


Abstract
The metabolism of delavirdine was examined using liver microsomes from several species with the aim of comparing metabolite formation among species and characterizing the enzymes responsible for delavirdine metabolism. Incubation of 10 μM [14C]delavirdine with either an S9 fraction from human jejunum or liver microsomes from rat, human, dog, or monkey followed by high pressure liquid chromatography analysis showed qualitatively similar metabolite profiles among species with the formation of three significant metabolites. The major metabolite was desalkyl delavirdine; however, the identity of MET-7 and MET-7a (defined by high pressure liquid chromatography elution) could not be unambiguously established, but they seem to be related pyridine hydroxy metabolites, most likely derived from 6′-hydroxylation of the pyridine ring. The apparent KM for delavirdine desalkylation activity ranged from 4.4 to 12.6 μM for human, rat, monkey, and dog microsomes, whereas Vmaxranged from 0.07 to 0.60 nmol/min/mg protein, resulting in a wide range of intrinsic clearance (6–135 μL/min/mg protein). Delavirdine desalkylation by microsomes pooled from several human livers was characterized by a KM of 6.8 ± 0.8 μM and Vmax of 0.44 ± 0.01 nmol/min/mg. Delavirdine desalkylation among 23 human liver microsomal samples showed a meaningful correlation (r = 0.96) only with testosterone 6β-hydroxylation, an indicator of CYP3A activity. Among ten human microsomal samples selected for uniform distribution of CYP3A activity, formation of MET-7 was strongly correlated with CYP3A activity (r = 0.95) and with delavirdine desalkylation (r = 0.98). Delavirdine desalkylation was catalyzed by cDNA-expressed CYP2D6 (KM 10.9 ± 0.8 μM) and CYP3A4 (KM 5.4 ± 1.4 μM); however, only CYP3A4 catalyzed formation of MET-7 and MET-7a. Quinidine inhibited human liver microsomal delavirdine desalkylation by about 20%, indicating a minor role of CYP2D6. These findings suggest the potential for clinical interaction with coadministered drugs that are metabolized by or influence the activity of CYP3A or CYP2D6.
Human immunodeficiency virus type-1 (HIV-1)2 reverse transcriptase catalyzes the transcription of viral RNA to proviral DNA, an essential step in the life cycle of HIV-1 and the progression to acquired immune deficiency syndrome in humans (Rosenberg and Fauci, 1991). Inhibition of reverse transcriptase has been a significant target for therapeutic intervention in the disease, and a number of nucleoside-based reverse transcriptase inhibitors, such as zidovudine, didanosine, and zalcitabine, have been shown effective in temporarily halting the progress of the disease (Mitsuya et al., 1990). Unfortunately, some nucleoside analogs produce serious clinical side effects, apparently related to their ability to also inhibit cellular DNA polymerases (Yarchoan et al., 1989). A number of structurally diverse non-nucleoside reverse transcriptase inhibitors have been discovered and are typified by pyridinone derivatives (Goldman et al., 1991) and nevirapine and aryl piperazine derivatives (Romero et al., 1991). These compounds seem to bind to sites adjacent to the polymerase domain of HIV-1 reverse transcriptase (Kohlstaedt et al., 1992) and display excellent selectivity for HIV-1 reverse transcriptase compared with other polymerases (Romero et al., 1991). Unfortunately, subsequent viral mutations can lead to viral resistance to non-nucleoside inhibitors (Declercq, 1994), although other mutations apparently sensitize the enzyme (Dueweke et al., 1993).
Delavirdine (PNU-90152T or Rescriptor) (fig.1) is a specific non-nucleoside inhibitor of HIV-1 reverse transcriptase (Dueweke et al., 1993) and was recently granted approval as an acquired immune deficiency syndrome therapeutic. It has demonstrated an IC50 of 0.26 μM against recombinant reverse transcriptase; at 3 μM, it halted the spread of virus in MT-4 cells and blocked replication of primary HIV-1 isolates in peripheral blood lymphocytes, including zidovudine-resistant variants (Dueweke et al., 1993). Kinetic analysis of delavirdine interaction with reverse transcriptase indicated that it was a mixed-type inhibitor and that it probably impairs the catalytic process subsequent to substrate binding (Althauset al., 1994).

Structure of delavirdine.
Analysis of plasma drug levels in male rats treated orally or intravenously with single doses of delavirdine hydrochloride showed obvious changes in the half-life and AUCs suggestive of saturation kinetics (Adams et al., 1996). Studies on the excretion of radiolabeled drug-related material in delavirdine-treated rats indicate that delavirdine was cleared primarily by metabolism and that the drug was apparently eliminated more rapidly from male rats than females.
This report describes the evaluation of delavirdine metabolism by subcellular tissue fractions from human, rat, and other species. We show that CYP3A is the primary enzyme responsible for delavirdine metabolism and that nonlinear pharmacokinetics observed in other studies might have been due in part to the lowKM for delavirdine metabolism. Delavirdine was also metabolized in a lower capacity pathway by CYP2D6.
Materials and Methods
Chemicals.
Delavirdine mesylate (1-[3-[(1-methylethyl)amino]-2-pyridinyl]-4-[[5-[(methylsulfonyl)amino]-1H-indol-2-yl]carbonyl]-piperazine, monomethanesulfonate), [14C]delavirdine mesylate (either carbonyl labeled or 2′-pyridine labeled), desalkyl delavirdine, furafylline, fluconazole, and dextrorphan (D-17-methylmorphan-3-ol) were prepared by Pharmacia & Upjohn (Kalamazoo, MI). Dextromethorphan was provided by Hoffmann-La Roche (Nutley, NJ), and levallorphan was provided by USP, Inc (Rockville, MD). Quinidine sulfate, α-naphthoflavone, dapsone, imipramine, chlorzoxazone, TAO, testosterone, β-NADP+, isocitrate dehydrogenase, trisodium isocitrate, and erythromycin were obtained from Sigma. Ketoconazole and sulfaphenazole were supplied by Research Biochemicals International (Natick, MA).(S)-Mephenytoin was obtained from Dr. W. F. Trager (University of Washington). Microsomes from either cDNA-transfected human B-lymphoblastoid cells or baculovirus-infected insect cells expressing specific P450 isoforms or FMO-3 were purchased from Gentest (Woburn, MA).
Tissues.
Male and female Sprague-Dawley rats were obtained from Charles River (Portage, MI). Male or female beagle dogs were obtained from Marshall Farms (North Rose, NY). Male cynomolgus monkeys were obtained from the Upjohn primate colony. Animals were euthanized, and livers were perfused with cold saline and, if necessary, frozen in liquid nitrogen and stored at −80°C. Hepatic microsomes were prepared by differential centrifugation of liver homogenates based on a standard method (vanderHoeven and Coon, 1974). All tissue manipulations were conducted at 4°C. Liver tissue was homogenized in 4 volumes of 1.15% KCl, 10 mM EDTA, pH 7.4, using a motor-driven Teflon mortar-glass pestle homogenizer. Cellular debris, nuclei, mitochondria, and lysosomes were removed by centrifugation at 10,000g for 20 min; the supernatant was then further centrifuged at 227,000g for 40 min. The resulting microsomal pellet was washed by homogenization in 100 mM sodium pyrophosphate, pH 7.4, 1 mM EDTA, and pelleted by centrifugation at 227,000g for 40 min. The microsomal pellet was finally homogenized in 0.25 M sucrose, 0.1 mM EDTA and stored at −80°C.
