


Abstract
Monkeys have been proposed as an animal model to predict the magnitude of human clinical drug-drug interactions caused by CYP3A4 enzyme induction. To evaluate whether the cynomolgus monkey can be an effective in vivo model, human CYP3A4 inducers were evaluated both in vitro and in vivo. First, a full-length pregnane X receptor (PXR) was cloned from the cynomolgus monkey, and the sequence was compared with those of rhesus monkey and human PXR. Cynomolgus and rhesus monkey PXR differed by only one amino acid (A68V), and both were highly homologous to human PXR (∼96%). When the transactivation profiles of 30 compounds, including known inducers of CYP3A4, were compared between cynomolgus and human PXR, a high degree of correlation with EC50 values was observed. These results suggest that cynomolgus and human PXR respond in a similar fashion to these ligands. Second, two known human CYP3A4 inducers, rifampicin and hyperforin, were tested in monkey and human primary hepatocytes for induction of CYP3A enzymes. Both monkey and human hepatocytes responded similarly to the inducers and resulted in increased RNA and enzyme activity changes of CYP3A8 and CYP3A4, respectively. Lastly, in vivo induction of CYP3A8 by rifampicin and hyperforin was shown by significant reductions of midazolam exposure that were comparable with those in humans. These results show that the cynomolgus monkey can be a predictive in vivo animal model of PXR-mediated induction of human CYP3A4 and can provide a useful assessment of the resulting pharmacokinetic changes of affected drugs.
Induction of cytochrome P450 (P450) enzymes can result in serious drug-drug interactions through loss of therapeutic efficacy or increased toxicity and is considered one of the major liabilities during the treatment of disease states involving multiple drug regimens (Lin, 2006). Drug-drug interactions involving CYP3A4 are of particular importance because of its significant involvement in metabolizing more than 50% of marketed drugs. Thus, the elimination of this liability through proper screening, as well as accurately predicting the magnitude of CYP3A4 induction in patients, has been a longstanding pursuit in preclinical development.
Nonhuman primates, including cynomolgus (Macaca fascicularis) and rhesus (Macaca mulatta) monkeys, have been used in pharmaceutical development as preclinical models of drug safety, drug metabolism, and pharmacokinetics as their absorption, distribution, metabolism, and ADME (elimination) properties are closely related to humans (Akahori et al., 2005). For example, it has been shown that both cynomolgus and rhesus monkey CYP2E1 and CYP2C activities are similar to the human isoforms (Sharer et al., 1995; Zuber et al., 2002). The recently cloned rhesus CYP3A64 was also characterized and found to be similar to human CYP3A4 in its amino acid sequence (93% homology) and enzyme kinetics (Carr et al., 2006). Monkey and human intestinal microsomes also show similar enzyme kinetic profiles for testosterone 6β-hydroxylation, suggesting a comparable intestinal first-pass metabolism by CYP3A substrates (Komura and Iwaki, 2008). Taken together, these findings have led to the use of monkeys as an in vivo model to estimate the absorption and first-pass hepatic or intestinal extraction of xenobiotics (Ward et al., 2004; Ogasawara et al., 2007). Monkeys have also been used to study CYP3A enzyme inhibition and the resulting drug-drug interactions in vivo between midazolam (MDZ) and CYP3A inhibitors, such as ketoconazole, erythromycin, diltiazem, and several preclinical development candidates (Kanazu et al., 2004; Prueksaritanont et al., 2006; Zhang et al., 2007).
The use of nonhuman primates as a surrogate animal model to study human P450 induction has also been gradually increasing. In vivo studies with cynomolgus monkeys revealed that hepatic CYP1A, CYP2B, and CYP3A enzymes are inducible in response to human P450 inducers (Bullock et al., 1995; Lee et al., 2006). In a recent study with rhesus monkeys, rifampicin (RIF), a human CYP3A4 inducer, markedly induced CYP3A64 mRNA and MDZ-1′-hydroxylase activity in primary hepatocytes and showed a significant reduction in MDZ exposure in vivo (Prueksaritanont et al., 2006). In addition, the induction profiles of CYP1A and CYP3A mRNA expression in primary cynomolgus monkey hepatocytes were closer to those in primary human hepatocytes compared with rat hepatocytes (Nishimura et al., 2007). The similarities in P450 induction responses, especially with CYP3A between rhesus monkeys and humans, can be explained by the fact that their ligand-binding domains (LBDs) of the pregnane X receptor (PXR, NR1a2) share relatively high sequence homology (∼96%) (Moore et al., 2002). Therefore, monkey PXR is expected to exhibit similar ligand specificity to human PXR (hPXR). This is in contrast to the rodent PXR, where the relatively large differences in sequence homology and ligand-binding affinities between human and rodent PXR have complicated the extrapolation of rodent induction responses to human drug-drug interactions (Gonzalez and Yu, 2006). Overall, these similarities in ADME, as well as induction properties, favor the use of the monkey as an in vivo animal model to assess the CYP3A4 induction potential of new chemical entities.
