU-1 N-OH was recovered after 24-h incubation with 1 mg/ml BSA. Similar results were obtained from the experiment in which heat-treated microsomes were used instead of BSA (data not shown). In contrast, FLU-1 was completely recovered regardless of incubation time and BSA concentration (Fig. 5B). Flutamide and OH-flutamide also did not show any interaction with BSA and FLU-1 (data not shown).
Furthermore, flutamide metabolite-protein adducts were detected by immunoblot analyses using a rabbit polyclonal antisera raised against a synthetic flutamide-keyhole limpet hemocyanin (KLH) conjugate. Hapten ({N-[4-nitro-3-(trifluoromethyl)phenyl]succinic acid}) was conjugated with KLH. The antiserum was developed by immunization of the hapten KLH conjugate. Several protein adducts were detected by the antisera between 37 and 75 kDa after incubation of microsomal protein with FLU-1 N-OH but not with FLU-1, flutamide, and OH-flutamide (Fig. 6). The adduct formation was prevented by addition of GSH in the reaction mixture (D. Nagai, R. Goda, E. Ichimura, Y. Akiyama, C. Nishimura, K. Nishikawa, M. Miyata, and Y. Yamazoe, manuscript submitted for publication).

Detection of flutamide metabolite-protein adducts using antiflutamide antiserum. FLU-1-N-OH, FLU-1, flutamide, or OH-flutamide (500 μM) was incubated with liver microsomal proteins (2 mg/ml) at 37°C for 2 h. Microsomal proteins were separated by SDS-PAGE with 10% acrylamide gel. Drug-protein complexes were detected by immunoblot analysis with antiflutamide antibody. Data are shown as two representative samples.
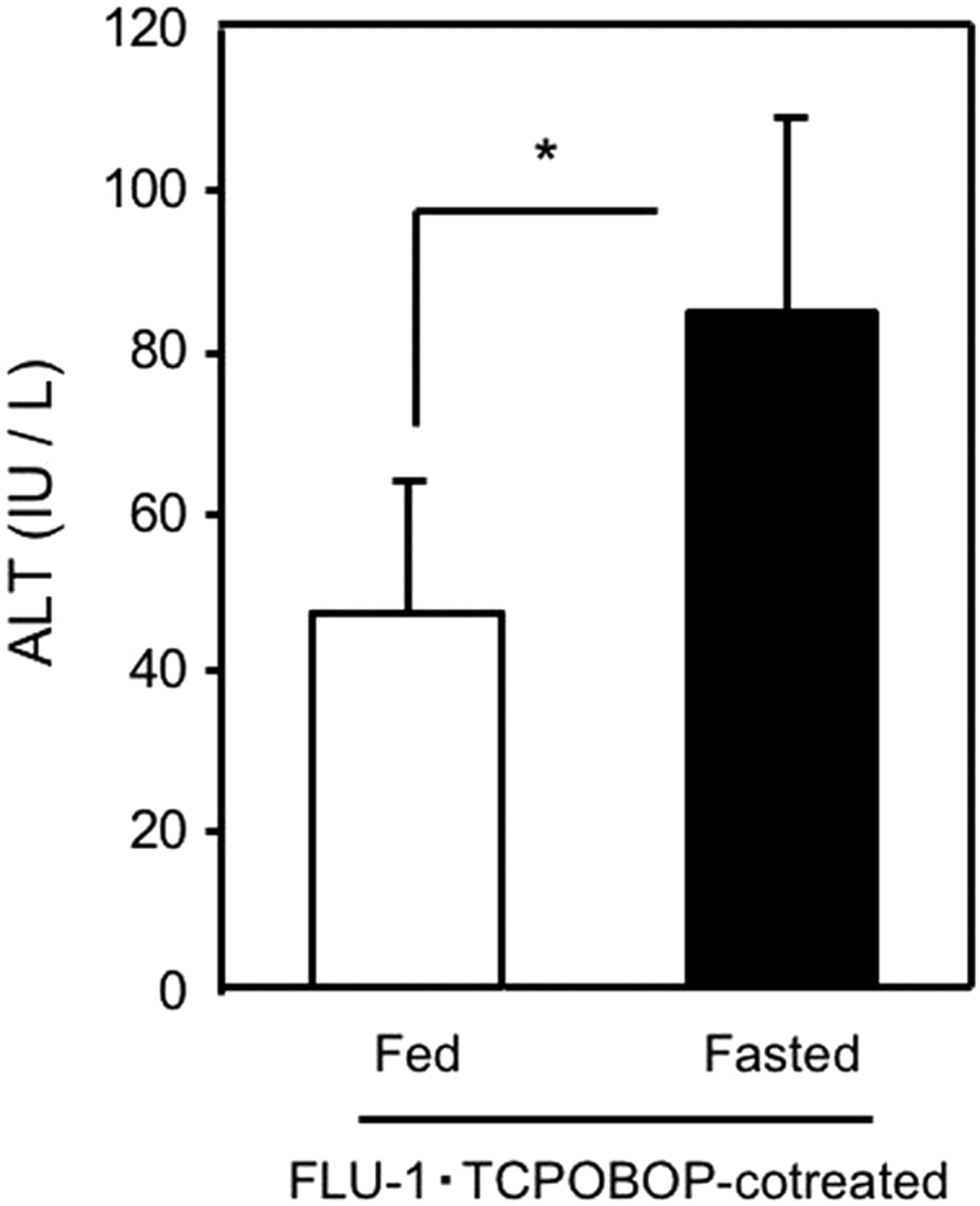
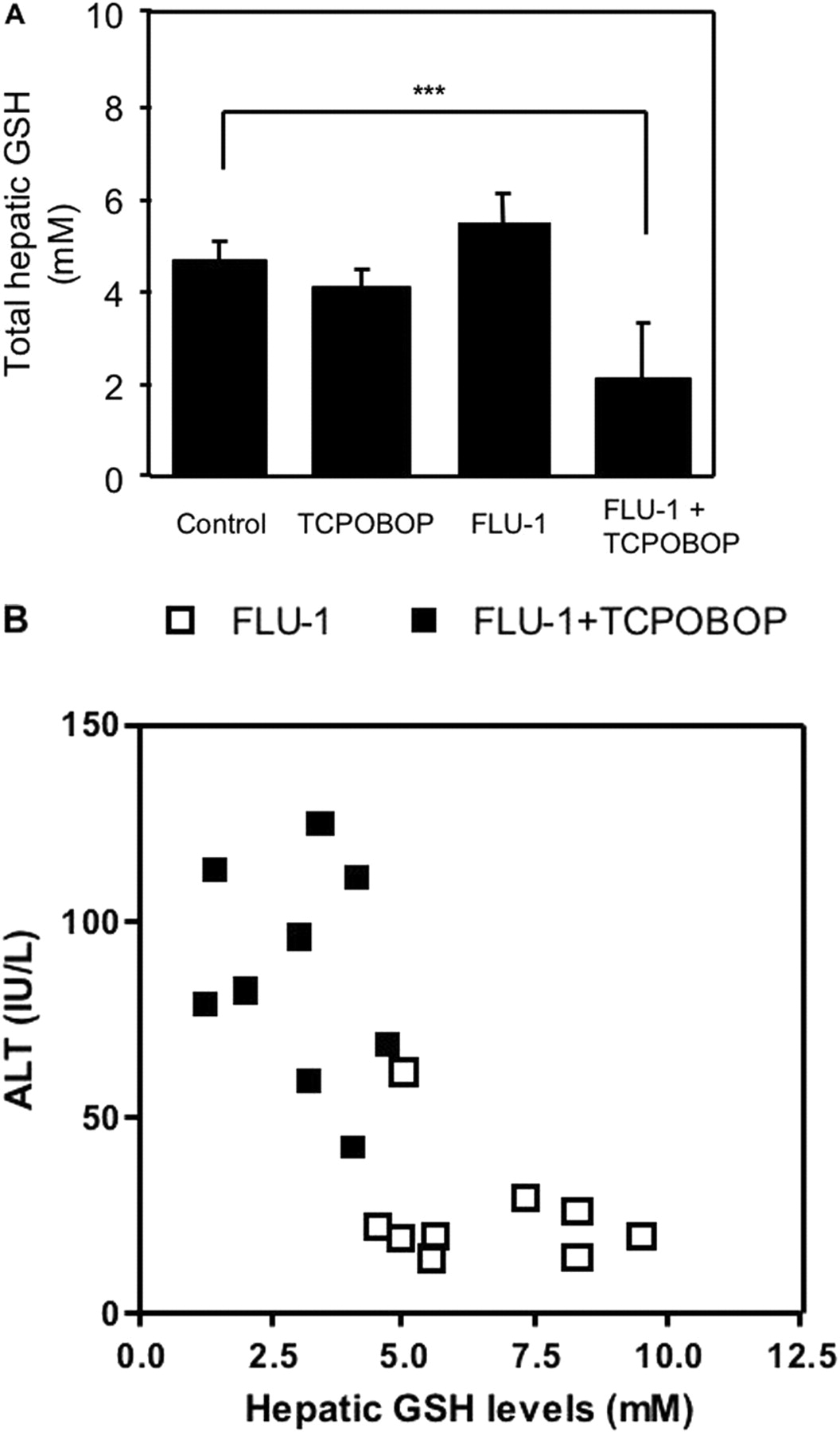
