


Abstract
Exposure to certain xenochemicals can alter the catalytic activity of the major drug-metabolizing enzyme, CYP3A4, either by enhancing expression of this cytochrome P450 or inhibiting its activity. Such alterations can result in adverse consequences stemming from drug-drug interactions. A simplified and reliable tool for detecting the ability of candidate drugs to alter CYP3A4 levels or inhibit catalytic activity was developed by stable integration of human pregnane X receptor and a luciferase vector harboring the CYP3A4 enhancers. Treatment of stable transformants, namely DPX-2, with various concentrations of inducers including rifampicin, mifepristone, troglitazone, methoxychlor, and kava produced dose-dependent increases in luciferase expression (between 2- and 40-fold above dimethyl sulfoxide-treated cells). Northern blot analyses of CYP3A4 mRNA in DPX-2 cells exhibited a good correlation to results generated with the reporter gene assay (r2 = 0.5, p < 0.01). Induction of CYP3A4 protein was examined by measuring catalytic activity with the CYP3A4 substrate, luciferin 6′ benzyl ether (luciferin BE). Metabolism of luciferin BE by DPX-2 cells was enhanced 5.2-fold above dimethyl sulfoxide-treated cells by treatment with rifampicin. Constitutive androstane receptor-mediated regulation of CYP3A4 protein was addressed by measuring catalytic activity in a separate cell line over-expressing this receptor. Phenobarbital and dexamethasone produced 1.5- and 2.0-fold increases, respectively, above control in luciferin BE metabolism. To determine the utility of DPX-2 cells for identifying inhibitors of CYP3A4 catabolism, luciferin BE activity was measured in the presence of various concentrations of ketoconazole, erythromycin, or kava. These agents exhibited dose-dependent decreases in CYP3A4 activity with IC50 values of 0.3 μM for ketoconazole, 108 μM for erythromycin, and 15.5 μg/ml for kava. Collectively, DPX-2 cells were used to identify xenobiotics that induce or inhibit CYP3A4 in a high throughput manner, demonstrating their applicability to early-stage drug development.
Drug interactions frequently occur when one drug modulates the metabolism of a second drug by inhibition or induction of a specific P450 enzyme. Given that CYP3A4 is the most abundant hepatic P450 enzyme and is responsible for the metabolism of a large number of currently used therapeutic agents, a major focus in determining the causes of drug interactions centers around identification of xenochemicals that alter the expression of CYP3A4 (Watkins, 1994). At least five categories of agents are considered CYP3A inducers: steroid hormones having either glucocorticoid or anti-glucocorticoid activities; phenobarbital and phenobarbital-like agents, such as polychlorinated biphenyls and organochlorine pesticides; macrolide antibiotics, including rifampicin; imidazole antifungal agents, such as clotrimazole; and receptor and enzyme antagonists, including nifedipine, troglitazone, and lovastatin (reviewed in Quattrochi and Guzelian, 2001).
CYP3A4 induction is frequently considered clinically less important than inhibition of its catalytic activity because induction is expected to reduce the efficacy, rather than cause toxicity, of coadministered CYP3A substrates. However, CYP3A4 inducers such as rifampicin and rifabutin can reduce plasma concentrations of certain drugs up to 40-fold, effectively abolishing their efficacy (Gillum et al., 1993; Grange et al., 1994). For example, enhanced metabolism, produced by rifampicin, of the CYP3A substrate cyclosporine has resulted in organ graft rejections (Lucey et al., 1990). Rifampicin and the widely used herbal antidepressant St. John's wort also attenuate the efficacy of HIV protease inhibitors (Cvetkovic and Goa, 2003), hindering AIDS therapy. Along similar lines, enhanced metabolism of antitumor agents can diminish the progress of chemotherapy. In epilepsy patients, CYP3A4 induction by carbamazepine and phenytoin results in low plasma concentrations of midazolam, reducing the hypnotic effect (Backman et al., 1996). Moreover, rifampicin treatment results in diminished hypnosis associated with another agent, triazolam, which can render this drug ineffective (Villikka et al., 1997). Induction of CYP3A4 can also produce enhanced metabolic activation of substrates catalyzed by this P450, resulting in hepatotoxicity. For example, autoinduction of CYP3A4 by troglitazone produces enhanced bioactivation and toxicity in HepG2 cells and human hepatocytes (Kostrubsky et al., 2000;Tettey et al., 2001). In vivo, troglitazone was implicated in severe and fatal hepatotoxicity in patients receiving this antidiabetic in the absence of other therapeutics (Gitline et al., 1998; Herrine and Choudhary, 1999). Collectively, induction of CYP3A4 can produce a decline in drug efficacy that in some instances equates to no therapy. Thus, screening new drugs for their ability to induce CYP3A4 should be considered as important as identifying inhibitors of this P450 when evaluating drug safety.
Enzyme inhibition is the most frequently encountered form of metabolically based drug-drug interactions. Agents that are clinically important CYP3A4 inhibitors include ketoconazole, itraconazole, erythromycin, clarithromycin, and nefazodone (Flockhart and Oesterheld, 2000). These inhibitors can cause marked increases in the plasma concentrations of drugs that are CYP3A4 substrates. For example, ketoconazole has been shown to produce 16-, 22-, and 73-fold increases in serum concentrations of midazolam (Tsunoda et al., 1999), triazolam (Varhe et al., 1994), and terfenadine (Honig et al., 1993), respectively. When the antihistamine terfenadine was administered simultaneously with ketoconazole, the combination produced serious and, in some cases, fatal cardiac arrhythmias (Wilkinson, 1996). Thus, the ability to rapidly screen new chemical entities for inhibition of CYP3A4 in cells, a situation more similar to the in vivo situation, holds inherent appeal.
To facilitate the identification of CYP3A4 inducers and inhibitors in a rapid screening assay, a stable cell line, DPX-2, over-expressing the nuclear receptor PXR and harboring corresponding response elements in the CYP3A4-regulatory region, was established. Based on the mechanism of PXR-mediated CYP3A4 regulation, we demonstrate here that this cell-based bioassay performed in 96-well plates will allow for the identification of mechanisms involved in CYP3A4 induction. Furthermore, an additional stable cell line integrated with human CAR (hCAR) demonstrated that this receptor does not appear to be as important as PXR in xenochemical regulation of CYP3A4. DPX-2 cells also exhibit dose-dependent induction of CYP3A4 by numerous compounds including known PXR activators, therapeutic drugs, environmental chemicals, and herbal products. We further used the stable transformants over-expressing PXR to develop cell-based bioassays for identifying inhibitors of CYP3A4 metabolism. Inhibition kinetics of a given compound on CYP3A4 enzyme activity in whole cells can readily be assessed in this high-throughput system. Results from metabolic assays in cells treated with various agents demonstrate that many inducers are also inhibitors of CYP3A4-mediated metabolism. Collectively, this screening system was designed for use during the early stages of drug development and has distinct advantages over other screening systems, such as primary hepatocytes, in that it is rapid, simple, and amenable to automated high-throughput formats.
Materials and Methods
Materials. LipofectAMINE 2000 and DMEM culture medium were purchased from Invitrogen (Carlsbad, CA). Mouse 18S rRNA was purchased from Ambion (Austin, TX). Restriction enzymes were obtained from New England Biolabs (Beverly, MA). Fetal bovine serum was obtained from Hyclone Laboratories (Logan, UT) and culture dishes were obtained from VWR (West Chester, PA). Natural products including, kava, grapeseed extract, garlic, and ginseng were purchased as TruNature products (IVC Industries, Inc., Freehold, NJ) and methoxychlor, mifepristone, and mevastatin were obtained from BIOMOL Research Laboratories (Plymouth Meeting, PA). Omeprazole, α-naphthoflavone (ANF), 2-acetylaminofluorene, pregnane 16α-carbonitrile (PCN), dexamethasone, androstane, clotrimazole, phenytoin, phenobarbital, rifampicin, chrysin, apigenin, resveratrol, and curcumin were obtained from Sigma-Aldrich (St. Louis, MO). RNeasy and plasmid purification kits were obtained from QIAGEN (Valencia, CA), and nylon membranes were purchased from Accelrys (San Diego, CA). The luciferase detection kit LucLite was from PerkinElmer Life and Analytical Sciences (Boston, MA). The P450-Glo Assay containing the CYP3A4 probe substrate, pGL3 vector, the dual luciferase detection kit, and the pRL-TK plasmid was purchased from Promega (Madison, WI). All other reagents used were of the highest quality available.