This study investigated whether the cynomolgus monkey can be an effective in vivo surrogate animal model for predicting human CYP3A4 induction potential. The first step to validate such an animal model was to evaluate human CYP3A4 inducers in in vitro assays derived from the cynomolgus monkey, namely, PXR transactivation and primary hepatocytes, to confirm that induction responses from these in vitro assays are comparable with the corresponding assays of human origin. Our laboratory cloned the full-length cynomolgus monkey PXR (cynoPXR) and incorporated it into a transactivation assay. Subsequently, 30 compounds including known inducers of human CYP3A4 were evaluated in both human and monkey PXR transactivation assays. In addition, the induction responses of two known CYP3A4 inducers, rifampicin and hyperforin, were evaluated in freshly isolated primary hepatocytes from cynomolgus monkey and human donors. Lastly, in vivo drug-drug interactions between CYP3A4/CYP3A8 substrate (MDZ) and inducers (rifampicin and hyperforin) were assessed in cynomolgus monkeys to establish whether the induction of CYP3A8 in monkey can result in MDZ pharmacokinetic changes comparable with those observed in humans at therapeutic doses and/or plasma exposures.
Materials and Methods
Chemicals and Reagents.
Hyperforin-DHCA was purchased from Alexis Biochemicals (Lausen, Switzerland). Celecoxib, rosiglitazone, and terbinafine were obtained from Sequoia Research Products (Oxon, UK). MDZ hydrochloride syrup (2 mg/ml) was obtained from Roxane Laboratories, Inc. (Columbus, OH). Both 1′- and 4′-hydroxymidazolam were purchased from BD Gentest (Woburn, MA). All the cell culture media and reagents were obtained from Invitrogen (Carlsbad, CA) including Lipofectamine 2000 and charcoal/dextran-treated fetal bovine serum. Alamar Blue reagent was purchased from Trek Diagnostics (Cleveland, OH). All the other chemicals, including rifampicin, were of analytical or high-performance liquid chromatography grade and were purchased from Sigma-Aldrich (St. Louis, MO). St. John's wort (SJW; 300 mg containing 0.3% hypericin) capsules were purchased from GNC Corporation (Pittsburgh, PA). Liquid chromatography/tandem mass spectrometry (LC/MS/MS) analysis of ethanolic extracts of SJW showed that each 300-mg SJW capsule contained 0.29 ± 0.02% (w/w) hyperforin or 0.87 mg/capsule.
Molecular Cloning of cynoPXR.
A cynomolgus monkey liver cDNA panel was purchased from the Biochain Institute (Hayward, CA). Forward (nucleotide 1-27) and reverse (1274-1305) primers were selected based on a rhesus monkey PXR (GenBank accession no. AF454671) sequence with the exception that CTG at the translation start site was substituted with ATG. Using an Advantage2 polymerase chain reaction (PCR) kit (BD Biosciences, San Jose, CA), the primary cDNA sequences were amplified by reverse transcription, and nine putative full-length (1.3 kilobase) cynoPXR clones were selected for sequencing. Raw sequence data were processed using an Applied Biosystems (Foster City, CA) 3730 Sequencer and imported into the Sequencher program (Genecodes, Ann Arbor, MI) for alignment and editing. The consensus cynoPXR was then cloned into the cytomegalovirus-based mammalian expression vector pcDNA3.1 from Invitrogen. Contigs were imported into the Vector NTI program (Invitrogen) where sequences were compared, translated, and aligned and displayed using the AlignX tool.
cynoPXR and hPXR Transactivation Assays.
hPXR and cynoPXR transactivation assays were performed following the protocol described previously (Zhu et al., 2007) with the exception that the cynoPXR was expressed in African green monkey kidney cells (CV-1) instead of HepG2 cells. In brief, cells were plated in T-175 flasks with Dulbecco's modified Eagle's medium containing 10% fetal bovine serum to achieve ∼80% confluency 1 day before transfection. On the day of transfection, 20 μg of CYP3A-luciferase vector and 1 μg of either cynoPXR or hPXR vector were premixed with Lipofectamine 2000 reagent for each flask and incubated for 30 min at room temperature. The transfection mixture was then added to the cells and incubated for 24 h. After the removal of transfection mixture, HepG2 and CV-1 cells were resuspended to a final concentration of 1.6 × 105 cells/ml in Dulbecco's modified Eagle's medium supplemented with 5% charcoal/dextran-stripped fetal bovine serum, 200 mM l-glutamine, 100 mM sodium pyruvate, and 10 mM nonessential amino acids. The transfected HepG2 and CV-1 cells were plated into 384-well plates at a density of 8000 cells/well. The plates already contained test articles that were serially diluted in dimethyl sulfoxide (DMSO) at a ratio of 1:3 to achieve 10 final concentrations ranging from 2.5 nM to 50 μM (0.5% DMSO v/v) in triplicate. Rifampicin (10 μM final concentration) was included in all the assay plates as a positive control. After an overnight incubation in a 5% CO2 incubator at 37°C, Alamar Blue reagent was added to each well. Plates were then incubated for 2 h at 37°C and then 1 h at room temperature, after which fluorescence was read at ex 525/em 598 nm to evaluate cytotoxicity of the test articles. The CC50 (concentration causing 50% cytotoxicity) was reported. The luciferase activities were then measured on a Viewlux (PerkinElmer Life and Analytical Sciences, Waltham, MA) after the addition of Steady-Glo reagent from Promega (Madison, WI). The raw data were normalized against the signal from 10 μM rifampicin and were expressed as a percent activation (%Act) at each concentration.