Protective Role of Hepatic GSH in Liver Injury. To determine the protective role of hepatic GSH against onset of liver injury, FLU-1 and TCPOBOP were coadministered to fasted or nonfasted mice. Coadministration of FLU-1 and TCPOBOP significantly increased ALT in fasted mice compared with nonfasted mice (Fig. 7). Furthermore, hepatic GSH levels were not changed by administration of TCPOBOP or FLU-1 alone in fasted mice, but hepatic GSH levels were significantly decreased approximately 50% in mice treated with both FLU-1 and TCPOBOP compared with FLU-1-treated mice (Fig. 8A), and an inverse relationship (r = 0.71; P < 0.001) was observed between hepatic GSH levels and serum ALT activities in those groups (Fig. 8B). These results suggest the protective role of hepatic GSH in flutamide-induced liver injury.
Enzymatic Reduction of FLU-1N-OH with GSH and NAD(P)H. When FLU-1 N-OH was incubated with hepatic cytosol, FLU-1 was detected as a metabolite in a time-dependent manner (Fig. 9A). Reduction of FLU-1 N-OH to FLU-1 accounted for approximately 40 to 60% loss of FLU-1 N-OH. The relative rates of FLU-1 formation to the disappearance of FLU-1 N-OH were decreased in a time-dependent manner. To study the influence of the presence of cofactors in the formation of FLU-1, FLU-1 N-OH was incubated with NADH or GSH in the presence of mouse liver cytosol. The formation of FLU-1 was not observed in a system of FLU-1 N-OH incubation with GSH or NADH alone (Fig. 9, B and C) or with GSH or NADH and heat-treated cytosols (data not shown). However, it was enhanced by addition of GSH or NADH and cytosols. Amount of FLU-1 N-OH did not change in the incubation with GSH or NADH alone.