Transplant quality human liver tissue, perfused with Belzer’s solution, was obtained through the International Institute for the Advancement of Medicine (Exton, PA). Tissue was flash frozen within 24 h of removal from donor and stored at −80 to −190°C. Microsomes were prepared essentially as described (Lu and Levin, 1972) and stored in 0.25 M sucrose at −80°C. The following assays were conducted essentially as described (Pearce et al., 1996) at saturating substrate concentrations: testosterone 6β-hydroxylation, ethoxyresorufin-O-deethylation, dextromethorphan demethylation, chlorzoxazone 6-hydroxylation, lauric acid 12-hydroxylation, S-mephenytoin 4′-hydroxylation, and tolbutamide hydroxylation. S9 fraction from human jejunum was obtained from the International Institute for the Advancement of Medicine.
Assays.
Protein was determined by the bicinchoninic acid assay using a 96-well plate format (Redinbaugh and Turley, 1986) and standardized relative to bovine serum albumin. Total cytochrome P450 content was determined spectrophotometrically (Omura and Sato, 1964). In most experiments, metabolism of delavirdine (1–100 μM final concentration) was determined with 0.5–1 mg/ml microsomal protein in 50 mM potassium phosphate or Hepes buffer, pH 7.4, 0.1 mM EDTA at 37°C. Preliminary experiments established conditions for reasonable substrate consumption and linear product formation with time and protein concentration. For measurement of enzyme activity, reactions were limited to 2–4 min. Microsomal suspensions were diluted in buffer (final volume 0.1 to 1.0 ml) followed by addition of drug in methanol (1% final concentration). The reaction was started by addition of an NADPH-generating system, which consisted of (final concentration) 1 mM β-NADP+, 5 mM trisodium isocitrate, 5 mM magnesium chloride, and 0.4 units/ml isocitrate dehydrogenase and stopped after 2–4 min, unless otherwise noted, by addition of an equal volume of acetonitrile containing an internal standard. For generation of microsomal metabolites for mass spectrometric analysis, approximately 5 mg of microsomal protein, from a human sample displaying high CYP3A activity (HLM 40), was diluted into 12.5 ml of phosphate buffer (above) in a T-25 flask followed by addition of 100 μM delavirdine, an NADPH-generating system, and incubated 60 min at 37°C. The incubation was terminated by addition of 1.25 ml of acetonitrile and centrifugation, and the supernatant was passed over a C18 solid phase extraction column (200-mg packing), which had been preconditioned with sequential acetonitrile and phosphate buffer washes. Experiments with radiolabeled drug showed complete retention of drug-related material on column. The column was then washed with 2 ml of water, and drug-related material was eluted with 0.5 ml of acetonitrile.
The following compounds were used as probes for specific P450 isoform involvement in delavirdine desalkylation: furafylline, CYP1A2 (Kunze and Trager, 1993); α-naphthoflavone, CYP1A1, -1A2, -2C8, -2C9, -2A6, -2B6 (Chang et al., 1994); sulfaphenazole and fluconazole, CYP2C9 (Baldwin et al., 1995; Cadle et al.,1994); quinidine and imipramine, CYP2D6 (Coutts et al.,1994; Guengerich et al., 1986); ketoconazole, dapsone, and TAO, CYP3A4 (Baldwin et al., 1995; Fleming et al., 1992); and (S)-mephenytoin, CYP2C19 (Yasumoriet al., 1993). TAO and furafylline were pre-incubated 10 or 15 min, respectively, with microsomes and NADPH prior to addition of delavirdine and were compared with appropriately treated controls.
Delavirdine and metabolites were measured by reversed phase HPLC using gradient or isocratic elution. For gradient elution, a YMC Basic column, 4.6 mm i.d. × 25 cm (YMC, Inc., Wilmington, NC), was used along with a mobile phase (1 ml/min) consisting of 10% acetonitrile for 5 min followed by a linear gradient over 35 min to 60% acetonitrile and finally a 5-min hold at 60% acetonitrile; the gradient was balanced to 100% with 0.1 M ammonium acetate, pH 4, containing 3% acetonitrile. For isocratic elution, a Zorbax SB-CN column, 4.6 mm i.d. × 15 cm (MAC-MOD Analytical, Inc., Chadds Ford, PA), was used with a mobile phase consisting of 26% acetonitrile and 74% 50 mM ammonium acetate, pH 5.8, containing 3% acetonitrile, flowing at 1.5 ml/min. Absorbance detection was by either UV at 295 nm or fluorescence with excitation at 305 nm and emission at 425 nm. For radiochemical detection, a Radiomatic model A-515 flow-through detector (Packard Instrument Co., Meriden, CT) was used and was equipped with a 0.5-ml cell. HPLC eluate was blended 1:3 (v/v) with Ultima-Flo M (Packard) scintillant.
The O-demethylation of dextromethorphan to dextrorphan was developed from a previously established method (Kronbach et al., 1987). Microsomes (1 mg protein/ml) were incubated, as described above, in a final volume of 100 μl containing 80 μM dextromethorphan. The reaction was started by addition of NADPH (see above) and stopped after 30 min by addition of 20 μl of 35% perchloric acid followed by addition of levallorphan as an internal standard. Formation of dextrorphan was determined by HPLC using a Zorbax Rx C8 column, 4.6 mm i.d. × 15 cm, and a mobile phase consisting of 68% 20 mM sodium perchlorate, pH 2.5, and 32% acetonitrile. Detection was by fluorescence (excitation 270 nm, emission 312 nm).
HPLC Mass Spectrometry.
HPLC ESI-MS was performed on a Finnigan-MAT TSQ 7000 triple quadrupole mass spectrometer (Finnigan-MAT, San Jose, CA) directly coupled to an HPLC system via a Finnigan atmospheric pressure ionization source operated in the electrospray mode. Tuning of the ESI source and MS was accomplished by flow-injecting a solution containing 1 mg/ml delavirdine into the mobile phase flow path post column via a manually operated rheodyne injection valve (Rheodyne Inc., Cotati, CA). Metabolite identification was accomplished while operating the MS in positive-ion mode, scanning the first quadrupole from 20 to 1000 amu in 3 sec. Product ion spectra were obtained using a collision cell offset of −30 V with 2.2 mtorr of argon (99.999% pure, AGA, Maumee, OH) as the collision gas while scanning the third quadrupole from 10 to 500 amu in 1 sec. The conversion dynode and electron multiplier were set to 15 kV and 1600 V, respectively. The capillary was operated at 250°C. The spray voltage was set to 4 kV, and nitrogen was employed as a drying gas at a sheath pressure of 70 psi and an auxiliary flow of 50 ml/min. HPLC separation of delavirdine metabolites was accomplished with a Hewlett Packard 1050 Series pump and autosampler (Hewlett Packard, San Fernando, CA) and a 4.6-mm i.d. × 25-cm YMC basic column. Metabolites were separated on a mobile phase gradient as described above. The column effluent was split 1:2 prior to entering the ESI source.