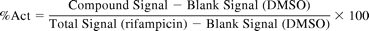 Emax and EC50 were reported from nonlinear regression of concentration-response (%Act) curves using the four-parameter Hill equation.
Emax and EC50 were reported from nonlinear regression of concentration-response (%Act) curves using the four-parameter Hill equation.
Cynomolgus Monkey and Human Primary Hepatocytes.
Freshly isolated cynomolgus monkey (three donors) and human primary hepatocytes (three donors) were obtained from either CellzDirect (Durham, NC) or Celsis (Chicago, IL) in 24-well collagen-coated plates. On receipt of the cells, the transport medium was replaced with serum-free Williams' E medium containing supplements provided by CellzDirect. After a 24-h acclimation, hepatocytes were treated for 3 consecutive days with DMSO, rifampicin, or hyperforin. The medium was replaced daily with fresh medium containing the test articles in DMSO (0.1% v/v). The concentrations for rifampicin ranged from 0.78 to 50 μM, and hyperforin was tested at concentrations ranging from 0.01 to 6.25 μM. The hepatocytes were then washed once with prewarmed Krebs-Henseleit buffer and incubated with 25 μM MDZ for 15 min (monkey hepatocytes) or 60 min (human hepatocytes). Metabolite formation was linear at the MDZ concentration and incubation times used in the assays. The medium was collected in microcentrifuge tubes containing a half volume of ice-cold acetonitrile containing an internal standard (0.5 μM terfenadine) to stop the enzymatic reaction. Protein was separated from the mixture by centrifugation at 10,000 rpm for 10 min, and the supernatant was used to quantitate the formation of 1-hydroxymidazolam by LC/MS/MS. The remaining monolayer of hepatocytes was incubated with lysis buffer from an SV-96 RNA purification kit (Promega), and total RNA was purified from the lysate following the manufacturer's protocol.
TaqMan Real-Time, Reverse Transcription-PCR.
TaqMan (Applied Biosystems) real-time, reverse transcription (RT)-PCR was used to measure expression levels of monkey CYP3A8 and human CYP3A4 mRNA using custom-designed primers and an FAM-MGB probe. The sequences used for monkey CYP3A8 were as follows: forward primer, TGCAGGAGGAAATTGATACAGTTTT; reverse primer, TC-GAGATACTCCATCTGTAGCACAGT; and probe, CCCAATAAGGCACCACCCACCT-ATGA. For human CYP3A4, the following sequences were used: forward primer, TGGT-GAATGAA-ACGCTCAGATT; reverse primer, CATCTTTTTTGCAGACCCTCTCA; and probe, TTCCCAATTGCTATGAGAC. Ribosomal 18S RNA was used as an endogenous control. Each RNA sample was analyzed in triplicate, and ∼10 ng/μl RNA was used in a one-step RT-PCR reaction. The RT reactions were performed for 30 min at 48°C. The PCR reactions were then started at 95°C for 10 min followed by 40 amplification cycles of 15 s at 95°C and 1 min at 60°C. Relative quantification was determined via the ΔΔCT method using the SDS 2.3 software (Applied Biosystems).
Pharmacokinetic Drug-Drug Interaction Studies in Cynomolgus Monkey.
The in vivo study, approved by Bristol-Myers Squibb Animal Care and Use Committee, was carried out in three male cynomolgus monkeys (4–8 kg). On days 0 and 7, each monkey received 2 mg/kg MDZ hydrochloride syrup by oral gavage followed by a sterile water rinse, and blood samples were collected at 0.25, 0.5, 0.75, 1, 2, 4, 6, 7, 8, and 24 h postdose in K2-EDTA-containing tubes. After the centrifugation of blood samples for 3 min at 13,000 rpm, the resultant plasma was stored at −20°C until analysis. On days 1 through 6, each monkey received an oral dose of 15 mg/kg rifampicin (as a suspension in polyethylene glycol 400) or a single capsule of SJW (300 mg) daily. On days 3 and 6, blood was withdrawn from each monkey to determine plasma levels of hyperforin (1 h postdose) or rifampicin (2 h postdose). Throughout the study, monkeys were fasted before each dose of MDZ, SJW, or rifampicin.
LC/MS/MS Analysis of MDZ, 1′- and 4′-Hydroxymidazolam, Hyperforin, and Rifampicin.
For the analysis of MDZ, 1′-hydroxymidazolam, 4-hydroxymidazolam, and rifampicin from monkey plasma, a 25-μl aliquot was extracted into 100 μl of quench solution (90% acetonitrile/10% isopropyl alcohol containing 0.5 μM terfenadine as internal standard), and a 50-μl aliquot of plasma was extracted into 200 μl of quench solution (100% acetonitrile with 0.1% formic acid containing 0.5 μM terfenadine) for the analysis of hyperforin. After protein precipitation using a strata impact protein precipitation plate (Phenomenex, Torrance, CA), the supernatants were injected into an API 4000 Q-Trap (Applied Biosystems) triple quadruple mass spectrometer. The high-performance liquid chromatography system consisted of two LC-20AD (Shimadzu, Norwell, MA) delivery pumps and an SIL-HTc autosampler or an Agilent Technologies (Santa Clara, CA) 1200 coupled with a HTS PAL autoinjector system (LEAP Technologies, Carrboro, NC). The mobile phase, which consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B), was delivered at a flow rate of 0.4 ml/min with the gradient condition as follows: the proportions of mobile phases A and B at 0, 3, 3.5, 4.5, 4.6, and 6 min were 95/5, 50/50, 0/100, 0/100, 95/5, and 95/5, respectively. LC/MS/MS analysis was carried out using multiple reaction monitoring transitions for each test compound: MDZ (326 → 291), 1′-hydroxymidazolam (342 → 324), 4′-hydroxymidazolam (342 → 297), rifampicin (823 → 791), hyperforin (535.2 → 313.2), and terfenadine (472 → 436). Standard curves were fit with a linear regression weighted by reciprocal concentration (1/x2) weighting. Calibration ranges were from 5 to 10,000 nM for all the analytes except hyperforin, which was 0.2 to 125 nM.
