


Abstract
The visible spectrum of a ligand-bound cytochrome P450 is often used to determine the nature of the interaction between the ligand and the P450. One particularly characteristic form of spectra arises from the coordination of nitrogen-containing ligands to the P450 heme iron. These type II ligands tend to be inhibitors because they stabilize the low reduction potential P450 and prevent oxygen binding to the heme. Yet, several type II ligands containing aniline, imidazole, and triazole moieties are also known to be substrates of P450, although P450 binding spectra are not often scrutinized to make this distinction. Therefore, the three nitrogenous ligands aniline, imidazole, and triazole were used as binding spectra standards with purified human CYP3A4 and CYP2C9, because their small size should not present any steric limitations in their accessing the heme prosthetic group. Next, the spectra of P450 with drugs containing the three nitrogenous groups were collected for comparison. The absolute spectra demonstrated that the red-shift of the low-spin Soret band is mostly dependent on the electronic properties of the nitrogen ligand since they tended to match their respective standards, aniline, imidazole, and triazole. On the other hand, difference spectra seemed to be more sensitive to the steric properties of the ligand because they facilitated comparison of the spectral amplitudes achieved with the drugs versus those with the standard nitrogen ligands. Therefore, difference spectra may help reveal “weak” coordination to the heme that results from suboptimal orientation or ligand binding to more remote locations within the P450 active sites.
When studying substrate binding to the cytochromes P450 (P450s), the heme prosthetic group often serves as a very useful chromophore that can be exploited for purposes of characterization. With P450s, the heme is bound as in a b-type cytochrome except that the iron atom is liganded to a single Cys side chain as found in only a few other heme-containing proteins (e.g., chloroperoxidase, nitric oxide-synthase, and prostacyclin synthase). Because oxidation of P450-bound substrate occurs at the heme, certain aspects of substrate binding can be monitored readily with a spectrophotometer. Most commonly, ligand titration experiments are carried out to determine a spectral dissociation constant (KS) for a P450 substrate or inhibitor, but little attention is paid to the signature manners in which the P450 heme spectra are altered (reviewed by Jefcoate, 1978). In particular, there seems to be an oversimplification regarding coordination of P450 substrates to the heme iron. This interaction stabilizes the low-spin iron configuration and thus is assumed to prevent catalysis since it is the high-spin iron that is more conducive to reduction by P450 reductase. [More specifically, P450 isoform differences arise because the rate of reduction is more dependent on reduction potential than spin state (Ost et al., 2003).]
Generally, nitrogen-containing heme ligands, which bind more avidly than oxygen-containing ligands, are more often inhibitors than substrates (Gigon et al., 1968). Hence, many substrates (i.e., type I ligands) actually displace the heme water ligand, shifting the iron spin equilibrium toward the high-spin form, whereas nitrogen heterocycles and anilines (i.e., type II ligands) can replace the water ligand to stabilize the low-spin form. However, some type II ligands are also substrates (Kunze et al., 2006; Pearson et al., 2006), whereas others derive little, if any, affinity for P450 from this interaction (Ha-Duong et al., 2001), even though it is commonly described as being tight or high affinity in nature.
Thus, the question remains, if it is assumed that the above examples are not just exceptions to the rule, can the substrate-bound P450 spectrum tell us more about the type II binding interaction? Recently, we were intrigued by the use of absolute P450 spectra by Atkins and coworkers (Roberts et al., 2005) in which CYP3A4 was saturated with a type II ligand (aniline) to shift the majority of the CYP3A4 sample to the nitrogen-liganded low-spin form. Based on this work, the question was raised whether absolute spectra or difference spectra reveal more insight into the different degrees of type II binding. The amplitude of any difference spectrum induced by a type II substrate should depend partly on the natural abundance of high-spin P450 present for the isoform in question (Kumaki et al., 1978). However, within the same isoform, different type II substrates can give different ΔAmax values, but it is not always clear whether this is due to a population of substrate binding in type I fashion, steric restraints that limit optimal nitrogen-iron interaction, electronic effects, or a combination of the aforementioned. It should be noted that ΔAmax values also are not indicative of binding affinity as shown with some anilines (Hodek et al., 2004). Therefore, both absolute and difference spectra of two major drug-metabolizing enzymes, CYP3A4 and CYP2C9, were recorded with drugs that elicit type II difference spectra and compared with the type II ligands aniline, imidazole, and triazole, which are functionalities commonly found in approved drugs. It is postulated that λmax in absolute spectra of type II binders is primarily determined by the electronic structure of the coordinating group. Alternatively, the ability of difference spectra to accentuate changes in the high-spin P450 makes them more apt to reveal multiple substrate binding modes and/or binding modes that are more peripheral to the P450 heme prosthetic group.
Materials and Methods
Materials. Potassium phosphate, glycerol, acetonitrile, HCl, and imidazole were purchased from Fisher Scientific (Fairlawn, NJ). Sulfaphenazole, aniline, 1,2,4-triazole, DMSO, itraconazole (mixed isomers), lyophilized isocitrate dehydrogenase (containing sodium citrate and manganous sulfate), and d-(+)-threo-isocitric acid monopotassium salt were purchased from Sigma (St. Louis, MO). [1-methyl]-1,2,4-Triazole was purchased from Alfa Aesar (Ward Hill, MA). PH-302 [2-((2-(1H-imidazol-1-yl)-6-methylpyrimidin-4-yl)(3-((benzo-[d][1,3]dioxol-5-ylmethyl)(methyl)amino)propyl)amino)acetamide] was provided by Pfizer, Inc. (St. Louis, MO). Chemically competent E. coli F′IQ cells were obtained from Invitrogen (Carlsbad, CA). Corning Costar 3904 96-well plates were purchased from Fisher Scientific (Hampton, NH).
Enzymes. Full-length human CYP2C9 containing the Barnes (1996) modified N-terminus was expressed in E. coli F′IQ from the pCW plasmid provided by Dr. A. E. Rettie from the University of Washington. Exact methods for expression and purification were described previously (Locuson et al., 2006, and references therein). Full-length expressed and purified human CYP3A4 and human cytochrome P450 reductase were purchased from Invitrogen. Both P450s contain a C-terminal histidine tag used for purification via nickel-chelate chromatography.
Visible Spectroscopy. All spectra were carried out in split-beam mode with an Olis upgraded Aminco DW-2000 spectrophotometer (Olis, Inc., Bogart, GA) with the settings as described previously (Locuson et al., 2006). The wavelength range typically encompassed 340 to 500 nm and readings averaged over 10 scans were taken in 1-nm steps. Some absolute spectra were also recorded through 600 nm to obtain the α (569-nm) and β (535-nm) bands.