Human Hepatocytes. Human hepatocytes plated on rat-tail collagen-coated flasks (T-25) in serum-supplemented media were obtained from the Liver Tissue Procurement and Distribution System (University of Minnesota, Minneapolis, MN). Hepatocytes were obtained from four donors, HH042, HH1065, HH1070, and HH1075. The characteristics of these donors are described in Table 1. Upon arrival of plated hepatocytes, medium was replaced with serum-free hepatocyte maintenance medium (Cambrex Bio Science Walkersville, Walkersville, MD) containing 10-7 M dexamethasone plus 10-6 M insulin, and the cells were maintained in an atmosphere of 95% air and 5% CO2 at 37°C as previously described (Raucy et al., 2002a, 2004; Raucy, 2003). After the 24-h equilibration period, hepatocytes were treated with 10 μM rifampicin or 1 mM phenobarbital for 36 or 60 h for the metabolism assays or with various concentrations of ANF (0.5, 2, 10, and 50 μM) and 10 μM rifampicin for 48 h for the induction studies. These agents were dissolved in dimethyl sulfoxide, which was added to the serum-free culture media at a final concentration of 0.1% (13.4 mM). Hepatocytes were then used for determining the metabolism of the P450-Glo substrate by CYP3A4 as described below or harvested for RNA isolation using the RNeasy procedure.
Characteristics of hepatocyte donors
Cell Culture and Transfection Assays. HepG2 cells were maintained at 37°C in 95% air and 5% CO2 in DMEM supplemented with 10% fetal bovine serum (FBS). For transient transfection experiments, 1 × 105 cells were split into 24-well plates a day before transfection. Transient transfection assays were performed using LipofectAMINE 2000 reagent as described by the manufacturer's protocol. In general, transfection mixtures contained 200 ng of luciferase construct harboring the CYP3A4 response element consisting of the proximal promoter, between +50 and -568 bp from the transcription start site (Quattrochi et al., 1995), a distal enhancer (XREM) between -7836 and -7106 (Goodwin et al., 1999), or both enhancers. The Renilla luciferase expression vector (pRL, 10 ng) was also added as an internal control for transfection efficiency. After transfection (24 h), chemicals were dissolved in dimethyl sulfoxide and added to the media to a final concentration not to exceed 0.1% (v/v). Media containing the test chemicals were changed at 24-h intervals. After a 48-h exposure to various xenochemicals, cells were harvested, lysed, and analyzed for both firefly and Renilla luciferase activities according to the manufacturer's instructions, and luminescence was detected with a LUMIstar luminometer (BMG Labtechnologies, Offenburg, Germany). Data are presented as the mean ± S.D. of duplicate experiments performed in triplicate.
Cell lines that Over-Express the Nuclear Receptors PXR and CAR. The expression vector containing human PXR (hPXR) was described previously (Raucy et al., 2002b; Raucy, 2003). hCAR was isolated by reverse transcription-polymerase chain reaction using the forward primer 5′-TAAAGAATTCATGGCCAGTAGGGAA-3′ and the reverse oligonucleotide 5′-ATTAGAATTCAGCTGCAGATCTCCT-3′ and cloned into an expression vector similar to that used to express PXR (Raucy et al., 2002b). After isolation and cloning, hCAR was subjected to sequence analysis. Results indicated that the hCAR isolated here was 100% identical to that previously described (Baes et al., 1994). The nuclear receptor CAR was transfected into HepG2 cells and stable transformants were obtained by antibiotic selection as previously described (Raucy et al., 2002b). Additional transformants consisted of HepG2 cells harboring PXR and the luciferase construct containing the CYP3A4 enhancer harboring both distal and proximal promoters as described above. This transformant was named DPX-2. The DPX-2 stable transformants were generated in DMEM supplemented with selection reagent. Stable transformants were maintained at 37°C in a CO2 incubator in DMEM supplemented with 10% FBS. Cell passages did not exceed 40, at which time cells were destroyed.
Luciferase Reporter Assay. For the CYP3A4 induction assay, DPX-2 cells were seeded in 96-well plates at 3 × 104 cells/well and recovered overnight, followed by 24-h treatment of various chemicals dissolved in dimethyl sulfoxide. The final concentration of dimethyl sulfoxide in the culture medium was 0.1% (v/v), and the negative control received 0.1% dimethyl sulfoxide. Transcriptional activation was monitored by luminescence, and data are presented as the mean (±S.D.) of two determinations in quadruplicate.
Northern Blot Analysis. Total RNA from human hepatocytes or DPX-2 cells was isolated using RNeasy and spectrally quantified at 260 nm. Purity was assessed from the 260/280 nm absorbance ratio and by integrity of the 28S and 18S bands on agarose gels. Northern blot analyses were performed by electrophoresis of total RNA (15 μg) through a 1% agarose gel containing 2.2 M formaldehyde, followed by blotting onto a nylon membrane (Shih et al., 1999; Allen et al., 2001). RNA was then cross-linked to the membranes which were hybridized to a random-primed cDNA probe encoding human CYP3A4 as described previously (Raucy et al., 2002a; Raucy, 2003) and visualized autoradiographically. The blots were subsequently washed and rehybridized to 18S rRNA for normalization. Autoradiographs of Northern blots were quantified by scanning autoradiograms with a ScanMaker II (Microtek) and digitizing with Un-Scan-It software (Silk Scientific Inc., Orem, UT). Exposure times used were in the linear range for the film, Kodak XAR-5 (Eastman Kodak, Rochester, NY).
CYP3A4 Metabolic Activity and Enzyme Inhibition Assays. Enzyme assays were conducted directly in cells over-expressing hPXR or hCAR cultured in 96-well plates or primary cultures of human hepatocytes in 24-well plates. Stable transformants were treated with various inducers and CYP3A4 activity was measured with a luminescent substrate (luciferin BE). Metabolism was assessed by measurement of luciferase activity according to the manufacturer's instructions. Briefly, the P450-Glo substrate (5 mM) was diluted to 50 μM with DMEM containing 10% FBS. When inhibitors were added, 20 μl of 1000× inhibitor stock at various concentrations was added with the substrate. Medium was removed from wells containing cells and wells were rinsed with 100 μl of PBS. After the PBS rinse, 50 μl of substrate medium was added to each well and plates were returned to the 37°C and 5% CO2 incubator for 3 h. At the end of the incubation period, 50 μl of luciferin detection reagent mixed with the P450-Glo buffer was added to each well and mixed thoroughly. After a 10-min incubation at room temperature, luminescence was detected. Inhibition kinetics were performed in DPX-2 stable cells cultured in 96-well plates and pretreated with rifampicin for 48 h. Coincubation of 50 μM luciferin substrate was performed with various concentrations of ketoconazole (0.001, 0.01, 0.1, 0.3, 1.0, 5.0, and 10.0 μM), erythromycin (0.01, 1.0, 10, 100, 200, and 500 μM), or kava (0.01, 0.1, 0.3, 1, 2, 10, and 50 μg/ml) in 50 μl of DMEM. Inhibitors were dissolved in 0.1% solvent (dimethyl sulfoxide/isopropanol/toluene, 2:1:1). After the 3-h incubation of substrate and inhibitor at 37°C in an atmosphere of 5% CO2, metabolism was monitored by luciferase activity in individual wells containing the DPX-2 cells. Results are expressed as relative light units (RLUs) ± S.D. of three experiments in triplicate.
Data Analysis. Luciferase activity in DPX-2 cells is the mean fold increase above dimethyl sulfoxide-treated cells ± S.D. of two to three experiments in quadruplicate. Luciferin BE results are the mean RLUs ± S.D. of three experiments in triplicate. RNA content in DPX-2 cells is expressed as CYP3A4 mRNA normalized to 18S rRNA and is the arbitrary units (AU)/μg of total RNA. In human hepatocytes, RNA is expressed as the CYP3A4 mRNA normalized to 18S rRNA and is the percentage of control (dimethyl sulfoxide-treated cells). Statistical significance was determined by Student's paired t test.