Pharmacokinetic and Statistical Analysis.
Pharmacokinetic analysis of plasma concentrations of MDZ and its metabolites versus time profiles was performed by noncompartmental analysis using Kinetica version 4.4 software (Thermo Fisher Scientific, Waltham, MA). Statistical analyses, including linear and nonlinear regression and calculation of confidence interval, were performed using GraphPad Prism version 4.0 for Windows (GraphPad Software Inc., San Diego, CA).
Results
Molecular Cloning and Sequencing of cynoPXR.
The full-length cynoPXR sequence has been deposited to GenBank and was assigned the accession number of GQ412289. The consensus cynoPXR sequence showed two amino acid residue differences at Cys182 and Glu189 in the LBD when it was compared with the previously published partial cynoPXR sequence (GenBank accession number EU153253), which has Tyr and Arg at the same positions, respectively (Milnes et al., 2008). It also showed two wobble base pair changes relative to the rhesus PXR sequence at nucleotide positions 354 and 1158 of the open reading frame (data not shown). One nonwobble base pair change in the DNA binding domain was also noted in relation to the rhesus PXR and hPXR and resulted in a conservative amino acid substitution (A68V) located within the DNA binding domain as shown in Fig. 1. Except for this change, both rhesus PXR and cynoPXR are identical in their amino acid sequences and are highly homologous to the human ortholog (96%). Within the LBD, 11 amino acids were found to be different between hPXR and monkey PXR (consensus between cynomolgus and rhesus): L184M, S200N, C207W, L209V, L213V, S231N, S256N, A280T, T311P, L357V, and P369Y.
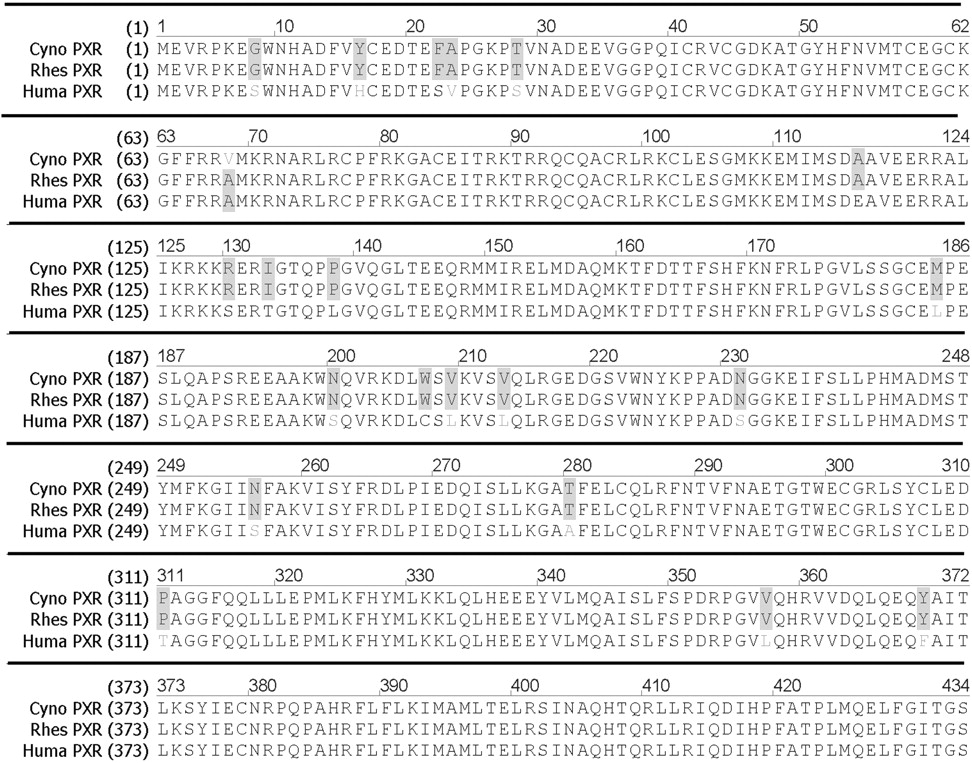
Open reading frame of cynoPXR; the protein sequence alignment with rhesus (rhes) and human (huma) PXR sequences was performed as described under Materials and Methods. The highlighted areas denote the difference between the sequences of cynomolgus, rhesus, or hPXR. The DNA binding domain spans amino acid residues 41 through 107, whereas the LBD spans amino acid residues 139 through 434.
Evaluation of Known CYP3A Inducers in cynoPXR and hPXR Transactivation Assays.