Substrate Preparation. PH-302 and sulfaphenazole were dissolved in DMSO and diluted in water to obtain the desired stock solutions. Aniline was used directly, or first dissolved in DMSO, and further diluted in water to achieve lower concentrations for stock solutions. Imidazole was prepared as a 2 M solution and the pH was adjusted to 6.0, 7.4, or 8.5 using 5 M HCl before being diluted further into buffer (0.1 M potassium phosphate, pH 7.4). Itraconazole (3.33 mM) was prepared in DMSO because it was found to be more soluble in this solvent than acetonitrile. Further dilution was carried out in DMSO, or in water, if the final concentration was <100 μM.
Substrate Titrations. All experiments were carried out at 30°C in matched micro quartz cuvettes (2 mm × 10 mm internal) containing 0.5 μM P450 and 20% glycerol (v/v) in 0.1 M potassium phosphate buffer, pH 7.4, unless otherwise noted. Substrates (Fig. 1) were then titrated into the cuvettes containing P450. To obtain difference spectra, a baseline was recorded with P450 in both the sample and reference cuvettes followed by substrate titration into the sample cuvette only and buffer titration of the same volume into the reference cuvette. In obtaining absolute spectra, a baseline was recorded with only the buffer and glycerol in the sample and reference cuvettes. Next, P450 was added to the sample cuvette followed by titration of the substrate into both cuvettes so that the majority of any interfering signal of substrate was subtracted except when high concentrations of ligand were needed to saturate the enzyme (e.g., aniline). All absolute spectra were normalized to the absorbance at 500 nm. Except for one case (itraconazole equilibration was not reached for ∼3 min), equilibration of samples after addition of substrate was reached immediately after mixing since no change in spectra could be detected upon repeated scans. Ranges of ligand concentrations used are presented in Table 1.
Binding spectra characteristics of CYP3A4 and CYP2C9 with type II ligands

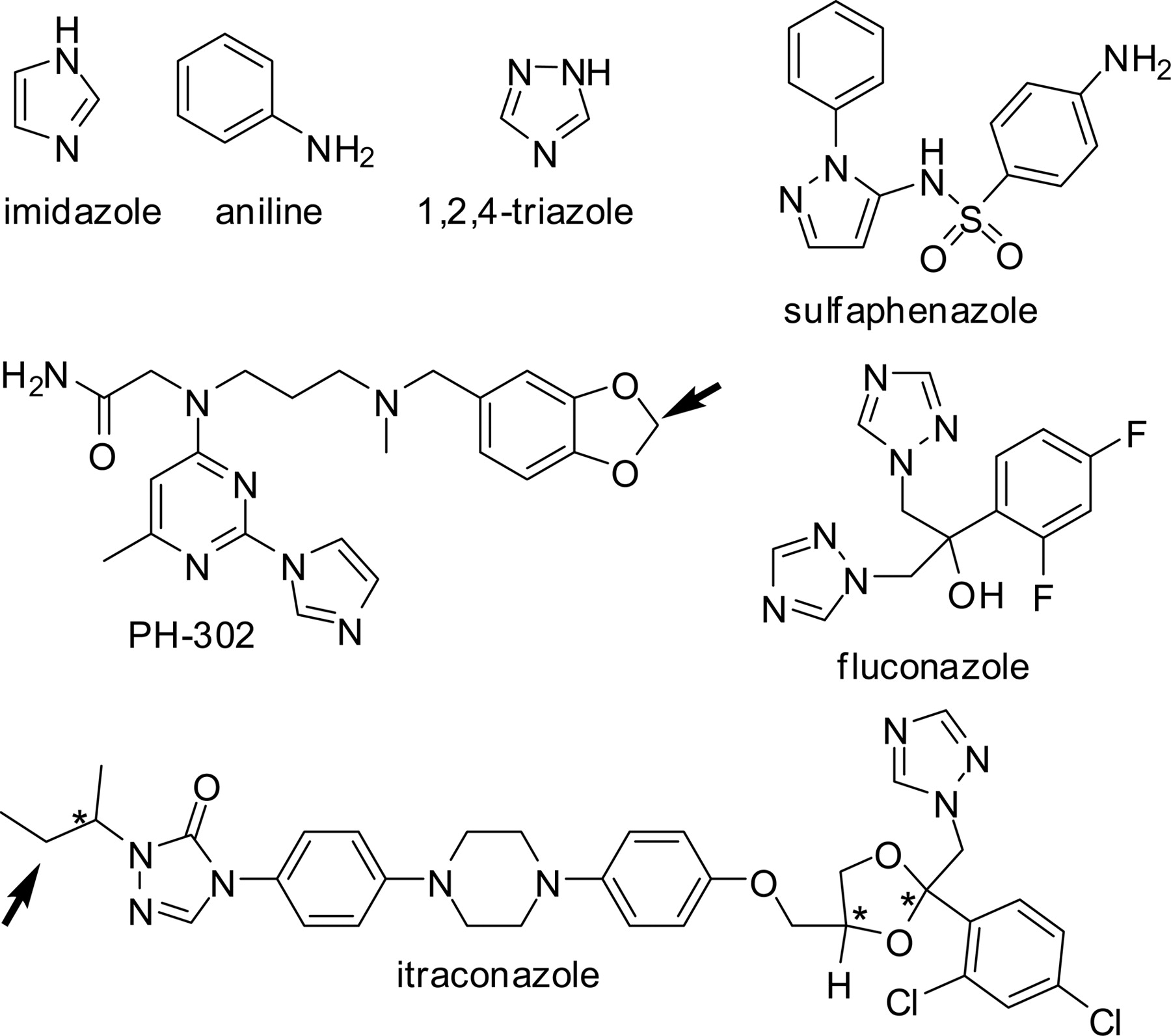
Structures of type II P450 ligands. Arrows denote the primary site of metabolism by CYP3A4 and asterisks denote chiral centers.