To further verify the electron donor for the formation of FLU-1, dialyzed cytosols were used to measure FLU-1 formation. This FLU-1 formation using dialyzed cytosols was reduced to 7% of native cytosols. FLU-1 formation was increased up to 6.9- and 8.3-fold by addition of NADPH (10 mM) or NADH (10 mM), respectively, to the reaction mixture containing the dialyzed cytosols (Fig. 10). Although addition of GSH (10 mM) alone did not increase such formation in the dialyzed cytosols, the addition of both GSH and NADH increased the formation compared with that of NADH alone.

Influence of fasting on liver injury induced by FLU-1 and TCPOBOP coadministration. Coadministration of FLU-1 and TCPOBOP was carried out as shown in Fig. 1. FLU-1- and TCPOBOP-coadministered mice were fasted for 2 days (fasted group) or continued to be fed ad libitum (nonfasted group). Blood samples were collected 4 h after euthanasia, and serum ALT activities were determined. Data are shown as the mean ± S.D. (n = 4–5). Data are analyzed by Student's t test. *, P < 0.05. (Significant difference between the groups.)
To establish which enzyme was involved in the formation of FLU-1, representative inhibitors of NAD(P)H-dependent cytosolic enzymes were added to the reaction mixture. The formation was strongly inhibited by addition of allopurinol (25 μM), a potent inhibitor of xanthine or aldehyde oxidase, whereas it was only weakly (less than 11%) inhibited by addition of pyrazole (5 mM), a potent inhibitor of alcohol dehydrogenase (Table 1). CDNB, a GST substrate, inhibited FLU-1 formation. The formation was inhibited approximately 30% by addition of CDNB (50 μM).