Among the 30 compounds tested in both hPXR and cynoPXR transactivation assays, hyperforin was the most potent activator of hPXR and monkey PXR, with EC50 values of 0.04 and 0.08 μM, respectively. SR-12813 was the second most potent with EC50 values of 0.16 (hPXR) and 0.63 μM (cynoPXR). All the other known activators of hPXR, such as clotrimazole, rosiglitazone, glimepiride, ritonavir, pioglitazone, verapamil, artemisinin, terbinafine, troleandomycin, troglitazone, mifepristone, and carbamazepine, activated both cynoPXR and hPXR equally, and their EC50 values are in good agreement between two species. Although all these compounds showed similar affinities to PXR for both species, rifampicin appeared to be the stronger activator of hPXR (EC50 = 0.84 μM) than cynoPXR (EC50 = 5.1 μM) as shown in Fig. 2 and Table 1. Similar differences (less affinity for cynoPXR) were observed with several other compounds, including 1,9-dideoxyforkskolin, paclitaxel, nifedipine, and celecoxib. However, the differences exhibited by these compounds were less prominent (2–4-fold) than rifampicin (∼6-fold). The most noticeable differences in terms of maximum PXR activation were exhibited by reserpine and sulfinpyrazone. The Emax values for reserpine and sulfinpyrazone were 103 and 73% for hPXR and 16 and 11% for cynoPXR. Thus, both compounds appear to be more potent hPXR activators among the compounds tested. As shown in Table 1, negative control compounds, methotrexate and probenecid, did not elicit any significant activation of PXR up to 50 μM, regardless of the species tested. Pregnenolone 16α-carbonitrile, a potent rodent CYP3A inducer, was shown to be a moderate PXR activator in both species with an EC50 of approximately 25 μM. Dexamethasone, another potent rodent CYP3A inducer, did not transactivate hPXR or cynoPXR. Phenytoin, meclizine, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene, and 6-(4-chlorophenyl)imidazo[1,3]thiazole-5-carbaldehyde-O-3,4-dichlorobenzyl)-oxime are all known as preferential constitutive androstane receptor activators; however, in our study all three compounds activated PXR in both species. Phenobarbital, another constitutive androstane receptor activator, did not transactivate PXR up to 50 μM. To assess which parameter would yield the best correlation of PXR transactivation between the two species, linear regression analyses were performed with EC50, Emax, and intrinsic induction activity (IIA; EC50/Emax) values obtained from both assays. The initial estimates of correlation coefficients were 0.71, 0.49, and 0.99 for EC50, Emax, and IIA, respectively. However, when both reserpine and sulfinpyrazone were excluded from the analyses because of their differential affinity for cynoPXR and hPXR, the correlation coefficients for EC50 and Emax improved to 0.93 and 0.64, respectively (Fig. 3). IIA values were found to be well correlated between hPXR and cynoPXR transactivation assays (R2 = 0.99) regardless of the inclusion of reserpine and sulfinpyrazone. As shown in Fig. 3, most of the data points were plotted within the 95% confidence interval lines, suggesting that most of the compounds tested have a reasonably similar affinity for PXR in both species. Most of the compounds tested in the assays were relatively noncytotoxic up to 50 μM (Supplemental Table 1). Hyperforin was tested from 0.025 to 1 μM because of cytotoxicity at concentrations greater than 1 μM.

Mean concentration-response profiles of rifampicin and hyperforin in both cynomolgus (cynoPXR) and human PXR (hPXR) transactivation (A and C, respectively) and Alamar Blue cytotoxicity assays (B and D, respectively). Each data point is expressed as average ± S.D.
PXR transactivation results for 30 compounds tested in human and cynomolgus monkey PXR transactivation assays

Linear regression analysis between cynomolgus monkey (cynoPXR) and human PXR (hPXR) transactivation profiles. EC50 (excluding reserpine and sulfinpyrazone) (A); Emax (excluding reserpine and sulfinpyrazone) (B); IIA (Emax/EC50) with all 30 compounds (C); and an enlarged view of inset square in C (D). The dashed lines denote 95% confidence intervals.
CYP3A Induction in Cynomolgus Monkey and Human Primary Hepatocytes with Rifampicin and Hyperforin.
Both rifampicin and hyperforin, known CYP3A4 inducers, increased CYP3A RNA expression and activity (MDZ-1-hydroxylation) in primary hepatocytes from cynomolgus monkey and human in a concentration-dependent manner (Fig. 4). The normalized rifampicin concentration-response curves for CYP3A8 RNA expression and the activity in three preparations of monkey hepatocytes were almost superimposable to those of CYP3A4 RNA expression and activity in human hepatocytes from three individual donors. Nonlinear regression analysis (Table 2) of the concentration-response curves showed that the maximum induction of CYP3A8 (100–151.1 and 101.2–104.5% RIF for RNA and activity, respectively) was within the range shown by three human donors (131–177.5% RIF and 107.5–116.8% RIF for RNA and activity, respectively). This result is consistent with PXR transactivation profiles where the maximum transactivation by rifampicin was similar between the species. Although rifampicin appears to be a more potent activator of hPXR, there were no significant differences in EC50 values between human and monkey hepatocytes for the induction of CYP3A RNA or activity. EC50 value for CYP3A8 RNA (3.38–12.18 μM) induction by rifampicin was within the range shown by human donors (3.06–14.23 μM), and the EC50 for CYP3A8 activity (0.67–2.34 μM) was also similar to EC50 values for human donors (0.76–1.94 μM). The comparison of concentration-response curves for hyperforin between monkey and human hepatocytes was complicated by the variable results at higher concentrations as shown in Fig. 4. This is probably because of the combined effects of previously reported cytotoxicity at concentrations greater than 1 μM, as well as the inhibitory potential against CYP3A4 (IC50 = 2.3 μM) at high concentrations (Obach, 2000). Cynomolgus monkey hepatocytes appear to be more resistant to these combined effects. Therefore, nonlinear regression analysis was performed using only the data from nontoxic concentrations of 0 to 1.65 μM. Human hepatocytes appeared to be more sensitive to the inductive effects by hyperforin for CYP3A RNA expression than monkey hepatocytes. EC50 values for CYP3A4 were within 0.37 to 1.27 μM compared with 2.64 to 3.38 μM for CYP3A8. On the contrary, there were no significant differences for the activity between the two species (EC50 = 0.23–0.32 μM for CYP3A4 versus 0.98–1.54 μM for CYP3A8).