Particle beam-electron ionization mass spectrometric data were collected on a VG Autospec Q hybrid mass spectrometer (Fisons, Manchester, UK) equipped with a ThermaBeam particle beam generator (Extrel Corp., Pittsburgh, PA). The data system was a VAX station 4000/60 running OPUS version 3.0AX. Electron ionization was used to generate the data at 70 eV with 500-ma emission current at 8 kV accelerating potential. The source temperature was 200°C. Calibration was done with perfluorokerosene-H (PCR Inc., Gainesville, FL). The particle beam nebulizer was set to 170°C, whereas the expansion region was held at 100°C. Helium head pressure was set to 120 psi, and the flow was restricted by 375 μm o.d. × 75 μm i.d. fused silica passing through 0.020 in nebulizer tubing. Scanning data were acquired operating the mass spectrometer at 8 kV accelerating potential, scanning from m/z 750 to 50 at 2 sec per decade. The resolving power was set at 1000 (10% valley definition). Metabolites were separated by HPLC using a YMC Basic column, 4.6 mm i.d. × 25 cm, and a mobile phase (0.5 ml/min) consisting of a linear gradient from 10% acetonitrile to 60% acetonitrile over 20 min, holding 60% acetonitrile for 20 min, and then a linear gradient to 80% acetonitrile over 10 min. The gradient was balanced to 100% by 0.1 M ammonium acetate, pH 4.0, containing 3% acetonitrile.
Data Analysis.
Electrophilic frontier values, a molecular orbital parameter, were calculated as described (Ackland, 1993). Kinetic parameters,KM
and Vmax, for delavirdine metabolism were estimated by nonlinear regression analysis of the Michaelis-Menten equation:
Results
Microsomal Metabolism of Delavirdine.
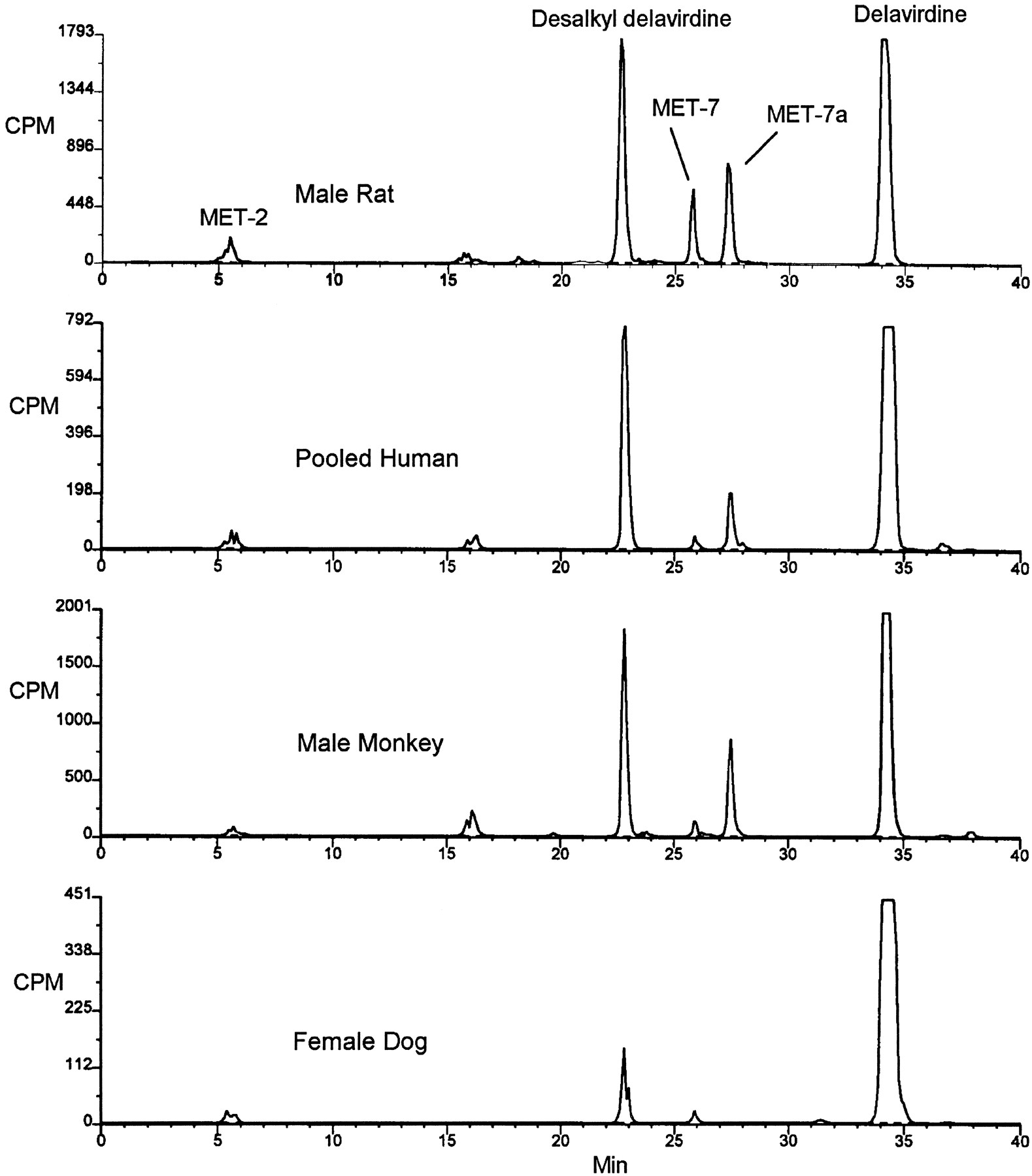
Chromatographic profiles of [14C]delavirdine and its metabolites generated by liver microsomes from human, rat, cynomolgus monkey, and dog are shown in fig.2. Examination of the profiles shows qualitatively similar formation of metabolites across all tested species. The major metabolite in each case had a similar retention time to desalkyl delavirdine. MET-2 had a similar retention time to despyridinyl delavirdine (Chang et al., 1997). MET-7 and -7a were not stable; over a span of hours MET-7a seemed to convert to MET-7 (not shown), which in turn converted to other metabolites, over a span of days, including despyridinyl delavirdine (MET-2).

