


Abstract
The Caco-2 cell line and its subclone TC7 are frequently used for studying human intestinal transport and metabolism of xenobiotics. We have investigated the expression of soluble sulfotransferases (SULT) in parental Caco-2 and TC7 cells by immunoblotting. SULT1A1, SULT1A2, SULT1A3, SULT1B1, SULT1C1, SULT1C2, and SULT2A1 were expressed in both cell lines. SULT2B1a, SULT2B1b, and SULT4A1 were absent. SULT1E1 protein was found in TC7 but not in Caco-2 cells. Other differences in SULT between the cell lines were minor. More important was the influence of differentiation. Expression of the various SULT forms was low or not detectable in cultures just reaching confluence but then increased strongly. Likewise, the rate of sulfation of the model substrate 3-hydroxybenzo[a]pyrene was increased with increasing culture duration. Benzo[a]pyrene-1-sulfate and -3-sulfate were formed in both cell lines when benzo[a]pyrene was used as a substrate. A further metabolite, 3-hydroxybenzo[a]pyrene-glucuronide, was detected in TC7 but not in parental Caco-2 cells. Cytochrome P450 inducers enhanced the conversion of benzo[a]pyrene to these metabolites without altering mRNA levels of major phenol-conjugating SULT forms (SULT1A1, SULT1A3, and SULT1B1). Overall, differentiated Caco-2 and TC7 cells are rich sources of SULT, as is human intestinal mucosa. The SULT pattern is most similar to that found in small intestine, although levels of SULT1A1 and SULT1B1 are lower, and those of SULT1C1 are higher in Caco-2 and TC7 cells than previously found in intestinal samples. The differentiation-dependent expression of SULT in the cultured cells reflects the in vivo situation, where SULT expression is focused to differentiated enterocytes.
Numerous drugs and food-borne xenobiotics enter the organism via the intestine. Therefore, the presence and characteristics of transmembrane transporters and xenobiotic-metabolizing enzymes in enterocytes may play an important role in the kinetics of xenobiotics. The cell line Caco-2, established from a human colon adenocarcinoma, is widely used as a model system for intestinal biotransformation and permeability (Artursson and Karlsson, 1991; Meunier et al., 1995). Despite its origin from a tumor, the Caco-2 cell line has retained the ability to differentiate into polarized epithelial monolayers and shows numerous biochemical and morphological characteristics of enterocytes (e.g., formation of microvilli, tight junctions and desmosomes, and expression of brush-border enzymes such as sucrase-isomaltase) (Hidalgo et al., 1989). Caco-2 cells express various phase 1 and phase 2 enzymes, e.g., CYP1A1 (Boulenc et al., 1992) and CYP1B1 (Buesen et al., 2002; Lampen et al., 2004) and sulfotransferases 1A1 and 1A3 (Tamura et al., 2001), as well as transport proteins of the ATP-binding cassette family, such as P-glycoprotein (Hunter et al., 1993; Hirohashi et al., 2000), multidrug resistance-associated proteins, and breast cancer resistance protein (Taipalensuu et al., 2001). However, levels of transporters (Lennernas et al., 1996) and xenobiotic-metabolizing enzymes (Caro et al., 1995) may substantially vary from those found in intestine. Therefore, alternative intestinal cell models have been developed. The Caco-2 subclone TC7 was generated by passaging Caco-2 cells 198 times (Chantret et al., 1994). This procedure led to a selection of faster growing cells from the inhomogeneous Caco-2 population. TC7 cells differ from the parental Caco-2 cells in their shorter population doubling time and higher cell density. They are fully differentiated after a shorter period of time, 14 days (Chantret et al., 1994) rather than 21 days for Caco-2 cells. Furthermore, TC7 cells show higher glucose consumption and express an elevated level of CYP3A4 (Caro et al., 1995), the predominant cytochrome P450 (P450) found in the small intestine (Lin et al., 1999). Because of their higher UDP-glucuronosyltransferase (UGT) activity (Münzel et al., 1999; Sabolovic et al., 2000) compared with the parental Caco-2 cells, TC7 cells have been widely used as a tool to investigate the metabolism and transport of conjugates of xenobiotics with glucuronic acid.