Data Fitting. Ligand binding curves were fit by nonlinear regression to one of four equations with SigmaPlot 8.0 (SSI, Inc., Richmond, CA). Equation 1 was used to determine KS from difference spectra where only one binding event was evident and the KS was higher than P450 concentration 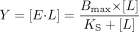 where Y is the spectroscopic signal, E is enzyme, and L is ligand. Equation 2 was used to determine KS when the plot of substrate concentration versus change in absorbance appeared hyperbolic, but for which the Eadie-Hofstee transform revealed curvature
where Y is the spectroscopic signal, E is enzyme, and L is ligand. Equation 2 was used to determine KS when the plot of substrate concentration versus change in absorbance appeared hyperbolic, but for which the Eadie-Hofstee transform revealed curvature 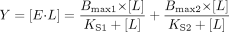 Equation 3 was used to determine KS for the high-affinity binding mode when multiple binding events were evident, but the high KS binding mode was not saturable
Equation 3 was used to determine KS for the high-affinity binding mode when multiple binding events were evident, but the high KS binding mode was not saturable 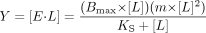 where m is the slope of the linear portion of the curve at high ligand concentrations. Equation 4 was used to determine KS when its value was lower than the concentration of P450 in the experiment
where m is the slope of the linear portion of the curve at high ligand concentrations. Equation 4 was used to determine KS when its value was lower than the concentration of P450 in the experiment 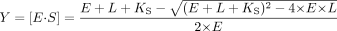 where E is P450 concentration. Equation 5 was used to determine KS when binding led to a sigmoidal response
where E is P450 concentration. Equation 5 was used to determine KS when binding led to a sigmoidal response 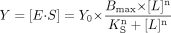 where Y0 is the signal at zero ligand concentration. The following term was multiplied by eqs. 1 to 3 when KS was determined from absolute spectra because the signal at zero ligand concentration was not zero
where Y0 is the signal at zero ligand concentration. The following term was multiplied by eqs. 1 to 3 when KS was determined from absolute spectra because the signal at zero ligand concentration was not zero 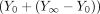 where Y0 and Y∞ are the signals at zero ligand and saturating ligand, respectively.
where Y0 and Y∞ are the signals at zero ligand and saturating ligand, respectively.
P450 Reconstitution. Purified CYP3A4 or CYP2C9 was incubated with two molar equivalents of human P450 reductase on ice for 5 min, followed by addition of extruded 1,2-dilauroyl-sn-glycero-3-phosphocholine to 1 μg/pmol P450. After 5 min, the reconstituted enzyme mixture was used in reaction mixtures containing substrate, an NADPH-regenerating system, and NADPH as described immediately below.
NADPH Consumption. The consumption of NADPH was monitored by a coupled enzyme method. Cytochrome P450 reductase uses NADPH to reduce P450 at two points in the P450 catalytic cycle. The conversion of NADP back to NADPH by isocitrate dehydrogenase then generates α-ketoglutarate, which is reacted with 2,4-dinitrophenylhydrazine to form a hydrazone chromophore (Friedemann, 1957). More recently, a similar method was used by Waskell and associates (Gruenke et al., 1995), but in the present case, the assay was adapted for use in 96-well plates. Reactions were carried out in 96-well plates in 50 mM potassium phosphate buffer, pH 7.4, using 20 pmol of reconstituted P450, the substrate concentrations shown in Table 1, 1 U of isocitrate dehydrogenase, and 10 mM d-(+)-threo-isocitrate in a total volume of 0.090 ml. Plates containing all the reaction components were preincubated in a water bath at 37°C for 3 min, and then 0.010 ml of NADPH (0.1 mM) was added to start the reactions. Trichloroacetic acid (70%, 10 μl) was used to quench the incubations after 30 min. Next, 0.05 ml of 1 mM 2,4-dinitrophenylhydrazine/1 M HCl solution was added and the plates were incubated at room temperature. Formation of the hydrazone of α-ketoglutarate was monitored spectroscopically at 390 nm to determine that 1 h was sufficient to ensure completion of the reaction. Next, 0.075 ml of 20% NaOH was added and the samples were monitored immediately at 450 nm along with standard curve samples in a Molecular Devices (Sunnyvale, CA) Thermomax microplate reader. Standard curves (duplicate samples) were prepared using α-ketoglutarate at 5 to 160 μM and processed at the same time as enzyme reaction samples.
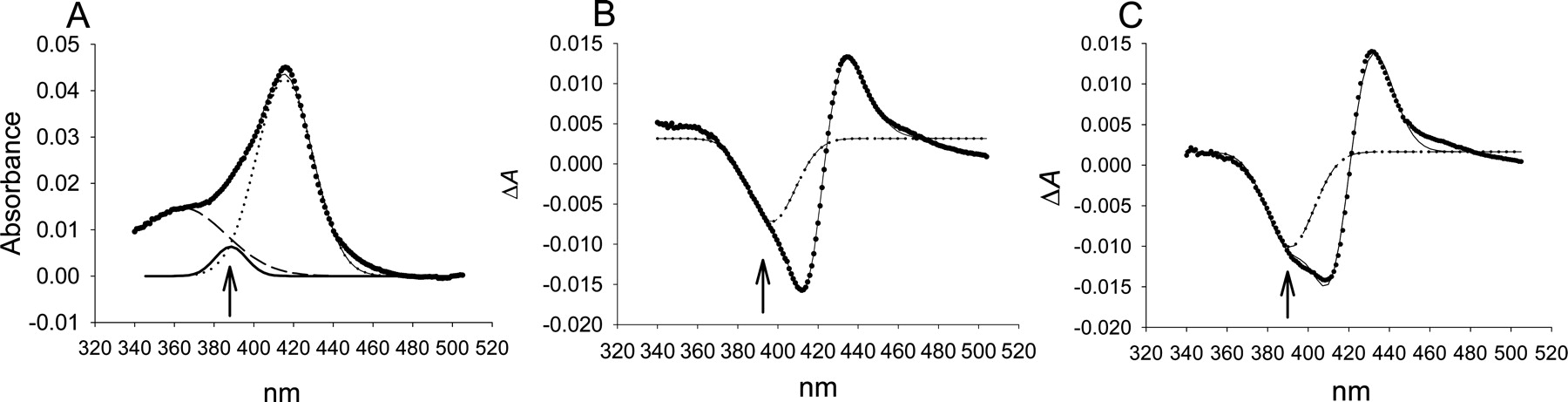
Gaussian Curve Fitting of P450 Spectra. Analysis was carried out using OriginPro version 7.5 (OriginLab Corp., Northampton, MA). All spectra were fit to three components. Absolute spectra were fit to three Gaussian curves at 360, 390, and 417 nm representing the delta, high-spin, and low-spin peaks, respectively. Difference spectra were fit to three Gaussian curves at 390, 410, and 430 nm. The shoulder of the trough at 390 nm represents the decrease in inherent high-spin P450, whereas the trough at 410 to 412 nm and peak at 425 to 432 nm result from the red-shift of the low-spin peak due to coordination of a ligand to the heme iron. The line through the data points in each plot represents the fit that is achieved using all three components, even though only the high-spin component is shown in difference spectra for clarity.