Results
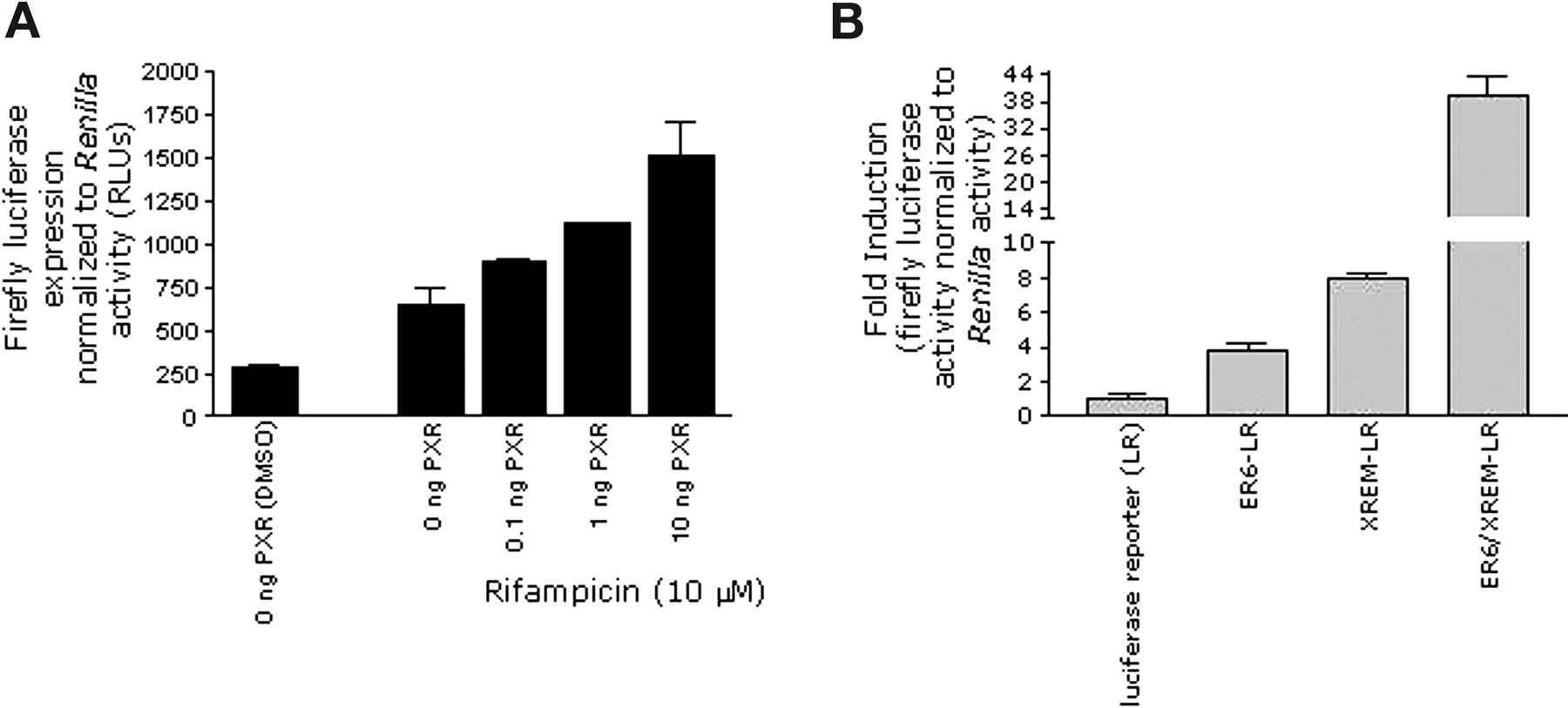
Construction of Stable Cell Lines to Assess CYP3A4 Induction. Before establishing stable cell lines, the responsiveness of reporter plasmids harboring CYP3A4 enhancer elements was determined by transient transfection assays. Initially, the requirement for PXR to elicit gene activation of the CYP3A4 proximal promoter (-568 to +50 bp from the transcription start site) was determined. Reporter gene activity was determined in HepG2 cells cotransfected with the CYP3A4 promoter plasmid in either the absence or presence of increasing amounts of hPXR (0.1-10 ng) and treated with vehicle alone (dimethyl sulfoxide) or 10 μM rifampicin (Fig. 1a). Reporter gene activity was dependent upon the amount of PXR transfected and increased with higher amounts of the cotransfected hPXR expression plasmid. These results demonstrated that hPXR activated transcription through the CYP3A4 proximal promoter plasmid constructed here. Similar results were obtained when the CYP3A4 distal enhancer (-7836 to -7106 bp upstream from the transcription start site) was cotransfected with various amounts of hPXR (data not shown). In addition, we determined which regions of the CYP3A4 gene produced the greatest transcriptional activation. HepG2 cells were cotransfected with 10 ng of hPXR and various CYP3A4-luciferase reporter gene constructs containing either the -568- to +50-bp region (proximal promoter region), the -7836- to -7106-bp segment of CYP3A4 (distal enhancer), or a combination of both regions. Cotransfected cells were then treated with dimethyl sulfoxide or rifampicin and luciferase activity was assessed. In the presence of the proximal promoter, rifampicin produced an increase in luciferase activity that was 3.8 ± 0.5-fold above that of dimethyl sulfoxide-treated cells, whereas the reporter construct harboring the distal enhancer produced a 7.9 ± 0.3-fold induction. Cotransfection of hPXR with the luciferase vector containing both CYP3A4 proximal and distal enhancers exhibited a substantial enhancement of rifampicin-mediated gene activation that was to 39.5 ± 4.3-fold above that of dimethyl sulfoxide-treated cells (Fig. 1b), demonstrating the cooperativity between both regions. Because of the greater rifampicin-mediated induction observed when both PXR response element motifs were present, the plasmid harboring the two enhancer elements was utilized for construction of stable cell lines.

Characterization of the CYP3A4 enhancer construct. a, HepG2 cells were cotransfected with various amounts (0, 0.1, 1, and 10 ng) of the hPXR expression vector, the CYP3A4 enhancer containing an ER6 motif in a luciferase reporter plasmid (ER6-LR), and the Renilla plasmid (pRL) as described under Materials and Methods. The transfected cells were treated with either 10 μM rifampicin or 0.1% dimethyl sulfoxide, and after a 48-h exposure, cell extracts were assayed for luciferase activity. Luciferase activity was normalized to that of Renilla to control for transfection efficiency. Results are expressed as RLUs for firefly luciferase activity normalized to Renilla activity. Data represent the mean ± S.D. of duplicate experiments performed in triplicate. b, HepG2 cells were cotransfected with the hPXR expression plasmid (10 ng), pRL, and either the control luciferase reporter plasmid (LR) or the plasmid containing various CYP3A4 enhancer motifs including ER6, XREM linked to LR, or both ER6 and XREM as described under Materials and Methods. After transfection, cells were exposed to 10 μM rifampicin for 48 h and lysed, and luciferase activity was assessed. Firefly luciferase expression was normalized to Renilla activity to control for transfection efficiency. Results are presented as the fold induction of the CYP3A4 enhancer constructs compared with the control luciferase reporter.
Characterization of Stable Transformants. The stable transformant, DPX-2, was constructed by stably integrating hPXR and the luciferase plasmid harboring both proximal and distal enhancers of CYP3A4. Following selection and purification, DPX-2 cells were characterized by exposure for 24 h to various concentrations of known CYP3A4 inducers including rifampicin, dexamethasone, clotrimazole, mevastatin, androstanone, mifepristone, troglitazone, omeprazole, phenytoin, and phenobarbital. In addition, the ligand for rodent PXR, PCN, was included as a negative control. Of the compounds tested, all exhibited dose-dependent activation of luciferase expression except for PCN. Because the difference in luciferase activity between control and xenochemical-treated cells was extensive, this sensitivity allowed for distinction of strong, moderate, and weak inducers of CYP3A4. For example, rifampicin-mediated luciferase activity ranged from 1.7 ± 0.1- to 30.6 ± 0.7-fold above dimethyl sulfoxide for doses ranging from 0.1 to 10 μM, respectively (Fig. 2a). In addition to rifampicin, omeprazole (100 μM) and phenobarbital (1000 μM) were strong inducers of CYP3A4-mediated luciferase expression and produced 25.9 ± 1.6- and 13.5 ± 0.7-fold increases above dimethyl sulfoxide. At the highest concentrations achievable without producing cytotoxicity, dexamethasone (250 μM), clotrimazole (10 μM), mevastatin (50 μM), androstanone (50 μM), mifepristone (10 μM), and phenytoin (1000 μM) were moderate inducers significantly (p < 0.001) increasing luciferase activity 7.1 ± 0.7-, 10.7 ± 1.1-, 11.0 ± 0.4-, 11.2 ± 0.5-, 9.3 ± 0.6-, and 11.2 ± 0.5-fold above dimethyl sulfoxide-treated cells, respectively. Troglitazone (50 μM) also acted as a moderate CYP3A4 inducer and produced 8.3 ± 0.3-fold activation of luciferase expression that was significantly different from that of dimethyl sulfoxide-treated cells (p < 0.001). In contrast, negligible to weak activation by PCN was observed by the doses examined here, consistent with interspecies variation of PXR ligands (Fig. 2a).