The polycyclic aromatic hydrocarbon (PAH) benzo[a]pyrene (B[a]P) is a ubiquitous contaminant with high carcinogenic potential, with the diet being the main source of exposure for the general population (Hattemer-Frey and Travis, 1991). Because of its lipophilicity, ingested B[a]P may passively penetrate cell membranes of the small intestinal enterocytes, where it could undergo monooxygenation mediated by P450 (e.g., CYP1A1 and CYP1B1). This phase 1 metabolism produces potential substrates for phase 2 enzymes. Soluble sulfotransferases (SULT, EC 2.8.2), which comprise a gene superfamily encoding at least 11 different protein forms in humans, are involved in the metabolism of small endogenous compounds (e.g., steroids, catecholamines) and xenobiotics (Glatt, 2002). As the resulting sulfo conjugates are charged, they are water-soluble and do not passively penetrate cell membranes, properties that facilitate excretion. However, the excretion requires vectorial transmembrane transport mediated by carriers. Depending on the localization of the carriers involved, metabolites may be transferred from enterocytes into the blood or the intestinal lumen.
Although sulfation facilitates the excretion of numerous xenobiotics, it is not free of risks. Thus, sulfate, generated via sulfonation of an oxygen atom, is a good leaving group in certain chemical linkages, e.g., if the resulting cation is resonance-stabilized. Such sulfo conjugates may bind covalently to macromolecules and subsequently induce cytotoxic, genotoxic, and carcinogenic effects (Glatt, 1997, 2005). In general, bioactivation at low substrate concentration requires the presence of a specific SULT form characteristic for a given progenotoxicant (Glatt, 1997, 2005). Therefore, knowledge of the expression sites of individual SULT forms may be useful to identify candidate target tissues of adverse effects for some chemicals.
Recently we studied the expression of SULT forms in mucosa along the intestinal tract, as well as the distribution within crypts and villi (Teubner et al., 2007). The objective of the present study was to elucidate the expression of the members of the SULT superfamily during the differentiation of Caco-2 and TC7 cells. Moreover, because several PAH are able to induce their own metabolism by up-regulation of enzymes (including phase 2 enzymes such as UGT) (Lampen et al., 2004), this study also addresses the effect of PAH on the mRNA expression of selected SULT.
With regard to the nomenclature of SULT, we follow the suggestions of Blanchard et al. (2004) except for the SULT1C family, for which we continue using the original names SULT1C1 [rather than SULT1C2 suggested by Blanchard et al. (2004)] and SULT1C2 (rather than SULT1C4), to be consistent with our previous articles (e.g., Glatt, 1997, 2002, 2005; Teubner et al., 2007).
Materials and Methods
Chemicals. B[a]P, indole-3-carbinol, and β-naphthoflavone (β-NF) were purchased from Sigma (Taufkirchen, Germany). Oltipraz was obtained from McKesson Bioservices (San Francisco, CA). Phenanthrene, pyrene, and chrysene were purchased from the National Cancer Institute Chemical Carcinogen Repository, Midwest Research Institute (Kansas City, MO). Their purity was higher than 99%, as checked by gas chromatography/mass spectrometry. Indeno[1,2,3-cd]fluoranthene was synthesized at the Biochemical Institute for Environmental Carcinogens (Grosshansdorf, Germany). Stock solutions of all the test compounds were prepared in dimethylsulfoxide and stored at –20°C until used. All the other reagents used were of analytical or high-performance liquid chromatography grade.
Cell Culture. The human colon adenocarcinoma cell line Caco-2 and the Caco-2 subclone TC7 were obtained from the European Collection of Animal Cell Cultures (Porton Down, UK). Caco-2 cells were maintained in Dulbecco's modified Eagle's medium (Gibco-Invitrogen, Karlsruhe, Germany) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere of 5% CO2 in air at 37°C. The cells were used at passages 32 through 46. TC7 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 20% fetal bovine serum, 1% nonessential amino acids, 100 units/ml penicillin, and 100 μg/ml streptomycin. TC7 cells were used at passages 26 through 30.
Effects of PAH on SULT mRNA Expression. Caco-2 cells were seeded at a density of 1.5 × 105/flask (25 cm2) and cultured for 18 days after reaching confluence. Subsequently, the cells were treated with culture medium containing various PAH (B[a]P, pyrene, phenanthrene, chrysene, benzo[k]-fluoranthene, dibenzo[a,l]pyrene). Controls were only treated with the delivering solvent (0.1% dimethylsulfoxide, v/v). After 24 h of incubation, the medium was removed, the cell monolayer was washed with ice-cold phosphate-buffered saline, and cells were harvested. Total mRNA was prepared from freshly isolated cells according to the method of Chomczynski and Sacchi (1987). RNA concentrations were determined spectrophotometrically at λ = 260 nm.