Mean concentration-response curves for RNA expressions and MDZ-1-hydroxylase activities in cynomolgus monkey and human primary hepatocytes: rifampicin RNA (A), rifampicin activity (B), hyperforin RNA (C), and hyperforin activity (D).
Donor information and nonlinear regression analysis of CYP3A4 induction in human and cynomolgus monkey hepatocytes
In Vivo Pharmacokinetic Study of MDZ with Rifampicin and SJW.
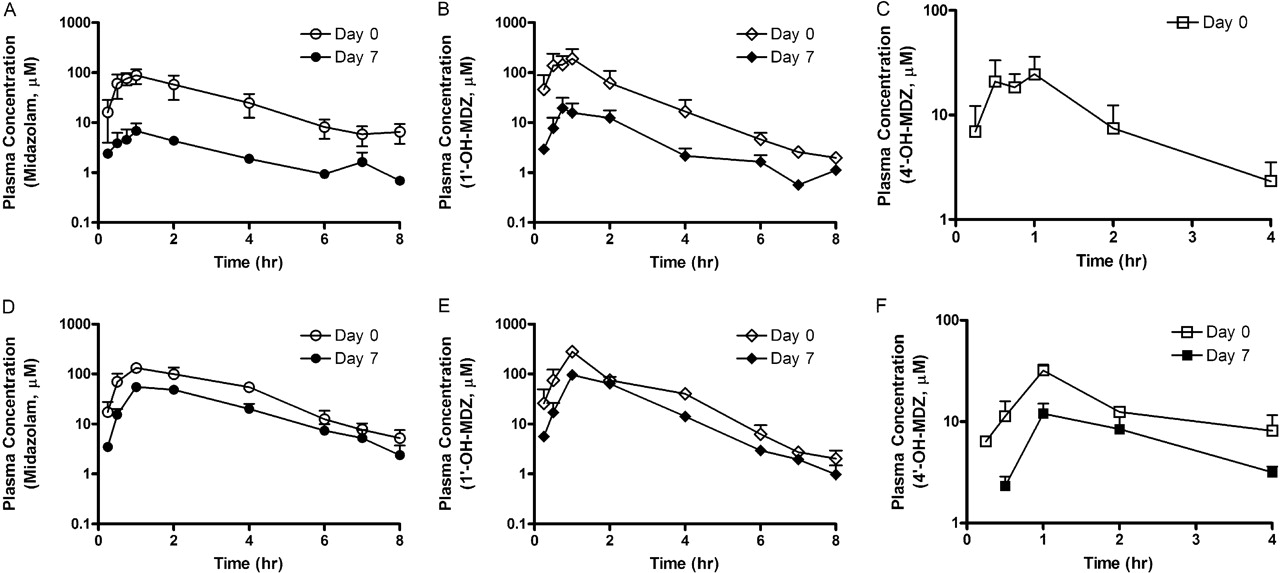
Drug-drug interaction studies were conducted in male cynomolgus monkeys using rifampicin and SJW as the inducing agents and MDZ as the probe substrate. The pharmacokinetic parameters of MDZ exposure were determined on days 0 and 7 before and after 6 days of daily oral administration of either rifampicin (10 mg/kg) or SJW (300 mg). The plasma concentrations of hyperforin and rifampicin were monitored on days 3 and 6 at 1 or 2 h after oral administration of the inducers, respectively. On days 3 and 6, the average plasma levels of rifampicin were 15.9 ± 2.4 and 4.2 ± 1.2 μM, respectively, and the hyperforin levels were 0.48 ± 0.14 and 0.96 ± 0.59 nM, respectively (the lower limit of quantitation for hyperforin was 0.2 nM). As shown in Fig. 5, the plasma concentration-time profiles of MDZ were significantly altered after 6 days of rifampicin or SJW treatment. Oral administration of rifampicin lowered the Cmax and area under the plasma concentration-time curve (AUC) of MDZ by 92.6 and 91.8%, respectively (Table 3). SJW also lowered these parameters, but the magnitude of change was less pronounced. The Cmax and AUC of MDZ were decreased by 62.3 and 58.0%, respectively, with SJW treatment. In contrast, the half-life of MDZ did not change significantly before or after treatment with rifampicin or SJW. The exposure changes of two MDZ metabolites, 1′-hydroxy (1-OH) and 4′-hydroxy (4-OH)-MDZ, appear to closely follow the changes observed for the parent compound. A similar extent of Cmax and AUC reduction in plasma for 1-OH-MDZ was observed in animals treated with either rifampicin (91.9 and 96.8%) or SJW (66.1 and 53.4%). In SJW-treated monkeys, the reductions in Cmax and AUC of 4-OH-MDZ were again similar (62.6 and 56.1%) to those of the parent compound. The plasma level of 4-OH-MDZ in rifampicin-treated animals was below the lower limit of quantitation, and the decreases in pharmacokinetic parameters could not be determined. The metabolite/parent ratio of 1- and 4-OH-MDZ was not significantly altered by SJW, whereas rifampicin increased the ratio from 1.3 to 2.1.