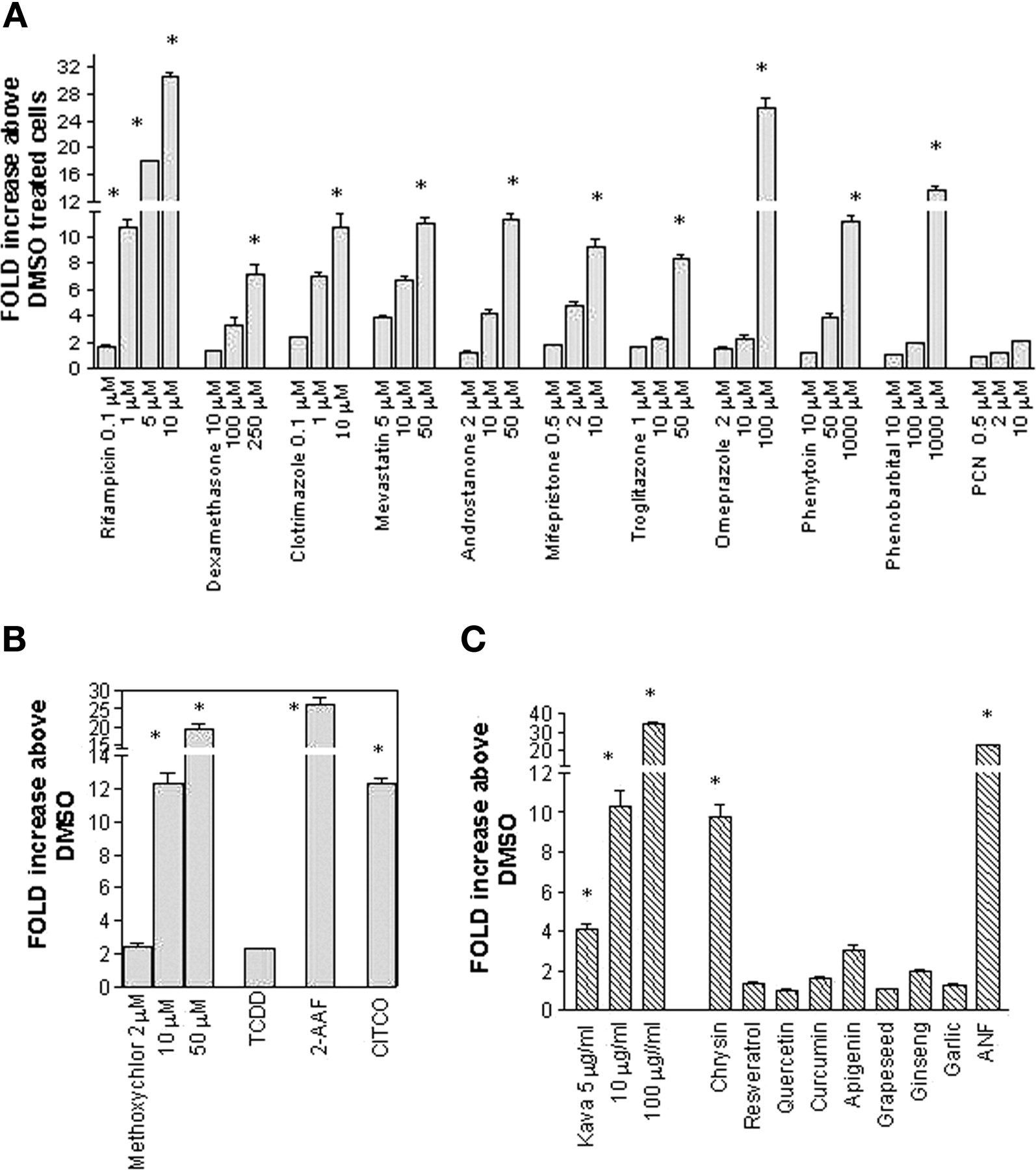
Characterization of the DPX-2 stable cell line. Stably integrated DPX-2 cells were developed by cotransfection of the hPXR expression vector and a luciferase reporter plasmid containing the CYP3A4-regulatory regions as described under Materials and Methods. Cells were plated in a 96-well plate format for characterization. After a 24-h exposure to various xenochemicals, luciferase activity was assessed. Results represent the fold increase in luciferase activity above dimethyl sulfoxide (DMSO) (0.1%)-treated cells. Values are the mean ± S.D. of two determinations performed in triplicate. a, to assess the ability of the DPX-2 cells to respond in a dose-dependent fashion, cells were treated with various concentrations of therapeutic agents including rifampicin (0.1, 1, 5, and 10 μM), dexamethasone (10, 100, and 250 μM), clotrimazole (0.1, 1, and 10 μM), mevastatin (5, 10, and 50 μM), androstanone (2, 10, and 50 μM), mifepristone (0.5, 2, and 10 μM), troglitazone (1, 10, and 50 μM), omeprazole (1, 10, and 100 μM), phenytoin (10, 50, and 1000 μM), phenobarbital (10, 100, and 1000 μM), and PCN (0.5, 2, and 10 μM). Following exposure, luciferase expression was determined. An asterisk indicates a significant difference from control (p < 0.001). b, DPX-2 cells were also treated with the environmental agents methoxychlor (2, 10, and 50 μM), TCDD (10 nM), and 2-acetylaminofluorene (100 μM). In addition, the synthetic CAR agonist, CITCO (10 μM), was examined for its ability to enhance luciferase activity in the DPX-2 cells. The asterisk denotes a significant difference from control (p < 0.01). c, stable DPX-2 cells were exposed to various herbals and flavonoids including kava (5, 10, and 100 μg/ml), chrysin (25 μM), resveratrol (10 μM), quercetin (10 μM), apigenin (10 μM), grapeseed extract (0.6 μg/ml), ginseng (1.1 μg/ml), garlic (1 μg/ml), and ANF (50 μM). Following incubation, luciferase activity was assessed. The asterisk denotes a significant difference from control (p < 0.01).
To demonstrate the utility of the DPX-2 cell line for identifying CYP3A4 inducers, cells were treated with various environmental chemicals (Fig. 2b) and phytochemicals (Fig. 2c). As shown in Fig. 2b, methoxychlor at 50 μM was a strong inducer exhibiting a dose-dependent response in luciferase activity (2.5 ± 0.2- to 19.2 ± 1.2-fold above dimethyl sulfoxide-treated cells) at concentrations ranging from 2 to 50 μM. At 10 and 50 μM, methoxychlor produced a significant increase in luciferase activity above control (p < 0.01). Interestingly, the Ah receptor ligand, 2-acetylaminofluorene, was also an efficacious inducer of CYP3A4 in the DPX-2 cell line, producing a 27-fold increase in luciferase activity that was significantly different from control (p < 0.01). CITCO, a synthetic compound that activates hCAR (Maglich et al., 2003), produced a moderate but significant increase (p < 0.01) in luciferase expression (12.3 ± 0.4-fold increase above dimethyl sulfoxide-treated cells) in the DPX-2 cells. In contrast, the Ah receptor ligand TCDD produced a negligible response (2.3 ± 0.1-fold induction) in PXR-mediated transcriptional activation. We also tested several flavonoids including dietary components and naturally occurring phytochemicals in the DPX-2 cells. Notably, kava exhibited a dose-dependent increase in luciferase activity that was significantly different from that of dimethyl sulfoxide (p < 0.01) in the DPX-2 cells (4.1 ± 0.2- to 34.1 ± 1.2-fold above control cells) with concentrations ranging from 5 μg/ml to 100 μg/ml. This nutraceutical was a strong inducer of CYP3A4 at a concentration of 100 μg/ml. The flavonoid chrysin (25 μM), also an Ah receptor ligand, produced a moderate increase in CYP3A4-mediated luciferase activity (9.8 ± 0.6-fold induction), whereas negligible responses were observed for the remaining flavonoids including resveratrol, quercetin, curcumin, and apigenin. Surprisingly, the Ah receptor antagonist ANF also caused a significant increase (p < 0.01) in luciferase activity at high concentrations (50 μM). Dietary supplements including grapeseed extract, ginseng, and garlic did not activate luciferase expression greater than 3-fold above dimethyl sulfoxide in the DPX-2 cells (Fig. 2c).