Results
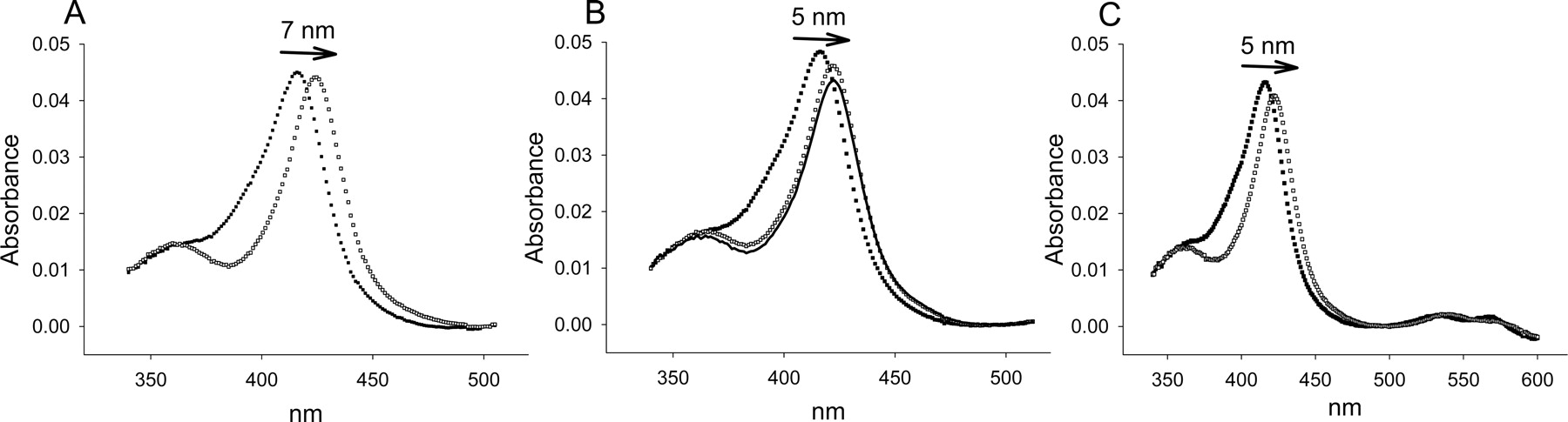
CYP3A4. The absolute spectrum of CYP3A4 displayed a low-spin Soret band with a λmax of 417 nm (Fig. 2A). A slight shoulder on the blue side of this peak (385–390 nm) was presumed to be the small percentage that exists in the high-spin state (Jefcoate, 1978) (Fig. 2A). High-spin CYP3A4 became more evident when the type II ligand imidazole was added to saturating concentrations (>20 mM) because the decrease at 385 nm was greater than would be expected if the spectrum was only red-shifted by imidazole (Fig. 2A). Saturating aniline (>20 mM) and 1,2,4-triazole (>40 mM) shifted the band 5 nm to 422 (Fig. 2, B and C), whereas imidazole ligation to the heme iron red-shifted the low-spin Soret band 7 nm to 424 nm. In addition, aniline decreased absorbance of λmax more than did imidazole (Table 1). No correction for dilution was made in this case since the total volume of the sample was changed less than 0.5%.
Previously, several nonaqueous solvents were shown to have effects on P450 spectra (Backes and Canady, 1981). Solvent effects can be identified in ligand titrations when reciprocal plots reveal nonlinearity (noncompetitive) or when a titration is performed with varying ligand/solvent ratios and different KS values result (competitive). In the current study, nonlinearity in Eadie-Hofstee-like plots was generally only observed with ligands that were not dissolved in DMSO. Addition of DMSO to 1% to a sample of CYP3A4 had no measurable effect on the λmax of the low-spin Soret band, but decreased its intensity 5.4% (Fig. 2B). KS values varied less than 1% when determined with 0 or 1% DMSO as demonstrated with imidazole and CYP3A4 (data not shown). Therefore, all subsequent ligand titrations were performed so that the final DMSO concentration remained under 1%.
Difference spectra were also obtained with CYP3A4 in the presence of the type II ligands aniline, imidazole, and 1,2,4-triazole (Fig. 3). These compounds acted as standards because there should be no steric restrictions in their ability to access the P450 heme. As noted with the absolute spectra, it was evident that imidazole coordination to the heme group differed from aniline and triazole since the ΔAmax between the peak and trough was ∼24% lower for aniline and ∼59% lower for triazole. In addition, the peak induced by aniline (429 nm) was located at a lower wavelength than that observed with imidazole (434 nm) as expected from the absolute spectra. All type II CYP3A4 spectra contained a significant shoulder in the trough that was not as intense as the lowest point at 410 nm, indicating that some high-spin P450 exists in the absence of substrate (i.e., mixture of type IIa and type IIb spectrum). If very little high-spin P450 was present before addition of type II ligand, only a trough centered around 410 nm would be observed (type IIb). Conversely, if very little low-spin P450 was present, a type II ligand would create a trough centered closer to 390 nm, representing the decrease in the high-spin Soret (type IIa).

Absolute spectra of 0.5 μM CYP3A4 in 0.1 M potassium phosphate buffer, pH 7.4, containing 20% glycerol (v/v) and type II ligands at 30°C. A, free (▪) and with 25 mM imidazole (□). B, 25 mM aniline (□) and 25 mM aniline after addition of 1% DMSO (—). C, 40 mM 1,2,4-triazole (□).
CYP3A4 Substrates. The compounds PH-302 and itraconazole were tested with CYP3A4 because they are not only type II binders, but also substrates that are metabolized in regions other than their nitrogen-containing rings. PH-302 is oxidized on the benzodioxole group to give a catechol (Hutzler et al., 2006), and itraconazole is metabolized at the alkyl side chain of the triazolone ring (Kunze et al., 2006). In addition, itraconazole is known to be a more potent inhibitor of CYP3A4 than of CYP2C9, whereas fluconazole was tested because it is a weaker inhibitor of CYP3A4 than CYP2C9 (Niwa et al., 2005). Hence, these differences in relative inhibition potency of the compounds toward individual P450s allowed assessment of isoform differences as well. Differences in electronic features could also be evaluated since PH-302 heme ligation involves binding of an imidazole group, whereas the antifungals itraconazole and fluconazole bind via their triazole groups.
PH-302 red-shifted the low-spin Soret band in the absolute spectra as effectively as imidazole, but the absorbance at λmax was decreased 7.6% (Fig. 4). In the difference spectra, the peak (431 nm) and trough (410 nm) induced by PH-302 were similar to the wavelengths given by imidazole, but ΔAmax was only ∼72% of that achieved with imidazole. Low-spin Soret band intensity was not affected as much with itraconazole and fluconazole compared with PH-302, but the red-shift of the Soret band of itraconazole and fluconazole was notably weaker in both forms of spectra, as were the ΔAmax values (Table 1). Titration of 1,2,4-triazole indicated that the degree of red-shift of the low-spin Soret is the same as that found for the triazole-containing drugs (Fig. 4B), as were the ΔAmax values from difference spectra.