Effect of inhibitors on cytosolic FLU-1 N-OH reduction
FLU-1 N-OH reduction was determined by HPLC (λ = 366 nm). FLU-1 N-OH (50 μM) was incubated in cytosol (0.1 mg/ml) at 37°C for 10 min. Inhibitors were added 3 min before start of incubation. Each value represents the mean of two independent experiments.
To test for the involvement of xanthine oxidase or GST in FLU-1 N-OH reduction, purified xanthine oxidase from bovine, GST proteins from human placenta (GSTM1 and GSTP1), equine liver, and rabbit liver were used. By using purified bovine xanthine oxidase, the FLU-1 formation was observed in the presence of NADH (37.7 nmol/mg of protein/min). This specific activity was 61-fold higher than that of cytosolic protein from mouse liver. The NADPH-dependent FLU-1 formation was 4-fold lower than that of NADH. The FLU-1 formation was not observed in the absence of NAD(P)H. By using any of GST proteins, FLU-1 formation from FLU-1 N-OH was not found. Furthermore, GSH consumption and GSSG formation during FLU-1 formation were not observed in the reaction mixture containing cytosol and FLU-1 N-OH.
The rate of FLU-1 N-hydroxylation per gram of liver was enhanced by TCPOBOP or FLU-1 administration (Table 2). On the other hand, the rate of FLU-1 N-OH reduction per gram of liver showed similar values (approximately 40 μmol/g of liver/min) among all the groups.
Comparison of FLU-1 N-hydroxylation with FLU-1 N-OH reduction in mouse livers
FLU-1 N-hydroxylation and FLU-1 N-OH reduction were determined by HPLC (λ = 366 nm). FLU-1 (200 μM) was incubated in microsomes (0.1 mg/ml) at 37°C for 10 min. FLU-1 N-OH (50 μM) was incubated in cytosol (0.1 mg/ml) at 37°C for 20 min. Data are shown as the mean of the activity per gram of liver ± S.D. (n = 4–5).
Discussion
The present study using a mouse model has suggested that N-hydroxy flutamide metabolite, FLU-1 N-OH, is involved in flutamide-induced liver injury. Furthermore, we show that FLU-1 N-OH is reduced to FLU-1 in the presence of cytosol and NAD(P)H. Coexistence of GSH increased the rate of the reaction.
Coadministration of FLU-1 and TCPOBOP elevated ALT in fasted mice (Fig. 3). On the other hand, FLU-1 administration alone or coadministration of flutamide and TCPOBOP did not increase ALT in fasted mice. These results suggest involvement of reactive intermediates from FLU-1, whose production is enhanced by treatment with TCPOBOP, in flutamide hepatotoxicity.

Influence of FLU-1 and TCPOBOP coadministration on hepatic GSH levels in fasted mice. A, FLU-1 and TCPOBOP were coadministered to mice as shown in Fig. 1. Mice were fasted for the last 2 days. Hepatic GSH levels were determined as reported by Tietze et al. (1969). Data are shown as the mean ± S.D. (n = 4–5) and analyzed by Tukey's test. **, P < 0.01; ***, P < 0.001. (Significant difference between the indicated groups.) B, relationship between hepatic GSH levels and serum ALT activities in FLU-1-administered mice and FLU-1- and TCPOBOP-coadministered mice. Data obtained from two separate experiments are shown. □, FLU-1; ▪, FLU-1 and TCPOBOP.
Formation of FLU-1 N-OH was detected in mouse liver microsomes (Fig. 4A) and previously reported in humans (Goda et al., 2006). Formation of FLU-1 N-OH by liver microsomes was enhanced approximately 5-fold after TCPOBOP treatment (Fig. 4B). Flutamide metabolite-protein adducts were detected by immunoblot analyses using antiflutamide antibody after incubation of microsomal protein with FLU-1 N-OH (Fig. 6). In contrast FLU-1 itself had little bonding to microsomal proteins and did not interact with BSA in a manner similar to that observed with flutamide and OH-flutamide. It is proposed that FLU-1 N-hydroxylation causes intracellular protein modification through the covalent binding to cause the liver dysfunction. Addition to FLU-1 N-OH, nitroso and quinine imine derivatives of FLU-1 N-OH are possible to react with proteins. Further study is required to understand the reaction between FLU-1 N-OH and macromolecules.