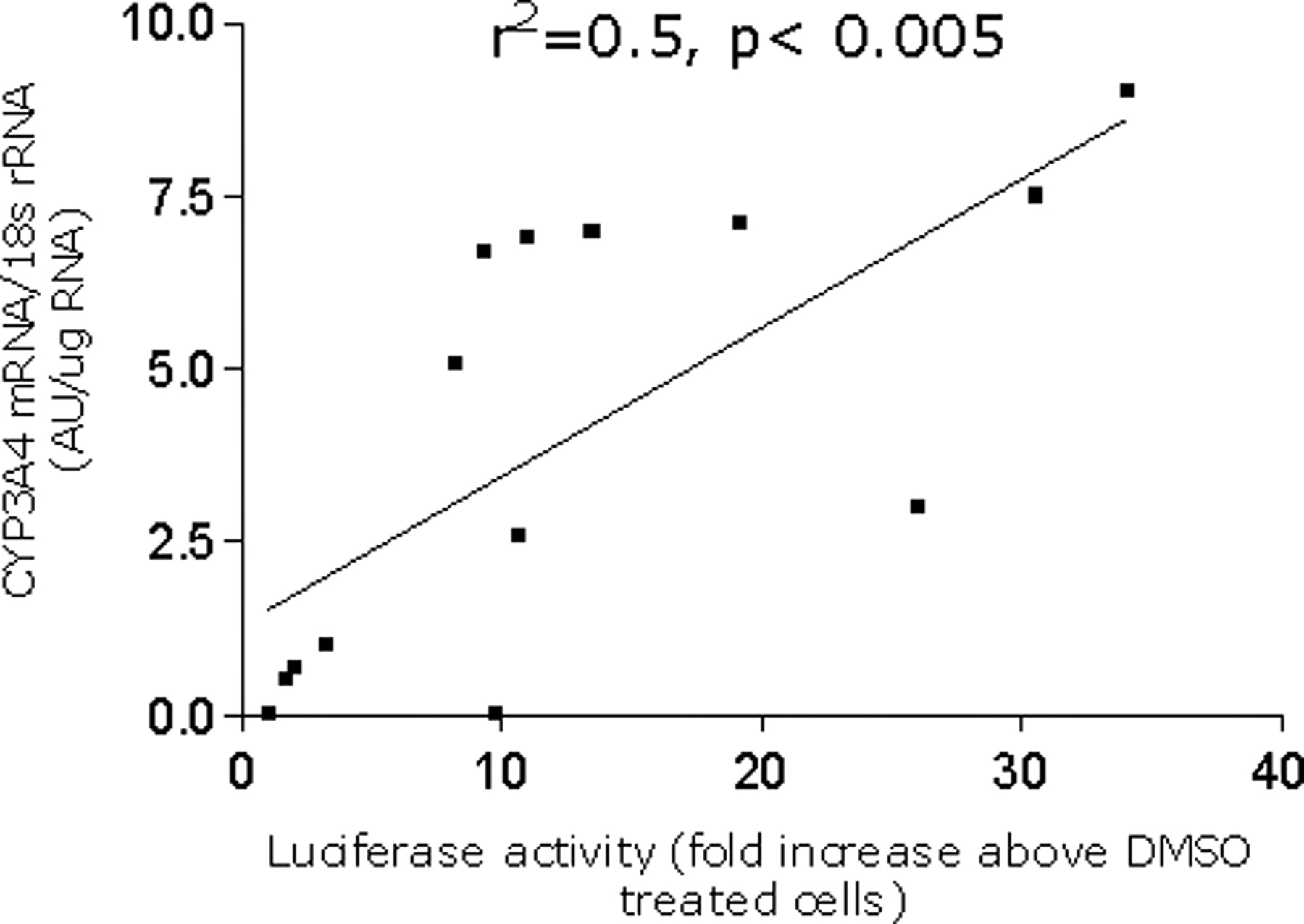
CYP3A4 mRNA Levels in DPX-2 Cells. To substantiate results generated in the reporter gene assay by the various agents described above, the effects of similar compounds on endogenous CYP3A4 mRNA expression in DPX-2 cells were determined by Northern blot analyses (Fig. 3a). Similar to the reporter gene assay, CYP3A4 mRNA expression was enhanced in a dose-dependent manner by various concentrations of rifampicin (0.1-10 μM). Furthermore, rifampicin exhibited strong enhancement of DPX-2 CYP3A4 mRNA levels at 10 μM, results similar to those obtained with the reporter gene assay (Fig. 2a). Of the other agents examined, kava (50 μg/ml), phenobarbital (1 mM), methoxychlor (50 μM), mifepristone (10 μM), and mevastatin (50 μM) were also strong inducers of CYP3A4 mRNA. Agents that exhibited moderate CYP3A4 induction included 2-acetylaminofluorene (100 μm) and troglitazone (50 μM), whereas dexamethasone (100 μM) and chrysin (25 μM) exhibited weak responses and PCN produced negligible enhancement of CYP3A4 mRNA expression (Fig. 3b). In general, a good correlation was observed between luciferase expression and CYP3A4 mRNA levels in DPX-2 cells by Northern blot analysis (r2 = 0.50, p < 0.005) (Fig. 4).

Northern blot analysis of DPX-2 CYP3A4 gene expression in response to various xenochemicals. DPX-2 cells were treated with various compounds for 48 h, followed by RNA isolation. Total RNA (15 μg) was subjected to Northern blot analysis as described under Materials and Methods. The upper blot was probed with a CYP3A4 cDNA and the lower panel was probed with 18S rRNA and used for normalization of RNA loaded onto agarose gels. Autoradiographs of Northern blots were scanned and quantified. Results are CYP3A4 mRNA normalized to 18S RNA expressed as AU/μg RNA. a, RNA from DPX-2 cells treated with: lane 1, 0.1% dimethyl sulfoxide; lane 2, 10 μM PCN; lane 3, 0.1 μM rifampicin; lane 4, 1 μM rifampicin; lane 5, 10 μM rifampicin; lane 6, 100 μM 2-acetylaminofluorene; lane 7, 50 μg/ml kava; lane 8, 25 μM chrysin; lane 9, 1000 μM phenobarbital; lane 10, 10 μM mifepristone; lane 11, 100 μM dexamethasone; lane 12, 50 μM methoxychlor; lane 13, 50 μM mevastatin; and lane 14, 50 μM troglitazone. b, graphical representation of the scanned and digitized blot in a.

Correlation analysis between the reporter gene assay and endogenous CYP3A4 mRNA in DPX-2 cells exposed to various xenochemicals. DPX-2 cells were exposed to xenochemicals and CYP3A4 mRNA levels were measured by Northern analysis as described in the legend to Fig. 3. In addition, DPX-2 cells treated with the same chemicals were analyzed for luciferase expression. A correlation (r2 = 0.5) was obtained between the two parameters with a significance level of p < 0.005.
The effect of various concentrations of ANF on DPX-2 CYP3A4 mRNA levels was also examined (Fig. 5a). Similar to results generated with the reporter gene assay, enhancement of CYP3A4 mRNA occurred in a concentration-dependent manner with 50 and 100 μM exhibiting the greatest increase (Fig. 5b). To further substantiate induction of CYP3A4 mRNA by ANF, human hepatocytes from subject HH1075 were treated with various concentrations, and a representative Northern blot of CYP3A4 mRNA is shown in Fig. 6a. Results indicate that mRNA levels were increased by exposure to various concentrations of ANF, but exhibited the greatest accumulation at 50 μM (Fig. 6b). Furthermore, hepatocytes treated with ANF from two additional subjects (HH1070 and 1042) exhibited similar findings (Fig. 6, c and d). When the mean from all three hepatocyte samples was compared with rifampicin (390 ± 80% of control CYP3A4 mRNA), 50 μM ANF produced a similar significant increase (p < 0.05) in CYP3A4 mRNA accumulation (340 ± 40% of control CYP3A4 mRNA) (Fig. 6e).