HPLC-radiochemical profiles of microsomal delavirdine metabolism among species. [14C]Delavirdine (10 μM) was incubated with microsomal samples (0.5 mg of protein/ml) shown for 20 min in the presence of NADPH. Reactions were quenched with acetonitrile and analyzed by HPLC with radiochemical detection.
Incubation of human liver microsomes in the absence of NADPH produced no significant metabolite formation as was true of the incubation of the complete system with boiled microsomes (not shown). Involvement of flavin monooxygenase was ruled out by showing metabolism by the complete system but with microsomes that had been pretreated by heating to 45°C for 4 min in the absence of NADPH, and there was no apparent metabolism by recombinant FMO-3 (not shown) (Lemoine et al.,1990). These experiments point to likely involvement of cytochrome P450 in the human metabolism of delavirdine, although participation of NADPH-dependent oxidoreductases cannot be ruled out.
Incubation of delavirdine with an S9 homogenate fraction of human jejunum established a metabolic profile identical to that of human liver microsomes (fig. 3), indicating the potential for presystemic metabolism of orally administered drug.

Delavirdine metabolism by S9 fraction from human jejunum.
Samples treated as in fig. 2, except S9 protein was 10 mg/ml.
Mass Spectrometry and Molecular Modeling.
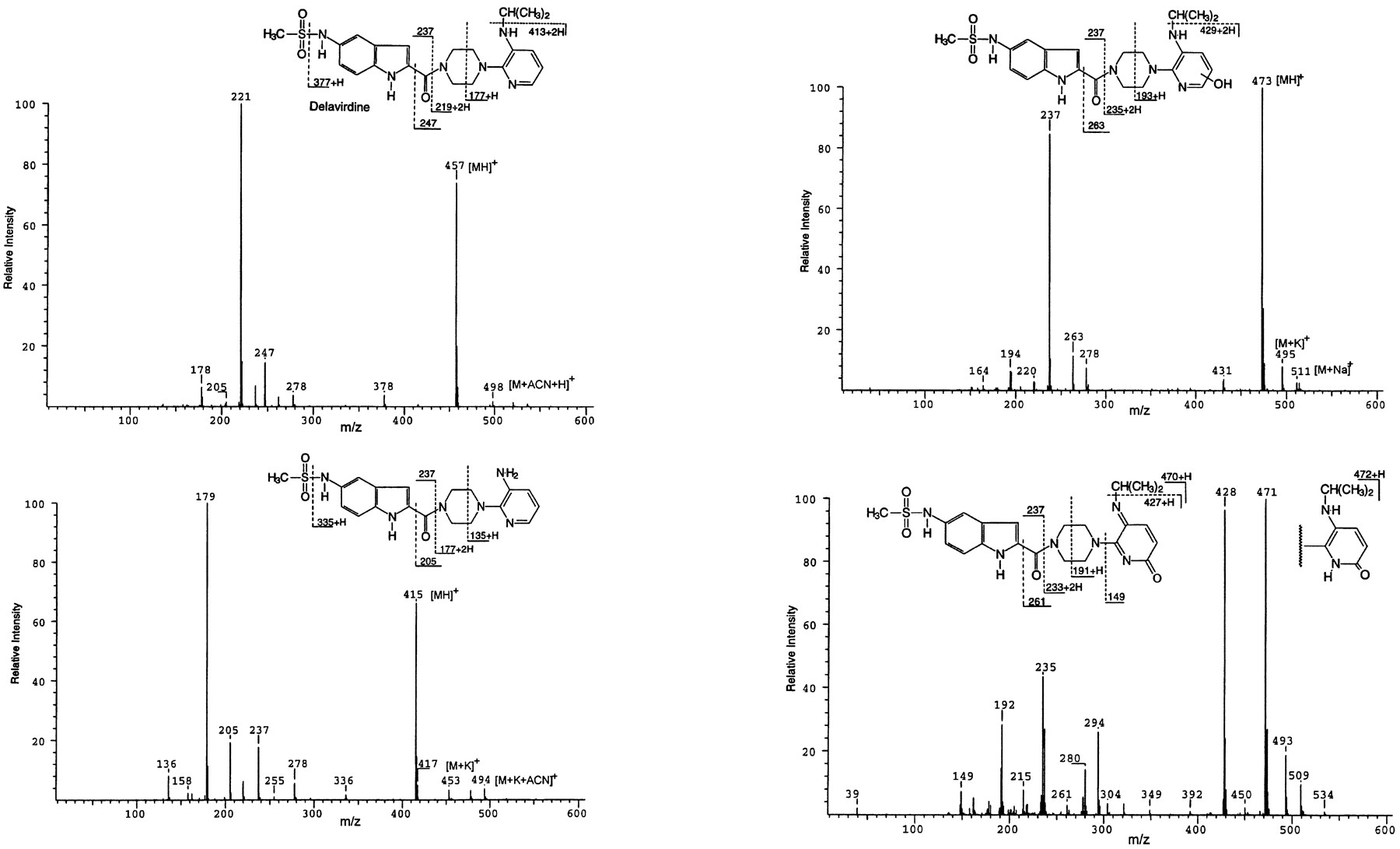
HPLC-ESI-MS characterization of delavirdine metabolites was carried out using chromatographic conditions identical to that used for comparison of microsomal samples (above). Delavirdine was characterized by a pseudomolecular ion (MH+) at m/z 457 with other major ions resulting from fragmentation across the piperazine group and on either side of the carbonyl group (fig.4). The major microsomal metabolite was confirmed as desalkyl delavirdine by appearance of the MH+ at m/z 415 (fig. 4) and chromatographic retention time corresponding to that of a synthetic standard. The mass spectrum of MET-7a displayed an MH+ at m/z 473, an increase of 16 amu over delavirdine, suggesting likely addition of an atom of oxygen; ions at m/z 194 and 431 point to probable oxygen addition to the pyridine ring, although the positional assignment cannot be made by mass spectrometry. HPLC-ESI-MS of MET-7 showed the major ion atm/z 471, a loss of 2 amu relative to MET-7a, with other ions at m/z 192 and 235, consistent with fragmentation across the piperazine ring and amide bond, respectively. Unlike delavirdine and MET-7a, the spectrum shows a relatively abundant ion at m/z428, probably a facile loss of the isopropyl group. Although the fragmentation suggests a quinone imine-like structure (shown derived from proposed 6′-hydroxylation), it should be noted that significant ions were also observed at m/z 473, 237, and 263, identical to MET-7a. Accordingly, the composition of MET-7 is not clear; it could be a mixture of two chemical entities (m/z 473 and 471), or it could be a single tautomeric variant of m/z 473, which undergoes source-induced auto-oxidation to the quinone imine. Single ion chromatograms show evidence of both m/z 471 and 473 in MET-7 but only m/z 473 in MET-7a (fig.5). Analysis of the microsomal metabolite profile by particle beam electron ionization mass spectrometry revealed molecular ions (M+) at m/z 472 for both MET-7a and MET-7, although there was clearly evidence for the loss of 2 amu (m/z 470) from MET-7 but not MET-7a (not shown).