Treatment with rodent Cyp3a or Cyp1a inducer, PCN or β-NF, respectively, also enhanced hepatic FLU-1 N-hydroxylation and TCPOBOP treatment (data not shown). Berson et al. (1993) and Fau et al. (1994) reported that flutamide-mediated cytotoxicity in rat hepatocytes and covalent binding to microsomal proteins were increased remarkably by preadministration of dexamethasone (an inducer of Cyp3a) and moderately by β-NF. These reports are consistent with our hypothesis that FLU-1 N-OH participates in liver injury through protein binding to intracellular components.
Coadministration of FLU-1 and TCPOBOP elevated ALT in fasted mice but not nonfasted mice (Fig. 7), suggesting that partial depletion of hepatic GSH during fasting enhances liver dysfunction associated with FLU-1 administration. Moreover, hepatic GSH levels were significantly decreased in mice treated with both FLU-1 and TCPOBOP compared with FLU-1-treated mice (Fig. 8A). An inverse correlation was observed between hepatic GSH levels and serum ALT in those mice (Fig. 8B). In in vitro experiments, adduct formation was prevented by addition of GSH in the reaction mixture (D. Nagai, R. Goda, E. Ichimura, Y. Akiyama, C. Nishimura, K. Nishikawa, M. Miyata, and Y. Yamazoe, manuscript submitted for publication). Coexistence of GSH enhanced NADH-dependent FLU-1 formation from FLU-1 N-OH (Fig. 10). It has been reported that two GSH adducts were detected in human liver microsomal incubation of FLU-1 in the presence of NADPH and GSH (Kang et al., 2008). These results suggest the protective role of hepatic GSH in flutamide-induced liver injury. Age-related decline of intracellular GSH is reported (Hazelton and Lang, 1980; Hernanz et al., 2000), although the mechanism is not fully identified. Loss of GSH biosynthesis may be associated with decrease of GSH levels in elderly persons (Nakata et al., 1996; Suh et al., 2004). It is speculated that these elderly patients tend to exhibit flutamide-induced hepatotoxicity because of lower hepatic GSH levels.
Unlike other N-hydroxy arylamines, FLU-1 N-OH did not spontaneously decompose in water as detected by HPLC analysis. This chemical contains nitro and amino groups in the molecule but is not mutagenic in the Ames test using Salmonella typhimurium TA98 (M. Ohbuchi, M. Miyata, and Y. Yamazoe, unpublished data). Bulky trifluoromethyl group ortho to nitro group causes perpendicular arrangement of nitro group to benzene ring. In addition to the electron-withdrawing effect of nitro and trifluoromethyl groups, these substitutions are likely to contribute to the stability of this N-hydroxylamine.
FLU-1 N-OH was enzymatically reduced into FLU-1 in the presence of NAD(P)H (Fig. 10). The cytosolic reduction was markedly inhibited by allopurinol. Furthermore, bovine xanthine oxidase catalyzed the reduction in an NAD(P)H-dependent manner. Thus, xanthine oxidase probably plays an important role in the cytosolic reduction of FLU-1 N-OH. Xanthine oxidase, in the presence of an adequate electron donor, can mediate the reduction of aromatic nitro compounds (Kitamura et al., 2006). In humans, several studies on the possibility of variation in levels of xanthine oxidase have been reported (Grant et al., 1983; Guerciolini et al., 1991; Kalow and Tang, 1991). In the Japanese population, 11% of subjects were determined to be putative poor metabolizers (Saruwatari et al., 2002). Further studies are necessary to understand the relation between xanthine oxidase polymorphism and susceptibility to flutamide-induced hepatotoxicity. Furthermore, it remains unclear how GSH enhances the cytosolic NAD(P)H-dependent reduction. The rate of FLU-1 N-OH reduction per gram of liver showed relatively high values among all the groups (Table 2). FLU-1 N-OH is expected to accumulate in the body and impair hepatocytes unless removed by enzymatic detoxification pathway. FLU-3 formation from FLU-1 is a major metabolic pathway in humans and mice compared with FLU-1 N-OH formation from FLU-1. Because FLU-3 is efficiently conjugated and removed from the body, FLU-1 formation from FLU-1 N-OH probably plays a crucial role in elimination of FLU-1 N-OH.