The effect of α-naphthoflavone on CYP3A4 mRNA expression in DPX-2 cells. a, DPX-2 cells were treated with various concentrations of the flavonoid, ANF, dimethyl sulfoxide, or, for comparison, rifampicin. Cells were exposed to each agent for 48 h and harvested, and RNA was isolated. Northern blot analyses were performed on the RNA samples and probed with a CYP3A4 cDNA. The upper blot represents CYP3A4 mRNA. RNA is from cells treated with: lane 1, dimethyl sulfoxide (DMSO); lane 2, rifampicin (10 μM)-treated cells; lane 3, 1 μM ANF; lane 4, 50 μM ANF; and lane 5, 100 μM ANF; and lane 6, 10 μM ANF. The lower panel in a is a scan of the agarose gel before transfer and hybridization. Both the autoradiograph and the agarose gel were scanned and digitized. b, the results of the scans, shown as CYP3A4 mRNA normalized to 18S rRNA (AU/μg RNA).

The effect of ANF on CYP3A4 mRNA expression in primary cultures of human hepatocytes. Isolated human hepatocytes from subjects HH1070, HH1075, and HH1042 were treated with various concentrations of ANF, dimethyl sulfoxide, and rifampicin. After a 48-h exposure, cells were harvested, and RNA was isolated and subjected to Northern blot analysis. a, a representative Northern blot of RNA from subject HH1075 demonstrates the effects of various concentrations of ANF on CYP3A4 mRNA expression. The upper blot was probed with a CYP3A4 cDNA and the lower panel is a scan of the agarose gel before transfer and hybridization. Lane 1, RNA from hepatocytes treated with 10 μM rifampicin; lane 2, RNA from cells treated with 50 μM ANF; lane 3, cells treated with 10 μM ANF; lane 4, cells were treated with 2 μM ANF; lane 5, hepatocytes treated with 0.5 μM ANF; lane 6, RNA from hepatocytes treated with dimethyl sulfoxide (control); and lane 7, cells that were untreated. Both the autoradiograph and the agarose gel were scanned and digitized. b-d, graphical presentations of the scanned and digitized Northern blots shown in a and blots containing RNA from hepatocytes treated with ANF from two additional subjects (HH1042 and HH1070). Results are expressed as the CYP3A4 mRNA normalized to the 18S rRNA (percentage of control). Control levels of CYP3A4 mRNA/18S rRNA expressed as AU/μg RNA were 0.17, 0.61, and 1.9 for HH1042, HH1070, and HH1075, respectively. The mean ± S.D. of results from the three blots are presented in e. An asterisk denotes a significant difference from control (p < 0.05).
CYP3A4-Mediated Metabolism in DPX-2 Cells. To examine CYP3A4 induction in response to chemicals at the protein level in DPX-2 cells, CYP3A4 enzyme activities were evaluated using the isoform-specific substrate, luciferin BE. Treatment of DPX-2 cells with 10 μM rifampicin for 48 h resulted in a significant 5.2-fold increase (p < 0.05) in CYP3A4-mediated metabolism compared with control (dimethyl sulfoxide-treated) cells (Fig. 7a). For comparison, CYP3A4-mediated metabolism of luciferin BE was also assessed in hepatocytes from a single subject, HH1065, treated with either 10 μM rifampicin or 1000 μM phenobarbital for 36 or 60 h. In contrast to results generated in DPX-2 cells, the increase in CYP3A4 enzyme activity in response to rifampicin was significantly increased (p < 0.05) but was only 1.6- and 1.7-fold, respectively, above dimethyl sulfoxide-treated hepatocytes (control) at both 36 and 60 h of exposure (Fig. 7b). Phenobarbital produced greater increases in CYP3A4 activity in these hepatocytes than did rifampicin, but rates were only significantly higher than control (p < 0.05) at 60 h of exposure to the barbiturate. The lower level of induction in hepatocytes may be due to the much higher basal levels of CYP3A4 activity when compared with the DPX-2 cells.

The effect of rifampicin on CYP3A4 catalytic activity in DPX-2 cells and primary hepatocytes. a, DPX-2 cells were treated for 48 h with 10 μM rifampicin or 0.1% dimethyl sulfoxide (DMSO). b, primary cultures of human hepatocytes from subject HH1065 were treated for 36 or 60 h with dimethyl sulfoxide, 10 μM rifampicin, or 1 mM phenobarbital. Following exposure, catalytic activity was determined using 50 μM luciferin substrate (luciferin BE), as described under Materials and Methods. After the 3-h incubation in the presence of substrate, luciferase activity was assessed. Results are expressed as luciferase activity in RLUs and is the mean ± S.D. of three determinations. The asterisk indicates a significant difference from control (p < 0.05).
Additional compounds identified as CYP3A4 inducers by the reporter gene assay were also examined for their effect on CYP3A4 protein in DPX-2 cells by monitoring enzyme activity. Only rifampicin (10 μM), phenobarbital (1 mM), dexamethasone (100 μM), and omeprazole (100 μM) were able to produce a significant increase (p < 0.05) in enzyme activity that was 4.0-, 5.8-, 2.6-, and 2.0-fold above control, respectively (Fig. 8a). In contrast, CYP3A4 enzyme activity failed to be significantly elevated by most compounds including kava (50 μg/ml), methoxychlor (50 mM), troglitazone (50 mM), 2-acetylaminofluorene (100 mM), and chrysin (25 μM). The observation that CYP3A4 activities in DPX-2 cells treated with the latter agents exhibited activities that were the same as control (dimethyl sulfoxide-treated) could be explained by inducer-mediated inhibition of activity. To demonstrate the inhibitory potential of each agent toward CYP3A4-mediated activity, metabolism of luciferin BE in the presence of methoxychlor, kava, troglitazone, omeprazole, 2-acetylaminofluorene, or chrysin was monitored in untreated DPX-2 cells and those treated for 48 h with 10 μM rifampicin (Fig. 8b). Results demonstrate that, with the exception of 2-acetylaminofluorene and methoxychlor, the aforementioned compounds are capable of inhibiting CYP3A4-mediated metabolism of luciferin BE in untreated cells. In cells treated for 48 h with rifampicin, all agents except methoxychlor inhibited CYP3A4 activity.

Effect of various chemicals on CYP3A4-mediated metabolism in DPX-2 and hCAR-HepG2 cells. DPX-2 cells exposed to inducers (a), untreated DPX-2 cells and those treated for 48 h with 10 μM rifampicin (R) (b), or HepG2 cells stably integrated and over-expressing hCAR (hCAR-HepG2) (c) were cultured in 96-well plates. DPX-2 cells in a and the hCAR-HepG2 cells were treated for 48 h with 0.1% dimethyl sulfoxide (DMSO) (control), or the following agents: 10 μM rifampicin, 1000 μM phenobarbital, 100 μM dexamethasone, 50 μg/ml kava, 50 μM methoxychlor, 50 μM troglitazone, 100 μM omeprazole, 100 μM 2-acetylaminofluorene, and 25 μM chrysin. CYP3A4 catalytic activity was determined using luciferin BE (50 μM) as substrate. Results are expressed as luciferase activity in RLUs and are the mean ± S.D. of three determinations. An asterisk denotes a significant difference from control (p < 0.05).