Mass spectral fragmentation of delavirdine and metabolites.
Upper left, delavirdine; lower left, desalkyl delavirdine; upper right, MET-7a; lower right, MET-7.

Selected ion monitoring of delavirdine metabolites using HPLC-ESI-MS.
Calculated electrophilic frontier values for the pyridine ring were 0.0228 for the 4′ position, 0.0392 for the 5′ position, and 0.109 for the 6′ position. A qualitative correlation has been observed between sites of oxidation and high electrophilic frontier values (Ackland, 1993), thus the model predicts hydroxylation on the pyridine 6′ position.
Kinetics of Delavirdine Metabolism.
Analysis of initial rate kinetics for delavirdine desalkylation produced linear Eadie-Hofstee plots, suggesting catalysis by a single enzyme or at least enzymes with similarKM values (fig.6). Estimated kinetic parameters for NADPH-dependent desalkylation of delavirdine by rat, human, monkey, and dog liver microsomes are shown in table1. Delavirdine desalkylation by microsomes pooled from several human livers was characterized by aKM of 6.8 ± 0.8 μM andVmax of 0.44 ± 0.01 nmol/min/mg. There was remarkably little variation in apparentKM values among the microsomal samples from different species, ranging from 4.4 to 12.6 μM, but there were substantial differences in Vmax resulting in large differences in intrinsic clearance (Vmax/KM ).

Eadie-Hofstee plots of delavirdine desalkylation by various hepatic microsomal preparations.
Data represent a single experiment.
Kinetic evaluation of delavirdine metabolism by liver microsomes from several species
Correlation Studies.
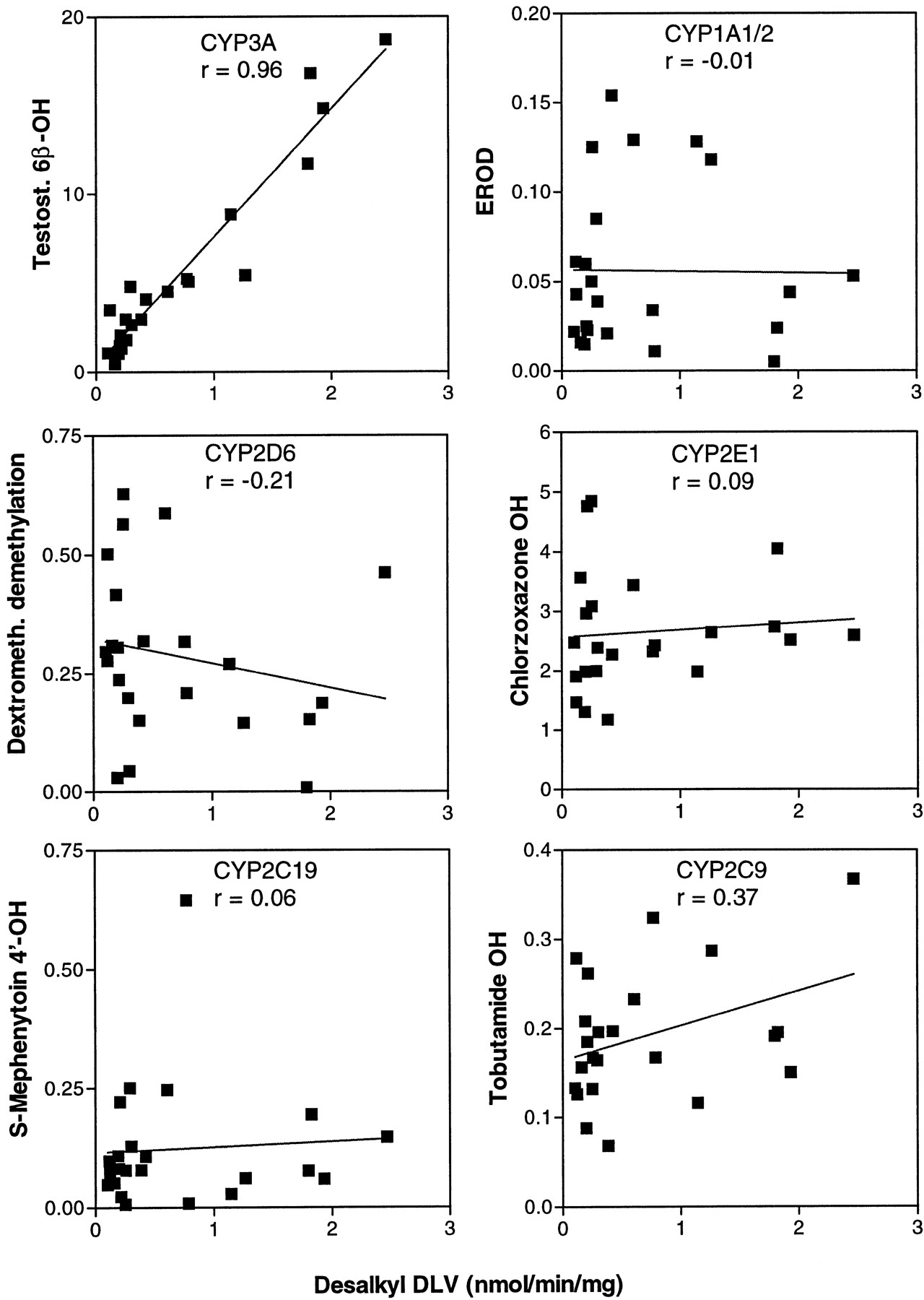
Delavirdine desalkylation rates were determined among 23 human liver microsomal samples, and rates were correlated with catalytic indicator activities for CYP1A1, -1A2, -2A6, -2D6, -2E1, -2C9, -3A4, and -4A11. Desalkylation rates displayed a wide sample-to-sample variation over a 24-fold range but were significantly correlated only with sample-to-sample variation in testosterone 6β-hydroxylase rates (r = 0.96) (fig. 7) and with coumarin 7-hydroxylase activity (r = 0.58), a marker for CYP2A6 (not shown). However, the latter activity was equally well correlated (r = 0.57) with testosterone 6β-hydroxylase activity. Similar correlation of these activities as well as inducibility by phenobarbital has been observed in monkeys, leading to the conclusion that CYP3A4 and CYP2A6 might be co-regulated (Bullock et al., 1995).

Correlation analysis of delavirdine metabolism with human CYP3A activity.
Delavirdine desalkylation was measured at 50 μM substrate among human liver microsomal samples (N = 23; 0.8 mg of protein/ml; 4-min incubation). Indicator CYP activities were determined by established methods. Sample-to-sample variation in rates of marker activities and delavirdine desalkylation were correlated. Rates are nmol/min/mg microsomal protein. Results represent mean of duplicate determinations.