KS values were determined from both types of spectra by plotting substrate concentration versus the change in absorbance between the peak and trough in difference spectra or the low- and high-spin Soret bands observed in absolute spectra (Fig. 5). KS values determined by the two methods were in agreement in most cases in which they could be compared (Table 1), except in the case of fluconazole, where the difference was roughly 2-fold. In the case of absolute spectra, the low-spin λmax absorbance used in fitting was 424 nm. Alternatively, use of the isosbestic point (420 nm) also led to similar KS values. As expected for type II binding compounds, which induce a decrease in high spin content, the ΔA from absolute spectra with CYP3A4 increased with ligand titration (e.g., itraconazole; Fig. 5).

Difference spectra of 0.5 μM CYP3A4 in 0.1 M potassium phosphate buffer, pH 7.4, containing 20% glycerol (v/v) and type II ligands at 30°C. Saturating concentrations of type II ligands (see Table 1) include imidazole (—), aniline (——), PH-302 (○), itraconazole (Δ), and fluconazole (•).
The substrate itraconazole produced a sigmoidal binding response (Fig. 5). Although this type of binding behavior has not been reported previously, differences in metabolism of specific itraconazole isomers in other studies (Kunze et al., 2006) suggest that our results may have arisen from using a mixture of isomers.
CYP2C9. Absolute spectra of CYP2C9 with and without the type II ligands aniline, imidazole, and triazole followed the same rank order with respect to the λmax of the low-spin Soret (imidazole > aniline ≅ triazole > no ligand) as observed with CYP3A4. However, slightly different values were obtained for the peak and trough in difference spectra compared with CYP3A4 (Table 1). In fact, the peaks for several type II ligands in CYP2C9 difference spectra were low in intensity and broad (Fig. 6). Also, the shoulder of the trough was less evident in difference spectra, suggesting that CYP2C9 in the absence of anything except buffer and glycerol exists with a smaller population of high-spin heme iron than does CYP3A4.

Absolute spectra of CYP3A4 with type II coordinating drugs. A, titration of PH-302 from 1 to 50 μM. B, saturating concentrations of fluconazole (□, 100 μM) and itraconazole (▴, 3 μM) compared with the free enzyme (▪). Conditions were identical to those in Fig. 1.

Itraconazole titration (0.1–3 μM) with CYP3A4 used to calculate the spectral dissociation constant, KS. Both the difference spectra (•) peak minus trough absorbance values and the difference between the low- and high-spin Soret bands in absolute spectra (○) were fit to eq. 5.
CYP2C9 Substrates. Similar to triazole and aniline, most of the drug substrates produced only minor red-shifts of the low-spin Soret band (Table 1; Fig. 6). In addition, the percentage change of the Soret band was greater than 5-fold more negative for CYP2C9 than for CYP3A4 on average. As a consequence, difference spectra typically possessed less intense peaks that were positioned at lower wavelengths. In several cases, this led to the difference spectra peaks being either very broad or not distinguishable from the baseline, as was the case with fluconazole (Fig. 6B). Unlike CYP3A4, fluconazole produced a much lower ΔA value compared with triazole with CYP2C9.
Sulfaphenazole was studied because it is a potent inhibitor of CYP2C9 that was initially thought to derive its affinity through type II binding via its aniline ring (Mancy et al., 1996). In concurrence with the results of others (Mancy et al., 1996), titration of sulfaphenazole in the present study resulted in type II difference spectra, although ΔAmax was only 4.8% of that obtained with imidazole (Fig. 6C). In addition, the λmax value (426 nm) obtained for the peak was similar to that reported by others (425 nm) (Ha-Duong et al., 2001), but at least 8 nm lower than that observed with imidazole. Absolute CYP2C9 spectra with saturating concentrations of sulfaphenazole resulted in a decrease in the absorbance of the low-spin Soret band and a very small red-shift of 1 nm.
Overall, CYP2C9 displayed more complex binding behavior than CYP3A4. Multiple binding events were evident in difference spectra with fluconazole and PH-302, which produced small ΔAmax values for the highest-affinity site (Fig. 7). The binding response for both drugs was best described as biphasic, containing a high-affinity/low-KS component and an unsaturable low-affinity/high-KS component. A biphasic response was also evident in absolute spectra for these compounds when the absorbance of the high-spin Soret band (385–390 nm) was subtracted from that of the low-spin Soret band (420–424 nm). The absorbance at 424 nm was used as the nitrogen-liganded low-spin Soret band because it represents the maximum red-shift observed (with imidazole), but use of the isosbestic point at 420 nm generally gave similar results.
When binding of drug was analyzed using absolute spectra (ΔA424–390) of CYP2C9, the ΔA value usually decreased during the titration, which would be expected for a type I binder (Table 1; Fig. 7B). It is proposed that this ΔA decrease resulted because the low-spin Soret band decreased in intensity more than it red-shifted, and more than the high-spin Soret band intensity decreased. With CYP3A4, which contained a higher population of inherent high-spin enzyme, ΔA424–390 always resulted in increasingly positive ΔA values upon type II ligand titration because the amplitude of the high-spin Soret band decreased more than that of the low-spin Soret band, and because the red-shift was usually larger.
KS Values. Equilibrium binding constants of the type II ligands were determined for both P450 isoforms using difference (ΔApeak-trough) and absolute spectra (ΔA424–390). The best model (i.e., r2) for describing the nonlinear fit and shape of the binding curve for a ligand using difference spectra was usually the same model giving the best fit of the corresponding absolute spectra. In the case of triazole, whose spectrum appeared hyperbolic, re-plots of ΔA/[L] versus ΔA (i.e., Eadie-Hofstee-like plot) were used to determine when a multiple-binding site model was required in nonlinear regression (e.g., eq. 2). The KS values obtained from the two forms of spectra differed significantly for the CYP2C9 substrates. Difference and absolute spectra KS values for fluconazole and sulfaphenazole binding to CYP2C9 differed by approximately 8.5- and 10-fold, respectively (Table 1; Fig. 7C). Also, KS values for imidazole, aniline, and triazole were generally higher than those for the drug ligands indicating a role for hydrophobic interactions in binding for drug-like type II ligands.

Visible spectra of CYP2C9 with and without a saturating concentration of type II ligand (see Table 1). A, imidazole difference spectrum. B, fluconazole absolute spectra (□). The inset shows the difference spectra at the concentration that is equal to KS1 (–) and the highest concentration tested (––). C, sulfaphenazole absolute spectra (□). The inset shows the difference spectrum under saturating concentrations.

Type II ligand titrations with CYP2C9 used to calculate the spectral dissociation constant, KS.A, ΔA obtained from difference spectra with fluconazole as the ligand. Eadie-Hofstee transform is shown in the inset. B, absolute spectra ΔA obtained with PH-302. C, fit of sulfaphenazole binding data using absolute (•) and difference spectra (□) ΔA values.