Because similar xenobiotics regulate expression of CYP3A and CYP2B and there is cross-talk between the PXR and CAR signaling pathways, we examined the effect of the same inducers of CYP3A4 enzyme activity in a stable cell line harboring the hCAR receptor. Rifampicin, phenobarbital, and dexamethasone produced only a slight but significant increase in enzyme activity (1.4-, 1.5-, and 2-fold, respectively) above that observed in dimethyl sulfoxide-treated cells (Fig. 8c). The remaining agents, kava, methoxychlor, troglitazone, 2-acetylaminofluorene, and chrysin, produced activities that were less than that in dimethyl sulfoxide-treated hCAR cells, suggesting inhibition of luciferin BE metabolism by the inducers. By comparison, enzyme activity was 2.8-fold greater in the hCAR (172,881 ± 4976 RLUs) than in the DPX-2 (61,209 ± 1980 RLUs) cell lines treated with dimethyl sulfoxide, consistent with constitutive activation of CYP3A4 by hCAR. Because of the elevated dimethyl sulfoxide activity and possible lack of induction, inhibition was more marked in the hCAR cells compared with the DPX-2 cells. More importantly, CYP3A4 metabolism in the PXR and CAR cell lines exhibited different inducer profiles. For example, dexamethasone was a stronger inducer of CYP3A4 activity than phenobarbital in the hCAR cell line, whereas phenobarbital was a stronger inducer than dexamethasone in the DPX-2 cells.
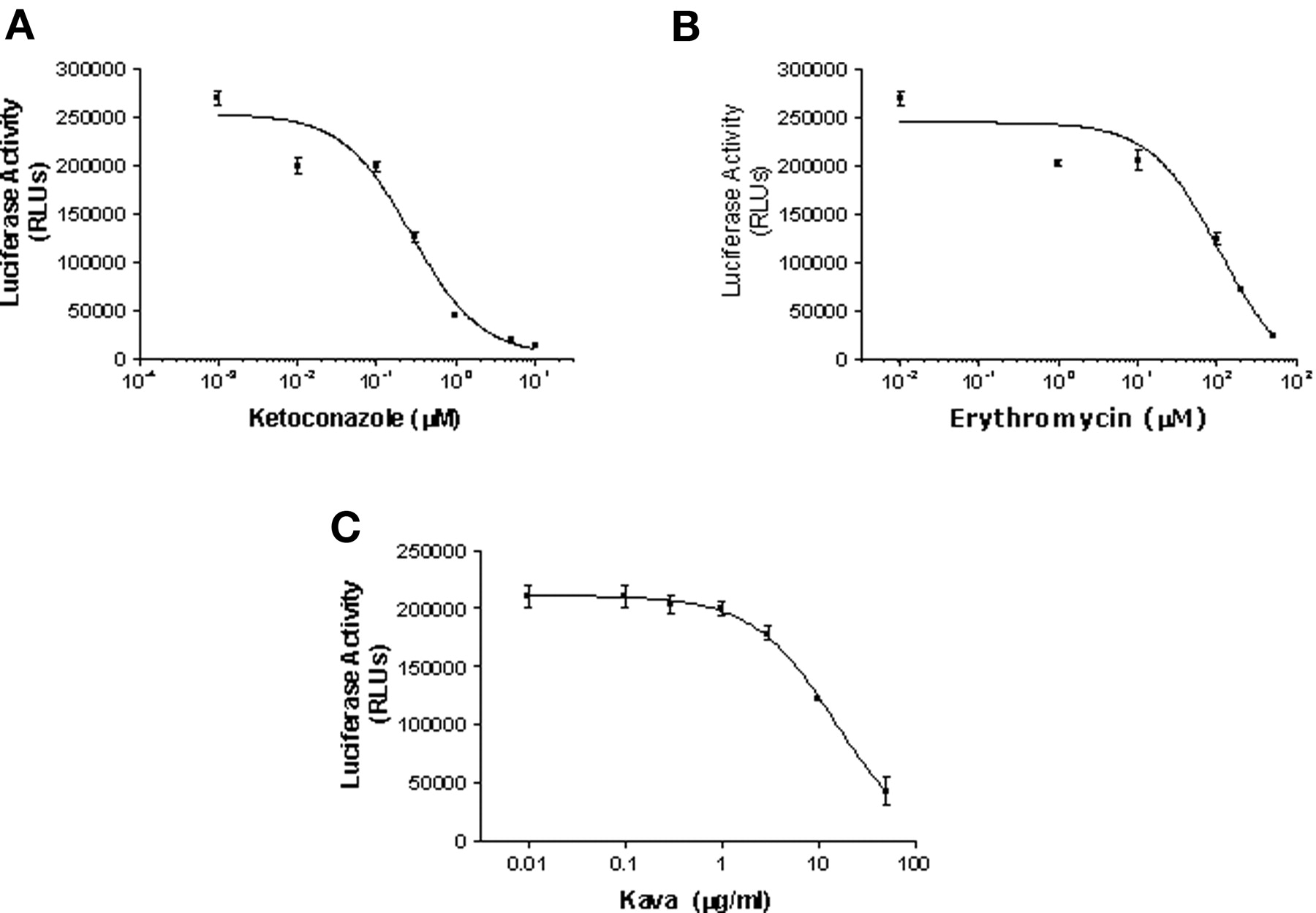
Identification of CYP3A4 Inhibitors in DPX-2 Cells. To determine whether DPX-2 cells could be used to identify inhibitors of CYP3A4 as well as inducers, cells were treated with 10 μM rifampicin for 48 h to achieve over-expression of CYP3A4. Following induction, inhibition kinetics were performed by coincubation of the luciferin substrate with various concentrations of inhibitors. Inhibition of CYP3A4-mediated metabolism of luciferin BE was conducted by using concentrations of ketoconazole that ranged from 0.01 to 10 μM (Fig. 9a), 1 to 500 μM erythromycin (Fig. 9b), and 0.01 to 100 μg/ml kava (Fig. 9c). These compounds inhibited CYP3A4-mediated activity in a dose-dependent fashion, with IC50 values that were calculated for ketoconazole, erythromycin, and kava of 0.3 and 108 μM and 15.5 μg/ml, respectively.

Inhibition of CYP3A4-mediated metabolism of luciferin BE by ketoconazole, erythromycin, and kava in DPX-2 cells. DPX-2 cells cultured in 96-well plates were exposed to 10 μM rifampicin for 48 h. Following induction, various concentrations of inhibitors were incubated in the presence of 50 μM luciferin BE for 3 h. Enzyme activities were determined by measuring luciferase activity, and results are expressed as RLUs. Values are the mean ± S.D. of three determinations. a, results of varying the concentration of ketoconazole from 0.001 μM to 10 μM; b, effects of various concentrations of erythromycin, ranging from 0.01 to 500 μM; c, effects of various concentrations of kava, ranging from 0.01 to 100 μg/ml, on CYP3A4-mediated metabolism of luciferin BE (50 μM).
Discussion
In response to a diverse array of compounds, PXR coordinately regulates target genes involved in the metabolism and transport of small molecules and hence is integral to their elimination. Among the genes regulated by PXR is CYP3A4, which catalyzes a broad range of substrates including 50% of the therapeutic agents. To study drug interactions mediated by CYP3A4, various experimental approaches have been developed, including primary cultures of human hepatocytes to identify inducers or cDNA-based heterologous expression to identify CYP3A4 inhibitors. In the present investigation, we utilized a reporter construct that contained the CYP3A4 promoter (-568 to +50 bp) and XREM (-7836 to -7106 bp) to establish a stable cell line, namely DPX-2. This construct was responsive to PXR when cells were treated with known activators such as rifampicin. In the DPX-2 cells, reporter gene expression was increased 30.6-fold above control by 10 μM rifampicin. This high magnitude of induction was a substantial advantage over our previous cell line that exhibited 8-fold induction for rifampicin (10 μM) (Raucy et al., 2002b). Other xenobiotics evaluated in DPX-2 cells included phenobarbital, mifepristone, omeprazole, mevastatin, dexamethasone, clotrimazole, troglitazone, and PCN. In addition to therapeutic agents, environmental chemicals and herbals were also screened for their ability to activate PXR and subsequently produce enhanced reporter gene activity mediated through the CYP3A4 enhancers. Of the compounds tested, the herbal kava and the DDT derivative methoxychlor were the strongest inducers. All agents examined, with the exception of PCN, exhibited dose-dependent increases in luciferase activity that would enable EC50 estimations, capabilities not readily feasible with human hepatocytes. Importantly, DPX-2 cells could distinguish between strong, moderate, and weak inducers. These attributes as well as the high degree of specificity toward PXR activators are strong arguments for the use of such cell lines.