Plasma-time profiles of MDZ, 1-OH-MDZ, and 4-OH-MDZ on day 0 (before inducer treatment) and day 7 (after inducer dose) in cynomolgus monkeys treated by either rifampicin (A, B, and C, respectively) or SJW (D, E, and F, respectively).
Pharmacokinetic parameters of MDZ, 1′-, and 4′-OH-MDZ in cynomolgus monkey plasma after oral administration of MDZ (2 mg/kg)
Discussion
Monkeys have been widely used in drug development as a species for toxicological evaluation and the characterization of ADME properties of new chemical entities. Recent advancements in our understanding of PXR as a master regulator of CYP3A expression and the high sequence homology between monkey PXR and hPXR led us to hypothesize that monkeys may be an effective surrogate in vivo animal model of human CYP3A4 induction. Such a model is important, especially in late-stage drug development, where the induction potential of a clinical candidate requires more extensive evaluation at therapeutically relevant in vivo concentrations to assess pharmacokinetic changes that may result from an interaction.
In a previous study, the rhesus monkey PXR was cloned and shown to have a high sequence homology (∼96%) to the LBD of hPXR. We report here the cloning of full-length cynoPXR and confirm that there are only minor differences in the nucleotide (354 and 1158) and amino acid sequences (A68V) between rhesus PXR and cynoPXR. This difference is not expected to affect the ligand-binding specificities of PXR between cynomolgus and rhesus monkey because the amino acid change is located in the DNA binding domain, not in the LBD of PXR. This was further confirmed in studies conducted in our laboratory where the PXR transactivation response of selected compounds was nearly identical between cynomolgus and rhesus PXR reporter assays (S. Kim and M. Sinz, unpublished results). These results suggest that, in PXR transactivation and binding assays, the PXR protein from both monkey strains may be used interchangeably. Within the LBD, our consensus sequence was different from the partial cynoPXR reported by Milnes et al. (2008) by two amino acid changes. These amino acid residues are conserved between human, rhesus, Japanese macaque, and our cynoPXR sequences but not in the cynoPXR reported by Milnes et al. (2008). Further studies are necessary to reconcile the difference between the two cynoPXR sequences. Excluding that difference, there are 11 amino acids within the LBD that are different between the monkey PXR and the hPXR, which may result in differential binding and transactivation profiles for certain PXR ligands between human and monkey.
Because there has been no comprehensive evaluation of monkey PXR reported in the literature, we investigated how well cynoPXR transactivation correlates to the hPXR activation response by testing 30 compounds in both cynomolgus monkey and human systems. When parameters representing intrinsic induction were used to compare PXR transactivation between cynomolgus monkey and human, Emax values between the two species showed a poor correlation, whereas EC50 and IIA (Emax/EC50) showed good correlations between the two species as shown in Fig. 3. Often, the maximum transactivation of PXR is obscured at high concentrations where cytotoxicity or poor solubility can interfere with the accurate determination of Emax. Therefore, it is not surprising that Emax values did not correlate well between the two assays. The most notable exceptions in transactivation response between the two species were reserpine and sulfinpyrazone where both compounds appear to be more potent activators of hPXR. When these two compounds were excluded from the linear regression analysis, the correlation of PXR transactivation between the two species was improved. Therefore, these results suggest that, although the cynoPXR transactivation assay can be predictive of human CYP3A4 induction, there are exceptions that require further evaluation. It is interesting to note that the amino acid change between human and monkey PXR at position 209 (L209V) did not alter the agonist properties of hyperforin despite the known interaction between Leu209 of PXR LBD and hyperforin (Teotico et al., 2008). Nonetheless, the close agreement of monkey and human in vitro data for the induction potential of the majority of these compounds gave us confidence that in vivo drug-drug interactions in monkeys would be predictive of human CYP3A4 induction.