In a related experiment, the desalkylation of delavirdine was measured among 10 human liver microsomal samples, selected to evenly span a wide range of CYP3A activity and run at concentrations of 5 or 50 μM delavirdine. Results show an excellent correlation of delavirdine desalkylation with testosterone 6β-hydroxylation at both 5 and 50 μM delavirdine (r = 0.92 and 0.95, respectively) and also show correlation of MET-7 with both delavirdine desalkylation and testosterone 6β-hydroxylation (table2). Noteworthy is the strong correlation (r = 0.96 and 0.98) of MET-7 and desalkyl delavirdine at substrate concentrations of 5 and 50 μM, respectively.
Correlation analysis of delavirdine metabolism (MET-7 and desalkyl-DLV) with testosterone 6β-hydroxylation by human liver microsomes (N = 10)
Effect of Isoform-Specific Inhibitors on Delavirdine Metabolism.
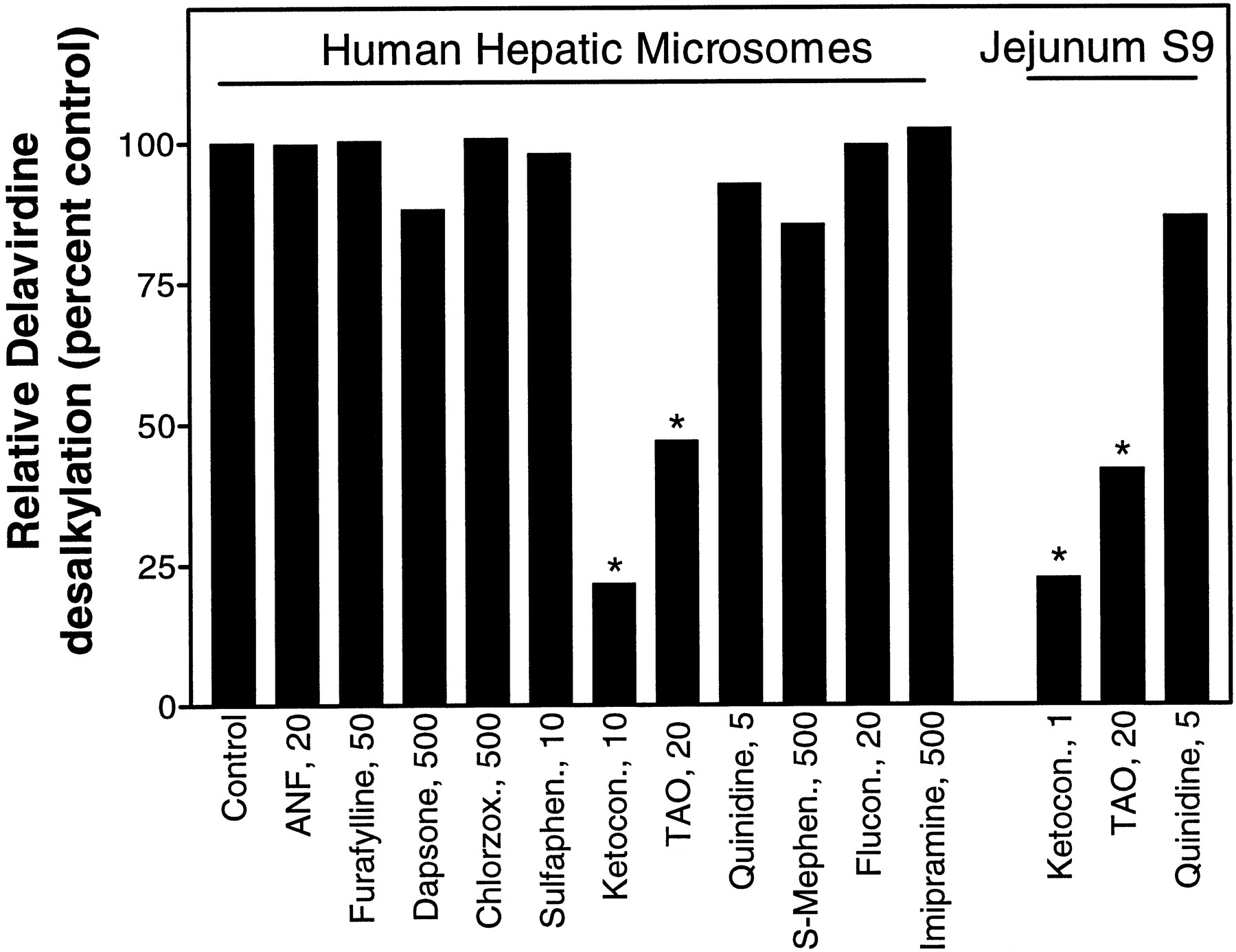
The involvement of specific CYP isoforms in delavirdine metabolism was assessed by examining human microsomal metabolism of delavirdine in the presence of certain known CYP inhibitors. Results, displayed as per cent of control, show that only ketoconazole and TAO demonstrated significant (>30%) inhibition of delavirdine dealkylation activity (fig. 8), supporting involvement of CYP3A in the metabolism of delavirdine by human liver microsomes. A related experiment using human jejunum S9 fraction showed that both TAO and ketoconazole produced significant inhibition of delavirdine desalkylation, whereas quinidine had only a small effect (fig. 8). This observation points to likely involvement of intestinal CYP3A in the metabolism of delavirdine.