The utility of any in vitro assay lies in its ability to reflect in vivo consequences of drug/chemical exposure and hence predict drug interactions. Of the agents examined here, we found several clinical correlates including autoinduction reported for troglitazone (Scheen, 2001), rifampicin-mediated increases in metabolism of HIV protease inhibitors and warfarin (reviewed in Borcherding et al., 1992), phenytoin and carbamazepine induction of midazolam and triazolam metabolism (Backman et al., 1996), enhanced metabolism of HIV protease inhibitors by statins (Fichtenbaum and Gerber, 2002), and increased metabolism of cyclosporine by antimycotics (Boissonnat et al., 1997). Thus, DPX-2 cells readily reflect in vivo responses and are good predictors of drug interactions. In further support of this interpretation, we subsequently determined correlations between luciferase activity from reporter assays and CYP3A4 mRNA levels in DPX-2 cells. The induction obtained from the reporter assay was generally consistent with the increases observed in mRNA expression. In addition to the aforementioned agents, we also determined the ability of the Ah receptor antagonist, ANF, to increase luciferase activity and CYP3A4 mRNA levels in the DPX-2 cells. Moreover, we found that this flavonoid was able to enhance CYP3A4 mRNA expression in human hepatocytes from three subjects. To our knowledge, this is the first report to demonstrate that ANF induces CYP3A4. Furthermore, DPX-2 cells demonstrated that the mechanism of induction involves the nuclear receptor PXR. Thus, ANF is a compound that is shared by both the Ah receptor and PXR. Other compounds that share these nuclear receptors and enhance expression of P450 enzymes include chrysin (Zhang et al., 2003; Sugatani et al., 2004), 2-acetylaminofluorene (Friedman et al., 1989; Sparfel et al., 2003), and omeprazole (Quattrochi and Tukey, 1993; Raucy, 2003).
Previous studies in our laboratory and others have primarily utilized isolated hepatocytes to determine induction of CYP3A4 by xenochemicals. Utilizing primary cultures has drawbacks including variable results due to interindividual differences and variations in culture conditions among laboratories. In addition, studies are time-consuming, costly, and labor-intensive. Depending on the drug applied, the fold induction of CYP3A4 by rifampicin can vary from 2 to 100-fold (Raucy, 2003). It should be taken into consideration that upon culturing, this induction is preceded by a marked decrease in the expression of CYP3A4 protein (42-65% decrease from unplated hepatocytes at 48-72 h in culture). Therefore, the magnitude of CYP3A4 induction must take into account the initial down-regulation and reflect the differential effect of cell culturing, rather than drug effects, on expression. This initial down-regulation does not occur in the DPX-2 cells. Thus, enhanced luciferase activity is a direct result of transcriptional activation of the CYP3A4 gene and may actually be more representative of in vivo regulation of the CYP3A4 gene by xenochemicals.
The stable transformants over-expressing hPXR, i.e., DPX-2, were further evaluated for their ability to metabolize CYP3A4 substrates. To perform metabolism studies in a 96-well format, we used a luciferin-based substrate that exhibited specificity toward CYP3A4. Rifampicin treatment of DPX-2 cells results in a 5.2-fold increase in enzyme activity. Interestingly, when human hepatocytes were treated with rifampicin or phenobarbital, less induction in enzyme activity was observed. This may be because hepatocytes from the individual examined here exhibited greater constitutive activity and, as such, induction by rifampicin was less than in hepatocytes from other individuals. Metabolism of luciferin BE was monitored in DPX-2 cells treated with a variety of CYP3A4 inducers identified both by the reporter gene assay and by assessing CYP3A4 mRNA accumulation. Of the agents examined, most failed to produce significant induction of catalytic activity. Only rifampicin, phenobarbital, and dexamethasone exhibited higher enzyme activities than those in DPX-2 cells treated with dimethyl sulfoxide (control). The lack of increased enzyme activity observed for troglitazone, kava, methoxychlor, omeprazole, 2-acetylaminofluorene, and chrysin was likely due to their ability to be metabolized by CYP3A4 and hence inhibit the enzyme. Thus, the inducers were also inhibitors and induction was masked by inhibition of luciferin BE metabolism. Troglitazone (Scheen, 2001), omeprazole (Andersson et al., 1993), methoxychlor (Hu and Kupfer, 2002), kava extract and lactones (Mathews et al., 2002), and chrysin (Nielsen et al., 1998) are all CYP3A4 substrates. The Km values for each of the aforementioned compounds was 50 μM or less, whereas that for luciferin BE is 50 μM. This finding suggests that CYP3A4 exhibited a greater affinity for the inducers and that competitive inhibition by each of the inducers could have occurred. Alternatively, methoxychlor and 2-acetylaminofluorene did not inhibit metabolism of luciferin BE in untreated DPX-2 cells. The reason for the decreased activity following induction of CYP3A4 may suggest that metabolites generated when CYP3A4 enzyme levels are elevated are responsible for the inhibition, rather than the parent compounds. The greater activity observed in DPX-2 cells treated with phenobarbital than with rifampicin suggests that rifampicin may also exhibit some weak inhibitory properties toward CYP3A4. Similar results were produced in the stable cell line harboring hCAR (Hep-CAR). However, induction of CYP3A4 was much less in this cell line than in DPX-2 cells. This may be because PXR transcriptionally activates CYP3A4, whereas CAR is mainly involved in constitutive expression of this P450. Such a role for CAR has been implicated for CYP2C9 and CYP2C19 (Ferguson et al., 2002; Chen et al., 2004).
In light of results presented here, where inducers act as inhibitors, performing metabolic activities to assess induction of CYP3A4 in the absence of mRNA or protein measurement could produce “false negatives.” Many of these false negatives can be alleviated by isolation of microsomes from cells or hepatocytes followed by performing catalytic analysis. However, with certain amines that form metabolic intermediate complexes, the inhibitor may remain bound to the enzyme even after microsomal isolation (Franklin, 1977), so that it is difficult to determine induction. However, compounds that form metabolic intermediate complexes, such as troleandomycin, may dissociate from the enzyme in vivo, enhancing CYP3A4-mediated activities (Pershing and Franklin, 1982). For example, after chronic administration of troglitazone to volunteers, its metabolism was increased, resulting in hepatotoxicity, indicating that this antidiabetic agent is a CYP3A4 inducer, but did not inhibit its own elevated catalysis in vivo (Scheen, 2001). Thus, it may be difficult to predict the net result of an inducer that is also an inhibitor, and caution should be exercised when selecting assays and should not be limited to catalytic analyses.
In summary, regulation of CYP3A4 expression in response to new chemical entities has been primarily examined in isolated cultures of human hepatocytes by measuring metabolism of probe substrates or by determining the levels of CYP3A4 mRNA and/or protein. Although primary cultures may be a system that in many ways represents the in vivo situation, the availability and interindividual differences limit their usefulness in predicting drug-drug interactions and demonstrate the need for alternative systems. Based on the mechanism of PXR-mediated CYP3A4-regulatory machinery, we demonstrate here cell-based bioassays, using DPX-2 cells, that are performed in 96-well plates and allow for rapid identification of compounds that exhibit the potential for drug interactions due to induction and/or inhibition. These assays can be used during the early stages of drug development to identify compounds that induce PXR-regulated genes or those that inhibit CYP3A4 catalysis. Importantly, these cell lines can be utilized to identify mechanisms involved in the regulation of drug-metabolizing enzymes.
Acknowledgments
We thank Lyndon Warfe and Scott Allen for technical assistance in constructing the DPX-2 cell line.
Footnotes
-
This work was supported by National Institutes of Health Grants GM58287 (M.-F.Y.), GM49511 (J.R.), and AA08990 (J.R.) and by the Liver Transplant, Procurement, and Distribution System (N01-DK-9-2310).
-
doi:10.1124/dmd.104.001594.
-
ABBREVIATIONS: P450, cytochrome P450; PXR, pregnane X receptor; hPXR, human PXR; CAR, constitutive androstane receptor; hCAR, human CAR; luciferin BE, luciferin-6′ benzyl ether; ANF, α-naphthoflavone; PCN, pregnenolone 16α-carbonitrile; XREM, xenobiotic response element module; rRNA, recombinant RNA; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; CITCO, 6-(4-chlorophenyl) imidazo [2,1-b][1,3] thiazole-5-carbaldehyde O-(3,4,dichlorobenzyl) oxime; HIV, human immunodeficiency virus; bp, base pair(s); RLU, relative light unit; AU, arbitrary unit(s); Ah, aryl hydrocarbon; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
- Received July 26, 2004.
- Accepted September 30, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}