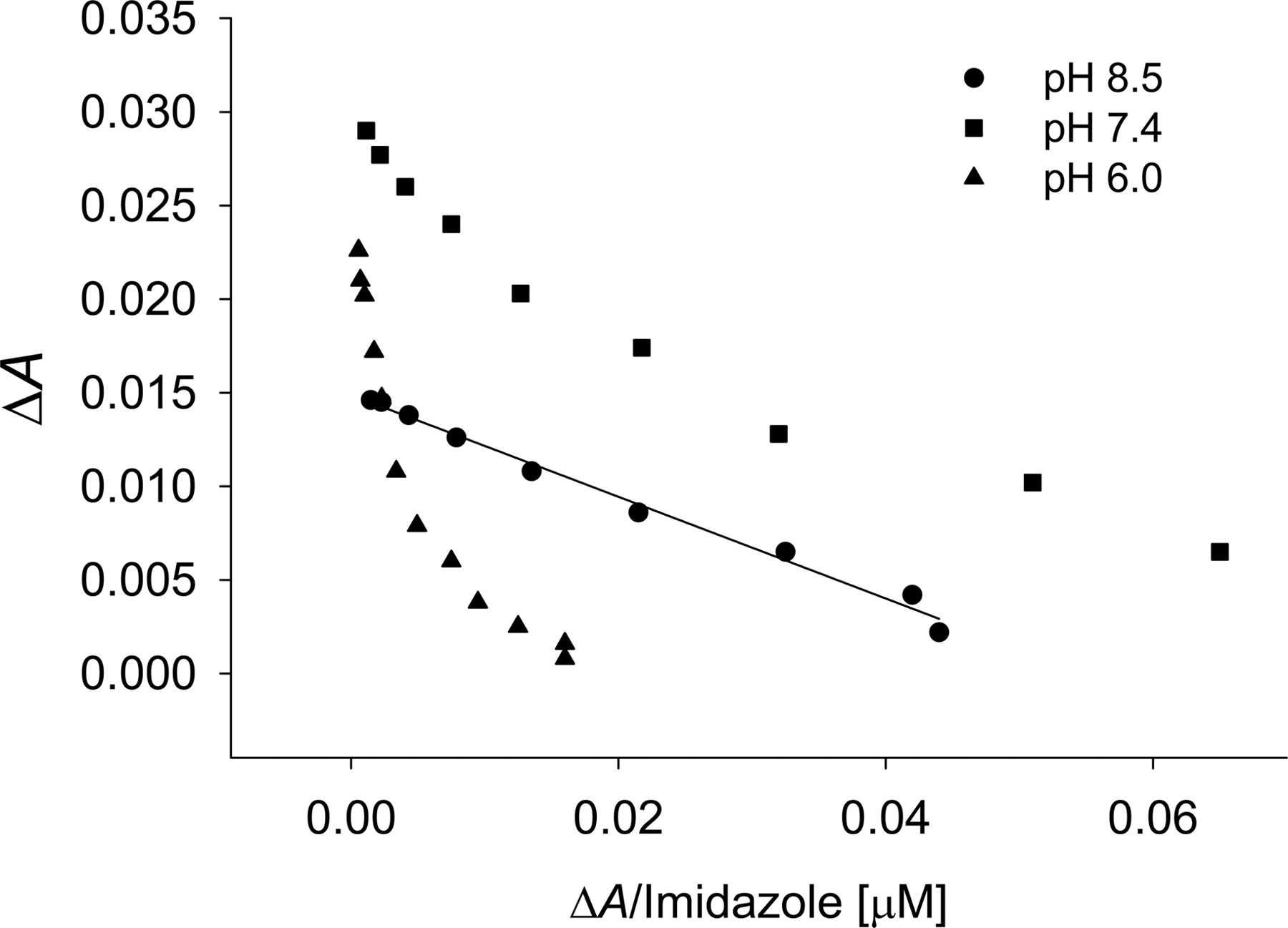
In an attempt to discern factors that may be responsible for the multiple binding phases of the type II ligands, two possibilities were considered: 1) multiple types of populated heme coordination modes, and 2) simultaneous occupancy of a P450 active site by multiple ligands that may have a cooperative effect on binding or the binding signal. Since triazole has three nitrogens that may produce different spectra, 1-methyl-1,2,4-triazole was used to mimic the 1-N-substituted drugs fluconazole and itraconazole. The apparent affinity was not altered, nor was the detection of multiple binding events attenuated with the methyl analog as compared with triazole with CYP2C9 (C. L. Locuson, J. M. Hutzler, and T. S. Tracy, unpublished data). This observation suggests that the multiple binding phases result from a single nitrogen or that the methyl group does not sufficiently hinder the nitrogens at positions 1 and 2. Next, the ionization state of the ligand was considered. Although the protonation of a second nitrogen in triazole should not occur near physiological pH (pKa = 2.45) (Satchell and Smith, 2002), pH produced noticeable changes in the spectra of imidazole-CYP2C9 complexes (Fig. 8). Under conditions where the majority of the imidazole should contain only one protonated nitrogen, a single binding event was observed (KS = 279 ± 10). Conversely, under more acidic conditions, multiple binding events became more evident, although the affinity of the higher-affinity binding event changed only modestly (KS = 274 ± 10).
NADPH Consumption. As an indication of the ability of the different type II ligands to stimulate the P450 catalytic cycle, the consumption of NADPH by cytochrome P450 reductase was monitored by a coupled enzyme assay. All enzyme incubations were carried out so that the same concentration of DMSO (2%) was present in all samples and controls, and the absorbance in samples containing everything but NADPH were used as blanks for each substrate. For both CYP3A4 and CYP2C9, aniline triggered the highest consumption of NADPH, and imidazole, the lowest, with the other type II drug ligands falling between these two extremes in ranking (Table 2; Fig. 9). Conversely, imidazole appeared to stabilize the low-spin P450 by type II heme coordination more effectively than any other ligand tested, as suggested by the low rates of P450 reduction by P450 reductase (Fig. 9). In the absence of substrate, the reconstituted CYP3A4-P450 reductase complex consumed more NADPH than CYP2C9, consistent with there being a higher population of high-spin P450 present in the CYP3A4 sample.
NADPH consumption rates associated with reconstituted CYP3A4 and CYP2C9 in the presence of type II ligands
P450s were reconstituted with human P450 reductase and lipid as described under Materials and Methods.