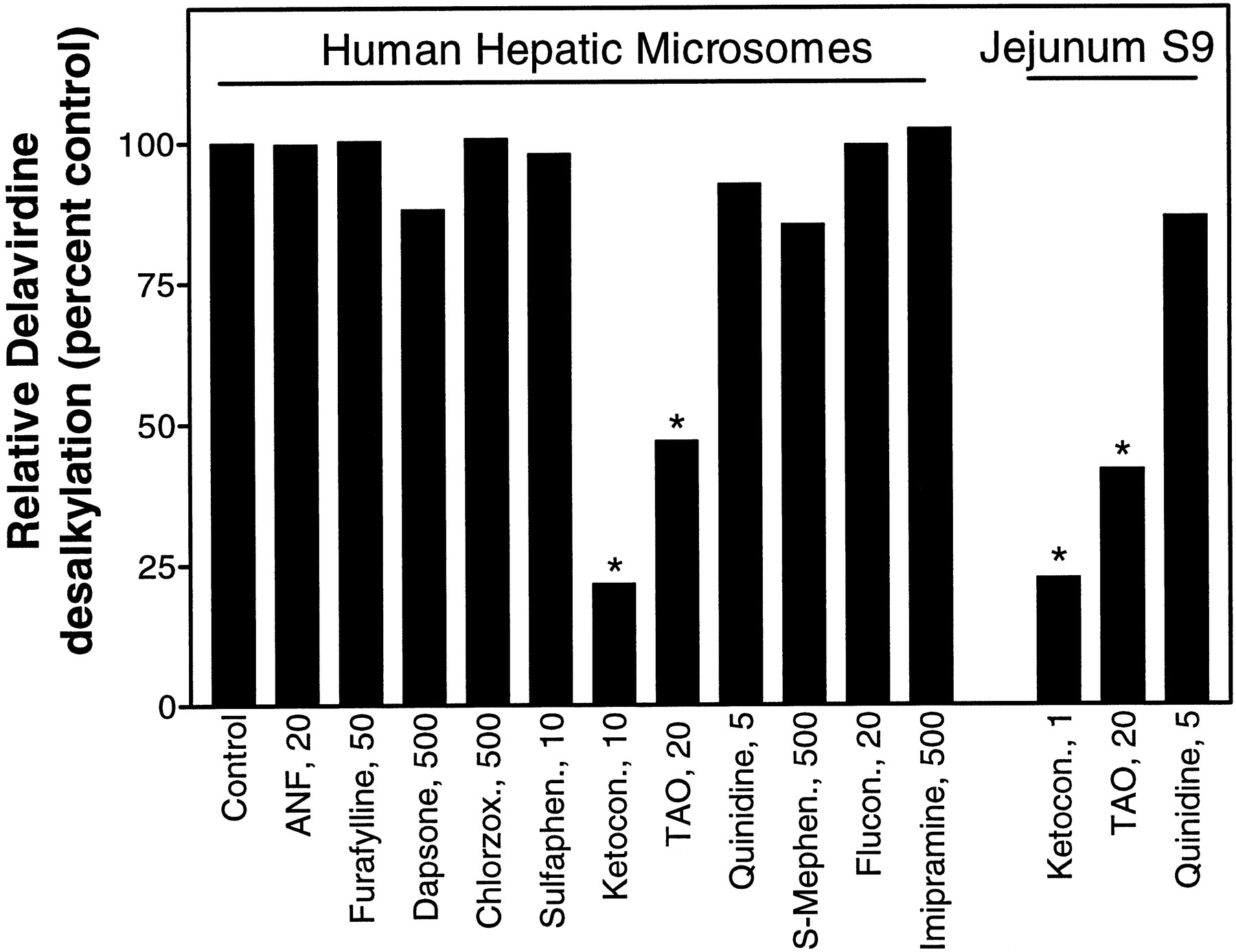
Effect of specific inhibitors on delavirdine desalkylation by human liver microsomes or jejunum S9.
Incubations were set up with pooled human liver microsomes (0.8 mg/ml) or jejunum S9 (10 mg/ml), 30 μM delavirdine, NADPH, and inhibitors at concentrations shown. Furafylline and TAO were preincubated with the metabolically complete system for 15 min before addition of delavirdine. Reactions were quenched after 10 min, and desalkylation was assayed by HPLC. Results are mean of duplicate determinations. Control values: human liver microsomes, 0.30 nmol/min/mg protein; jejunum S9, 0.0050 nmol/min/mg protein.
Metabolism of Delavirdine by cDNA-expressed P450 Isoforms.
The selectivity of certain human P450 isoforms for delavirdine metabolism was evaluated by incubation of delavirdine with cDNA-expressed CYP1A1, -1A2, -2A6, -2C8, -2C9, -2C18, -2C19, -2D6, -2E1, -3A4, and -3A5. Results showed that only CYP2D6 and CYP3A4 demonstrated significant desalkylation of delavirdine (fig.9). Incubation of delavirdine with increasing concentrations of CYP3A4 showed increased formation of desalkylated delavirdine as well as the formation of MET-7 and MET-7a (not shown). Desalkyl delavirdine was also catalyzed by CYP2C8 and CYP3A5 but only after prolonged incubation at high enzyme concentration (not shown). These experiments suggest that delavirdine desalkylation may be catalyzed by both CYP3A4 and CYP2D6 but that formation of MET-7 and -7a seems to be catalyzed only by CYP3A4.

HPLC-radiochemical profiles of delavirdine metabolism by cDNA-expressed P450 isoforms. [14C]Delavirdine (50 μM) was incubated with microsomal samples (0.5 or 1.0 mg protein/ml) for 30 min in the presence of NADPH.
Reactions were quenched with acetonitrile and analyzed by HPLC with radiochemical detection.
Evaluation of the kinetics of delavirdine metabolism by cDNA-expressed CYP2D6 revealed an apparent KM of 10.9 ± 0.8 μM and Vmax of 0.23 ± 0.01 nmol/min/mg microsomal protein. Metabolism by CYP3A4 showed aKM of 5.4 ± 1.4 μM andVmax of 0.14 ± 0.01 nmol/min/mg protein for production of desalkyl delavirdine; kinetics of MET-7 could not be estimated owing to its formation being below the limit of sensitivity under the assay conditions used.
Evaluation of Delavirdine Metabolism by CYP2D6.
The contribution of CYP2D6 to overall microsomal delavirdine metabolism was evaluated by measuring the desalkylation of delavirdine in the presence and absence of quinidine, using three microsomal samples that were phenotypically deficient in CYP2D6 and three samples that were phenotypically normal (table 3). Owing to the wide range of CYP3A activity among the samples, there was no apparent relationship between desalkylation and CYP2D6 activity. Addition of 1 or 10 μM quinidine to the incubations showed very little effect on the CYP2D6-deficient samples (approximately 2–9% loss in desalkylation) and only a moderate effect on the CYP2D6 containing samples, resulting in about a 15–21% loss in desalkylation activity. The results of this experiment suggest that CYP2D6 probably plays a minor role in the metabolic elimination of delavirdine in humans.
Effect of quinidine on delavirdine desalkylation catalyzed by human liver microsomes
Discussion
Hepatic microsomal metabolism of delavirdine was qualitatively similar among species and consistent with results of human and animal studies that showed that desalkyl delavirdine was the major circulating metabolite in plasma (Cheng et al., 1997; Chang et al., 1997). Desalkyl delavirdine is not active against HIV-1 reverse transcriptase (Genin et al., 1996). MET-7 and MET-7a were also formed by each species, with the possible exception of dog, and seem to be structurally related pyridine hydroxy compounds.