Reduction of FLU-1 N-OH to FLU-1. A and B, FLU-1-N-OH (50 μM) was incubated with GSH (10 mM) and/or hepatic cytosolic protein (0.1 mg/ml) prepared from mouse livers of control group. Reaction mixture was analyzed by HPLC (λ = 366 nm). Formation of FLU-1 and residual FLU-1 N-OH were determined for indicated incubation period. C, FLU-1-N-OH (50 μM) was incubated with NADH (10 mM) and hepatic cytosolic protein (0.1 mg/ml) prepared from mouse livers of control group. Formation of FLU-1 was determined for the indicated incubation period.

Reconstitution of reductase activity. Cytosolic fraction was dialyzed three times with 0.1 M potassium-phosphate buffer, pH 7.4, containing 4.8 mM MgCl2. FLU-1-N-OH (50 μM) and dialyzed cytosol (0.1 mg/ml) were incubated with NAD(P)H (10 mM) and/or GSH (10 mM) for 10 min. Cont indicates cytosol alone.
Fasted mice coadministered with FLU-1 and TCPOBOP showed slight increase of ALT (approximately 100 IU/l). The mouse model developed in the present study is considered to reproduce mild hepatotoxicity, which is often observed in patients taking flutamide. Idiosyncratic hepatotoxicity induced by flutamide may require somewhat of a “second hit,” such as cytokine- and immune-mediated mechanism, followed by binding of reactive intermediate of flutamide to the hepatocellular macromolecule.
In conclusion, we have investigated the hepatotoxicity of FLU-1 N-OH and the metabolism of FLU-1 and FLU-1N-OH. Proposed mechanism of flutamide-induced liver toxicity is shown as Fig. 11. Our in vivo study suggests that FLU-1 N-OH is involved in the onset of flutamide-induced liver injury. Binding of FLU-1 N-OH to intracellular proteins is proposed as one of the mechanisms by which flutamide induces hepatotoxicity. The present study shows that FLU-1 N-OH is reduced into FLU-1 by cytosolic enzyme, xanthine oxidase in the presence of NAD(P)H. This reduction of FLU-1 N-OH is proposed as one of the important detoxification pathways of FLU-1 N-OH in flutamide-induced liver injury. The present study suggests that the balance between hepatic FLU-1 N-OH production and reduction is a critical factor for flutamide-induced liver injury.

Proposed mechanism of flutamide-induced liver injury. FLU-1 hydrolyzed from flutamide or OH-flutamide is oxidized mainly by CYP3A and partially CYP1A into FLU-1 N-OH, which possesses a capacity to bind to intracellular proteins. FLU-1 N-OH is detoxified by NAD(P)H-dependent reduction, but in the case that FLU-1 N-OH formation is increased and/or NAD(P)H-dependent FLU-1 N-OH reduction is decreased, FLU-1 N-OH escapes these detoxification pathways and binds to intracellular proteins to induce hepatotoxicity. GSH depletion probably suppresses FLU-1 N-OH reduction and also facilitates protein binding.
Footnotes
-
This study was supported by a grant-in-aid from the Ministry of Education, Science, and Culture, Japan [Grants 17390039 and 17590114]; and a grant-in-aid from the Ministry of Health, Labor, and Welfare, Japan [Grant H17-toxico-ippan-001].
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.021964.
-
ABBREVIATIONS: FLU-1, 5-amino-2-nitrobenzotrifluoride; FLU-3, 5-amino-2-nitro-4-hydroxybenzotrifluoride; FLU-1 N-OH, N-hydroxy-5-amino-2-nitrobenzotrifluoride; P450, cytochrome P450; GSH, glutathione; GST, glutathione S-transferase; GSSG, glutathione (oxidized form); CDNB, 1-chloro-2,4-dinitrobenzene; TCPOBOP, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene; β-NF, β-naphthoflavone; PCN, pregnenolone-16α-carbonitrile; BSA, bovine serum albumin; ALT, alanine aminotransferase; HPLC, high-performance liquid chromatography; PAGE, polyacrylamide gel electrophoresis; PBS, phosphate-buffered saline; KLH, keyhole limpet hemocyanin.
- Received April 21, 2008.
- Accepted September 29, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}