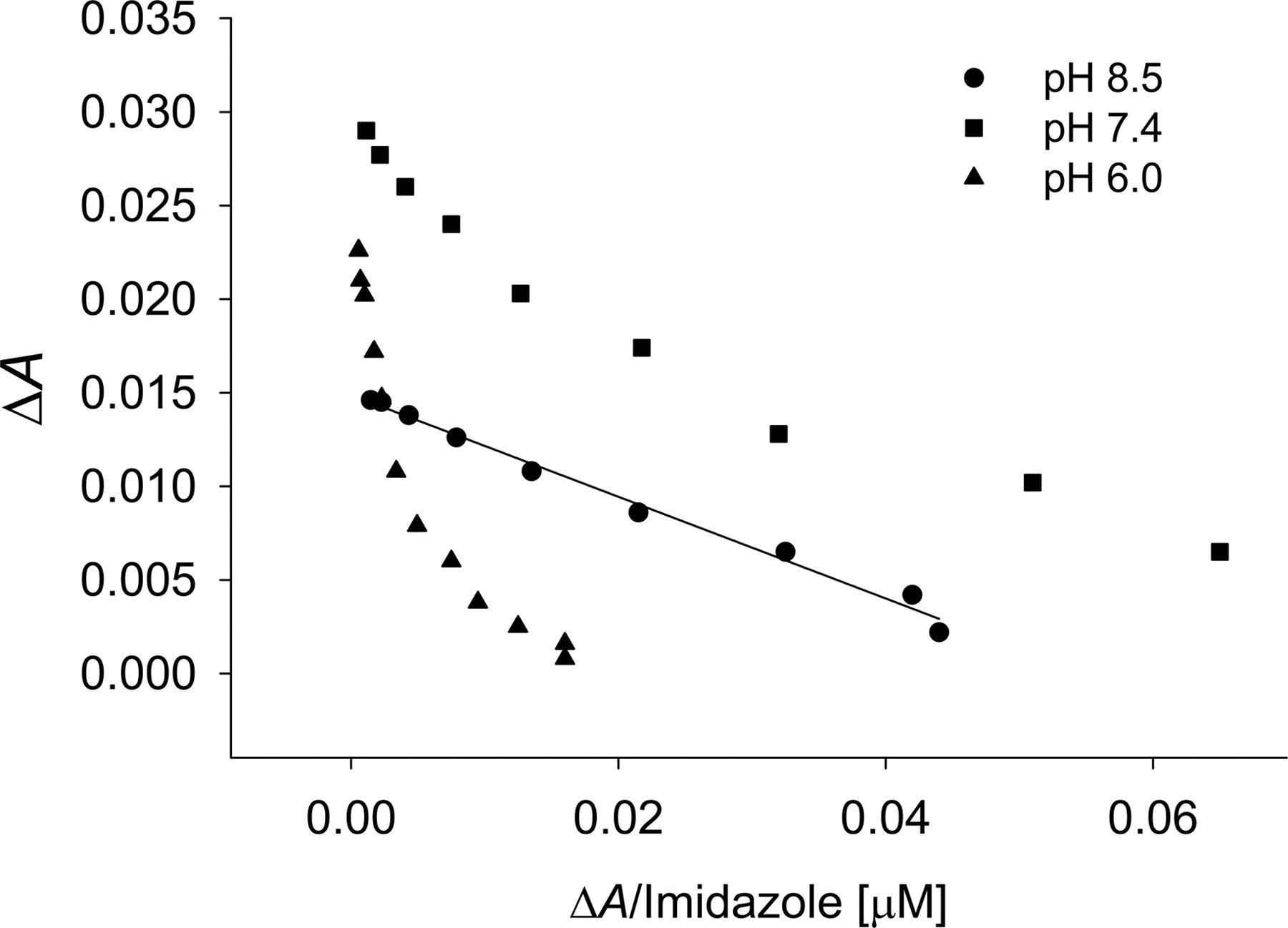
Effect of pH on the detection of multiple binding modes of imidazole with CYP2C9 as shown by Eadie-Hofstee plots. Imidazole concentrations ranged from 1 to 500 μM.

Evaluation of Gaussian curve fitting of spectra to determine relative changes in high-spin P450 upon addition of type II ligands. A, CYP3A4 absolute spectrum displays a small high-spin component in the absence of ligand near 390 nm. B, difference spectrum of CYP2C9 with a saturating concentration of imidazole demonstrates a mixed type II response with a moderate amount of a type IIa shoulder near 390 nm. C, difference spectrum of CYP3A4 with a saturating concentration of imidazole demonstrates a mixed type II response with a higher amount of type IIa than CYP2C9 according to the shoulder near 390 nm. See Materials and Methods for a description of the fitting procedure.
Discussion
Type II complexes result from P450 heme coordination of low-pKa nitrogen-bearing ligands. This binding directly to the heme of P450s can inhibit metabolism of a coadministered drug and result in a drug-drug interaction. Furthermore, drugs designed to target P450s have been made more potent by adding imidazole groups to them to enable type II coordination (Hackett et al., 2005). Interestingly, some type II ligands may also be substrates and some aniline- or imidazole-containing drugs do not always contribute to binding energy even though they give type II spectra, suggesting the possibility of different degrees of type II binding. The present studies compared the difference and absolute spectra of purified P450s with type II ligands to determine the spectral characteristics of varying degrees of type II binding.
Type II difference spectra of nitrogen-containing drugs were characterized with CYP3A4 and CYP2C9 enzymes. All compounds tested produced smaller ΔAmax values in difference spectra than imidazole alone (Table 1). Several compounds also decreased the amplitude of the low-spin Soret band. Interestingly, PH-302 and itraconazole are both metabolized by CYP3A4 at positions removed from the imidazole group, requiring binding of these compounds in multiple modes (Fig. 1). Conversely, fluconazole is oxidatively metabolized, to a small extent (Brammer et al., 1991), on the heme-coordinating triazole to give an N-oxide. If carried out by P450, fluconazole oxidation also argues for the existence of multiple binding modes, except that the motion of the substrate relative to the heme would be translational rather than rotational. These findings suggest that clues about multiple binding modes, steric factors (Chiba et al., 2001), and/or metabolism might be detected by comparing binding spectra to a standard spectrum, such as that of imidazole or triazole.
Although some of the spectral differences appear minor (Table 1), the case of CYP2C9 and sulfaphenazole provides evidence that difference spectra may indeed indicate which type II ligands have higher drug-drug interaction potential. Sulfaphenazole produces a weak type II signal with CYP2C9, but binds with equal affinity when the aniline group is replaced with a tolyl group (Rao et al., 2000). Given this information, compounds like sulfaphenazole can reveal the spectral characteristics of a weak type II ligand. In general, the ΔAmax is weaker than changes induced by saturating amounts of the standard, in this case, aniline. Further studies are needed to confirm this hypothesis.