The present data set does not allow assignment of the position of pyridine monooxygenation; hydroxylation on the 4′, 5′, or 6′-positions as well as N-oxide formation are all possible. However, because rat metabolism experiments unambiguously established 6′-O-glucuronyl delavirdine as a major metabolite (Changet al., 1997), it seems likely that MET-7a could be the 6′-hydroxylated metabolite of delavirdine. This was supported by electrophilic frontier values (Ackland, 1993), which show that the 6′ position would be most electron dense, affording hydrogen abstraction and radical rebound, as proposed for P450-catalyzed aromatic hydroxylation (Ortiz de Montellano, 1989). Although the structures and relationship of MET-7 and MET-7a are not clear, it is possible that MET-7a and MET-7 are tautomers, with MET-7a representing the enol form, which slowly converts to MET-7, the keto form. As the pyrid-6′-one with the oxygen para to the isopropylamine substituent, MET-7 might be more prone to oxidation to the quinone imine, affording the observed stable desisopropyl ion (m/z 428) under ESI conditions. It has been shown that slowly interconverting tautomers can be chromatographically resolved (Kohl and Gescher, 1996; Lhoestet al., 1993). The chromatographic conditions used in this study (pH 4) would be sensitive to changes in metabolite pKa, and because the pyridine nitrogen would have an amide-like character in the keto form, it is possible the chromatographic system could separate the proposed tautomers. A 6′-hydroxydelavirdine standard could not be synthesized owing to chemical instability, perhaps like that observed here. Metabolism of pyridine by rats results in pyrid-2- and -4-one excretion, among other metabolites (Damani et al., 1982), suggesting the pyridone tautomer might be the more stable form of hydroxy pyridine, and a pyrid-6-one metabolite of nicotine has been proposed (Hochstein and Rittenberg, 1959). It has been shown that 2- and 4-oxygenated pyridines exist preferentially as pyridones (Katritzky, 1963). Furthermore, it has been proposed that despyridinyl delavirdine subsequently arises from MET-7 (as a pyridone), probably after addition of water and loss of the pyridine functionality (Chang et al., 1997). Alternatively, MET-7a could be the sulfate conjugate (MET-8) observed in delavirdine-treated rats that elutes with the same retention time as MET-7a; MS analysis of MET-8 revealed a spectrum-like MET-7a, owing to the lability of the sulfate group (Chang et al., 1997). However, such conjugation would be highly unlikely in a microsomal system that would lack cytosolic sulfotransferase and activated sulfate.
Evaluation of microsomal delavirdine desalkylation kinetics showed that most species, including human, displayed an apparentKM for desalkylation in the range of 10 μM or less. Owing to the instability of MET-7, lack of a MET-7 standard, and its greatly reduced formation relative to desalkyl delavirdine, the kinetic parameters for its formation could not be measured. Nevertheless, its formation, catalyzed by CYP3A, should be characterized by the same KM as desalkylation, as it has been implicitly demonstrated that for two or more products derived from a single substrate binding to a single enzyme, a single KM must characterize the entire system (Porter et al., 1977). No attempt was made to develop scaling factors to predict in vivo plasma clearance of delavirdine in either rats or humans based on the observed clearance. Although scaling factors are available for prediction of plasma clearance based on microsomal kinetic measurements (Houston, 1994), the present situation contains significant variables that would quickly invalidate such calculations. As will be described in another report, we observed significant irreversible binding of radiolabeled delavirdine metabolites to microsomal protein, and the metabolism of delavirdine results in progressive loss of metabolic capacity. Both factors would cause unpredictable changes inVmax.
Nevertheless, the kinetic determinations described in this report may be used advantageously to predict or explain characteristics of observed in vivo human plasma pharmacokinetics of delavirdine. Because therapeutic concentrations of delavirdine in plasma are expected to be in the range of approximately 10 μM, hepatic concentrations of the drug might normally exceed itsKM value, resulting in a tendency toward capacity-limited plasma clearance. Such conditions could potentially interfere with the clearance of co-administered drugs, particularly those with KM values comparable with or higher than delavirdine. Examination of metabolic profiles in rats treated with increasing doses of delavirdine showed a tendency toward capacity-limited metabolism as plasma concentrations exceeded 10 μM (Adams et al., 1996).
All experiments designed to identify the primary human P450 isoforms involved in delavirdine metabolism pointed to a catalytic role for CYP3A. Metabolism was inhibited by ketoconazole and TAO, metabolism was catalyzed by cDNA-expressed CYP3A4, and metabolism correlated with sample-to-sample variation in CYP3A-dependent activity in a panel of human liver microsomal samples. The strong correlation of MET-7 and desalkyl delavirdine formation supports the notion of common metabolite origin, that MET-7 formation and delavirdine desalkylation are both catalyzed by CYP3A and should share the sameKM , with theVmax of each reaction proportional to enzyme expression (Porter et al., 1977). Because CYP3A is involved in the metabolism of numerous drugs, the potential exists for delavirdine to be involved in adverse drug interactions. CYP3A5 also catalyzed the desalkylation of delavirdine but very slowly relative to CYP3A4 and remains to be fully characterized. CYP3A5 is a polymorphically expressed isoform that seems to be similar to CYP3A4, although substrate selectivity differences have been described (Wrighton and Stevens, 1992).
Interestingly, there was no significant correlation with CYP2D6 activity in the panel of human liver microsomes even though CYP2D6 carries out delavirdine desalkylation, and three of the human microsomal samples used were apparently deficient in CYP2D6. This points out a limitation of correlation analysis in that small but possibly significant enzymatic contributions can be masked.
Quinidine showed only limited inhibition of delavirdine metabolismin vitro, consistent with a minor contribution of CYP2D6-deficient microsomal samples; however, CYP2D6-dependent hepatic microsomal metabolism was demonstrated by showing that quinidine had almost no inhibitory effect on delavirdine metabolism by hepatic samples deficient in CYP2D6. Because CYP2D6 catalyzes the metabolism of many drugs and delavirdine showed a KM of 10.9 μM for CYP2D6, delavirdine has the potential to competitively inhibit the metabolism of some CYP2D6 substrates.
The reported experiments provide little characterization of the rat CYP isoforms involved in delavirdine metabolism. Although it seems likely that rat CYP3A might be involved (and unpublished experiments showed that cDNA expressed rat CYP2D1-catalyzed delavirdine desalkylation), we could not test for involvement of rat CYP2A, -2B, or -2C family members, owing to lack of commercially available cDNA-expressed rat CYP isoforms. The rat microsomal metabolites we observed are consistent with the rat in vivo metabolism scheme (Chang et al., 1997) with the exceptions that we did not observe amide bond cleavage, secondary metabolites, or conjugates; however, those reactions might not be expected in a microsomal system.
Acknowledgments
We gratefully acknowledge the help of A. Parkinson (University of Kansas Medical Center, Kansas City, KS) for preparation and evaluation of the human liver microsomal samples and the International Institute for Advancement of Medicine for supplying human liver tissue. We thank R. C. Mann for technical assistance and M. Chang, R. C. Steenwyk, and L. C. Wienkers for helpful discussions. We thank R. S. P. Hsi, J. A. Easter, and E. H. Chew for synthesis of radiolabeled delavirdine.
Footnotes
-
Send reprint requests to: Richard L. Voorman, Ph.D., Drug Metabolism Research, Bldg. 300-3, Pharmacia and Upjohn, Kalamazoo, MI 49007.
-
↵1 Present address: Department of Drug Metabolism, Pfizer Central Research, Sandwich, Kent, UK.
-
This work was presented in part at the 9th International Conference on Cytochrome P450, Zurich, Switzerland, 1995.
- Abbreviations used are::
- HIV-1
- human immunodeficiency virus type-1
- ESI
- electrospray ionization
- LC
- liquid chromatography
- MS
- mass spectrometry
- TAO
- troleandomycin
- Received December 4, 1997.
- Accepted March 4, 1998.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}