The in vitro-in vivo correlation was further tested with rifampicin and hyperforin in primary hepatocytes and in vivo. In primary hepatocytes isolated from both humans and cynomolgus monkeys, rifampicin and hyperforin induced both RNA expression and enzyme activities of CYP3A as predicted by their PXR transactivation results. However, some quantitative differences were also observed between PXR transactivation and CYP3A induction in hepatocytes by both inducers. Although rifampicin appeared to be a less potent activator of PXR, it was an equally potent inducer of CYP3A in both cynomolgus monkey and human hepatocytes. In contrast, hyperforin appears to be a less potent inducer of CYP3A8 RNA expression compared with that of CYP3A4 RNA expression in primary hepatocytes, although the cytotoxicity and potential CYP3A inhibition complicated the quantitative comparison between human and monkey induction responses. This species difference in the induction of CYP3A RNA expression did not significantly impact the activity changes mediated by hyperforin. MDZ-1-hydroxylase activities in hepatocytes from both species were induced in a similar fashion (excluding the data from concentrations greater than 1.65 μM). These quantitative differences in the prediction of CYP3A induction can be attributed to the differences in either metabolic capacity or transporter functions between the host cell lines in PXR assay (HepG2 and CV-1) and primary hepatocytes, which can result in differential intracellular concentrations of the inducers.
Nonetheless, the authors concluded that both compounds were still appropriate candidates to be evaluated in a monkey in vivo model because the most important in vitro parameter for CYP3A induction (activity changes in primary hepatocytes) is similar between the two species, and the differences are minimal considering the therapeutic concentrations that these inducers reach during an established standard of care treatment (i.e., EC50 ≪ Cmax). When 15 mg/kg rifampicin was administered, the plasma concentrations in monkeys were 15.9 and 4.2 μM 2 h postdose on days 2 and 6, respectively. The decrease in C2h values after multiple days of dosing may be the reflection of autoinduction by rifampicin as reported previously (Zhang et al., 1998; Wilkins et al., 2008), but these levels are close to the reported Cmax values in patients (2.9–12.2 μM) after a 600-mg oral dose of rifampicin (Israili et al., 1987). The results show that the rifampicin dose chosen for the monkey study produced a similar plasma exposure to that seen in patients. At these levels, rifampicin induction of CYP3A8 resulted in significant changes in MDZ pharmacokinetics. MDZ Cmax and AUC on day 7 decreased from day 1 levels by 92.6 and 91.8%, respectively, similar to the reported exposure changes in human subjects after a 600-mg daily administration of rifampicin for 5 days (94 and 96%, respectively) (Backman et al., 1996). On the other hand, the plasma concentrations of hyperforin 1 h postdose were 0.48 and 0.96 nM in monkeys, which are far lower than the reported Cmax values (∼51.5 nM) after a single dose of 900 mg of SJW in humans (Cui et al., 2002). Unlike rifampicin where the Tmax value in monkeys is known (Prueksaritanont et al., 2006), the Tmax for hyperforin in monkeys is not known. Assuming that hyperforin was rapidly absorbed, a Tmax of 1 h was chosen; however, it is not known whether this is correct. Therefore, a better estimation of hyperforin exposure (Cmax) is necessary with additional sampling times to understand whether there is an actual plasma exposure difference between monkeys and humans at these doses. Despite the potentially lower exposure, oral administration of 300 mg of SJW (0.87 mg of hyperforin) to monkeys resulted in Cmax and AUC decreases in MDZ exposure of 62.3 and 58%, respectively. The magnitude of decreases in MDZ Cmax and AUC was similar to those in patients (42.5 and 52.3%, respectively) given daily administration of 900 mg (300 mg three times per day) of SJW for 14 days (Wang et al., 2001). It is interesting to note that rifampicin and SJW treatment caused profound decreases in both 1- and 4-OH-MDZ plasma concentrations. A similar finding for the MDZ metabolites was also reported in the rhesus monkey CYP3A64 induction study (Prueksaritanont et al., 2006). Because PXR can also regulate the expression of phase II enzymes and transporters in addition to CYP3A, rifampicin and hyperforin may have simultaneously induced pathways involved in the elimination of the MDZ metabolites, such as glucuronidation and/or transporters. This secondary induction, in turn, may have contributed to the rapid metabolism of 1- and 4-OH-MDZ metabolites or enhanced elimination via transporters.
In summary, the induction data from both in vitro (PXR transactivation and primary hepatocytes) and in vivo (MDZ AUC reduction) experiments from cynomolgus monkeys have been shown to be predictive of human CYP3A4 induction responses. Given the positive qualitative and quantitative correlations between monkeys and humans, the cynomolgus monkey may serve as an important animal model to more accurately assess CYP3A4 induction potential and pharmacokinetic changes occurring as a result of drug-drug interactions mediated by PXR during preclinical drug development.
Acknowledgments.
We thank Drs. A. David Rodrigues and Kenneth Santone for critical review of the manuscript. We also thank Sandra Matson and Dr. Tatyana Zvyaga for help with the hPXR transactivation assay.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.109.029637
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
- P450
- cytochrome P450
- ADME
- absorption, distribution, metabolism, and elimination
- MDZ
- midazolam
- LBD
- ligand-binding domain
- PXR
- pregnane X receptor
- hPXR
- human pregnane X receptor
- cynoPXR
- cynomolgus monkey pregnane X receptor
- SJW
- St. John's wort
- LC/MS/MS
- liquid chromatography/tandem mass spectrometry
- PCR
- polymerase chain reaction
- RIF
- rifampicin
- DMSO
- dimethyl sulfoxide
- RT
- reverse transcription
- IIA
- intrinsic induction activity
- AUC
- area under the plasma concentration-time curve
- SR-12813
- 4-[2,2-bis(diethoxyphosphoryl)ethenyl]-2,6-ditert-butyl-phenol.
- Received August 11, 2009.
- Accepted October 7, 2009.

- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}