In several cases where very different binding modes must exist to achieve metabolism, the nitrogen-bearing drug produced a red-shift in absolute spectra similar to that of the respective imidazole or triazole. The red-shift induced by imidazole was most similar to that observed with the imidazole-containing PH-302, and likewise for triazole compared with fluconazole and itraconazole. Since the small imidazole and triazole standards should not be hindered in accessing the heme group, these results suggest that the P450 absolute spectrum may be useful in identifying the electronic structure of the ligand. This is in agreement with previous work (Sono and Dawson, 1982) characterizing the Soret red-shifting ability of P450 ligands containing different coordinating atoms following the order: sulfur > nitrogen > oxygen.
Alternatively, differences in steric properties or in binding that is, on average, distant from the heme, may be more detectable with difference spectra. In all but one case (CYP3A4 with itraconazole), the ΔA amplitude for the type II drug ligands did not match that achieved with imidazole, aniline, or triazole, although this was sometimes complicated by multiple binding phases in CYP2C9. These ΔAmax differences between ligand standard and drug may occur because distal heme ligands exhibit strong preferences for orientation (Roberts et al., 2001; Scott et al., 2004). The observed differences in ΔAmax are much greater than the electronic effects reported earlier (Kulkarni et al., 1974; Mailman et al., 1974) supporting a possible relationship between low ΔAmax values and suboptimal coordination orientation or hindered binding. Thus, the trough amplitude in difference spectra may be more important than peak intensity, which should be somewhat dependent on the degree of low-spin Soret red-shift (Fig. 6B).
Field strength of the ligand and the planarity of the heme (Roberts et al., 2001) may also affect the λmax and ΔAmax values in absolute and difference spectra, respectively, of type II P450 complexes. However, the most basic and probably the most nucleophilic compound, imidazole, produced the most significant changes in binding spectra, agreeing with earlier work by Dawson (Sono and Dawson, 1982). Since hydrophobic forces play a large role in P450 ligand binding (Regev-Shoshani et al., 2004), the hydrophilic nature of the nitrogen ligand standards may also contribute to the lack of correlation between the affinity of nitrogen ligands and spectral characteristics. Donation of charge from the proximal cysteine axial ligand to the iron may also disfavor coordination at the distal axial position regardless of resulting spectral properties (Sono and Dawson, 1982). Thus, the nucleophilicity that allows imidazole to stabilize low-spin P450 more effectively than triazole may also explain its lower affinity.
Reduction of P450, indirectly monitored as NADPH consumption, was also used to probe the ability of a type II ligand to stabilize the low-spin state. Again, imidazole was alone in its ability to stabilize low-spin P450 (Table 2; Fig. 9). Total NADPH consumption also includes any uncoupled reactions carried out by P450 or P450 reductase in which a metabolite is not produced from the type II ligand. However, since the rate with imidazole is actually lower than that obtained in the absence of any substrate, the net effect observed with imidazole is interpreted here as fewer P450 reduction events. It should be noted that type II complexes are still reducible, so that the measured rates will be dependent on multiple factors.
Human P450 isoform differences were also observed, as CYP2C9 displayed more complex binding behavior than CYP3A4. Except in the case of triazole, this was not a consequence of the type of spectra used for the nonlinear regression analysis of the binding constant, because both difference and absolute spectra usually fit best to the same binding model. By altering the pH of the titration experiment buffer and the ligand solution, the binding of imidazole, but not triazole, could be “normalized” to a single binding event. The multiple nitrogens in triazole may give rise to a population of different heme complexes. Alternatively, the multiple binding phases may result from a population of enzyme with triazole directly coordinated to the heme iron and another population bound to the heme water ligand. Recently, fluconazole was found to occupy two distinct binding modes in CYP121 crystals and in electron paramagnetic resonance experiments even though, spectrally, hyperbolic binding was observed (Seward et al., 2006).
Another intriguing observation was that the CYP2C9 low-spin Soret band in type II ligand complexes exhibited decreased intensities compared with CYP3A4. This finding suggests that the deconvolution of absolute spectra obtained with type II ligands into low- and high-spin components may not be straightforward with some P450 isoforms. For instance, even though the fraction of low-spin enzyme increases with type II ligands, the low-spin complexed enzyme apparently has a lower extinction coefficient, as shown for CYP2C9 with several ligands (Fig. 6B; Table 1). Moreover, the decrease in high-spin enzyme, which is already low before addition of ligand, makes accurate deconvolution of the high-spin component difficult (Fig. 9A). Both of these observations argue that absolute spectra are less than optimal for making quantitative comparisons. The peaks of difference spectra are also suspect in their representation of low-spin enzyme because ligand-specific extinction coefficients and λmax values sometimes hinder peak detection altogether (Fig. 6B, inset). Conversely, the trough region of a difference spectrum does not appear to suffer as much from the abovementioned problems. In particular, it is hypothesized that ΔA500–390 would be useful when comparing a type II difference spectrum obtained with drug to that of a standard chemical with the same heme-coordinating group (Fig. 9, B and C). The intensity of the 390-nm region reflects the ability of a ligand to decrease the fraction of inherent high-spin enzyme present and, therefore, is not expected to produce ligand-specific effects on trough intensity or λmax. Deconvolution of the negative high-spin component of CYP3A4 and CYP2C9 produced with imidazole was accomplished readily, but the use of ΔA500–390 would eliminate the error resulting from having two baselines due to substrate interference at lower wavelengths (Fig. 9). However, the degree of type IIa spectrum (i.e., inherent high spin) exhibited by a P450 ultimately determines the analytical sensitivity in this approach unless a type I ligand is first added to the sample.
In conclusion, type II ligand-P450 complexes display varying spectral fingerprints based on their physicochemical properties and the P450 isoform under study. Several drugs have been described that undergo P450 oxidation at regions distinct from moieties containing type II-coordinating nitrogens, and other nitrogen-containing heterocycles may be preferentially metabolized by P450s (Dowers et al., 2004). Hence, P450 heme coordination by a drug lead does not necessarily imply drug-drug interaction potential. Furthermore, recent studies demonstrating substrate-dependent inhibition by the type II ligands ketoconazole and miconazole (Kumar et al., 2006) suggest that inhibition studies involving type II ligands may not always be sufficient for assessing inhibition potential. Thus, further exploration of type II binding characteristics may result in better decisions during the drug development process.
Acknowledgments
We thank Dr. Peter M. Gannett for thoughtful discussions.
Footnotes
-
This work was supported in part by a grant from the National Institutes of Health to T.S.T. (GM063215).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.012609.
-
ABBREVIATIONS: P450, cytochrome P450; DMSO, dimethyl sulfoxide; PH-302, 2-((2-(1H-imidazol-1-yl)-6-methylpyrimidin-4-yl)(3-((benzo[d]-[1,3]dioxol-5-ylmethyl)(methyl)amino)propyl)amino)acetamide.
- Received August 31, 2006.
- Accepted January 23, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}