Reverse transcription of 0.1 μg of RNA using oligo(dT)15 was performed for 120 min at 37°C with 200 units of Superscript II reverse transcriptase (Gibco BRL, Karlsruhe, Germany) in 50 mM Tris, pH 8.3, 75 mM KCl, 3 mM MgCl2, 20 mM dithiothreitol, and 0.2 mM deoxyadenosine triphosphate, deoxyguanosine triphosphate, deoxycytidine triphosphate, and deoxythymidine triphosphate. The polymerase chain reaction (PCR) was performed using 1 μl of the resulting cDNA solution. Primers for human β-actin were nucleotides (nt) 371 through 391 for sense and nt 800 through 820 for antisense (GenBank accession number X00351). Primers for human SULT1A1 were nt 39 through 58 for sense and nt 1006 through 1025 for antisense (GenBank accession number L10819). Primers for human SULT1A3 were nt 139 through 158 for sense and nt 1106 through 1125 for antisense (GenBank accession number L19956). Primers for human SULT1B1 were nt 343 through 362 for sense and nt 878 through 897 for antisense (GenBank accession number U95726). PCR was performed with 0.5 units of Taq polymerase (Qiagen, Hilden, Germany) in an automatic DNA thermal cycler (PerkinElmer/Cetus, Norwalk, CT). Briefly, 50 μl of a PCR master mixture was added to the cDNA samples containing PCR buffer, MgCl2 (to a final concentration of 1.5 mM), and 30 pmol of each primer. For amplifications of SULT1A1, SULT1A3, and β-actin sequences, 25 to 28 cycles were used (45 s at 94°C, 30 s at 57°C, 60 s at 72°C) followed by an additional 10 min at 72°C, whereas 30 cycles (60 s at 94°C, 45 s at 57°C, 60 s at 72°C) followed by an additional 10 min at 72°C were used for SULT1B1. In each experiment, water was used as a negative control. Under these conditions, all the amplifications produced single products within a linear range (data not shown). Each amplified product was checked by sequencing. BLAST 2.1 computer analyses (National Center for Biotechnology Information) showed that the products corresponded to the expected sequences. Furthermore, the length of the product was verified by performing the PCR with plasmid DNA containing the cloned cDNA.
Genotyping ofSULT1A1andSULT1A2in Caco-2 and TC7 Cells. The polymorphic SULT1A1 and SULT1A2 sequences were amplified in separate reactions using the forward primer SULT1A1-F (5′-GGT TGA GGA GTT GGC TCT GC-3′) and the reverse primer SULT1A1-R (5′-ATG AAC TCC TGG GGG ACG GT-3′) for SULT1A1 (Ozawa et al., 1998) and SULT1A2-F (5′-GGT CGA GGA GCT GGC TCT AT-3′) and SULT1A2-R (5′-CCT CAT GAA GGG GGA GAT GC-3′) for SULT1A2. With the exception of the amount of template DNA, PCR were performed using the conditions described by Engelke et al. (2000) in a T-gradient thermocycler from Biometra (Göttingen, Germany). Primers were obtained from BioTeZ (Berlin-Buch-GmbH, Berlin, Germany). Using these primers, PCR products were obtained with lengths of 281 base pair for SULT1A1 and 309 base pair for SULT1A2. They were quantified by electrophoresis in a 2% agarose gel (Agarose Ultra Pure, Invitrogen, Paisley, UK) using pUC19/MspI marker (MBI Fermentas, St. Leon-Roth, Germany) as standard.
Genotyping was performed by restriction fragment length polymorphism analyses after incubation of 70 ng of PCR product with 1 unit of Bsp143II for SULT1A1 and 1 unit of BpiI for SULT1A2 at 37°C for 3 h and subsequent separation of the digestion products on a 4% agarose gel (NuSieve 3:1 agarose, FMC Bioproducts, Rockland, ME). Electrophoresis was performed at 60 V.
Immunoblot Analysis. Caco-2 and TC7 cells were seeded in large culture flasks (75 cm2, 4 flasks for each group, 3 × 105 cells/flask) and allowed to grow for up to 19 days (Caco-2) or 14 days (TC7) after reaching confluence (day 0 is designated the day of confluence). The cell monolayers then were washed with ice-cold phosphate-buffered saline and scraped into 400 μl of ice-cold homogenization medium (0.1 M KH2PO4, 0.1 M K2HPO4, 0.1 M KCl, 1 μM ethylene glycol tetraacetic acid, 0.1 μM dithiothreitol, and 10 μM phenylmethylsulfonyl fluoride, pH 7.4). The cells were homogenized by ultrasonication on ice for 2 min. Subsequently, cell debris was removed by centrifugation (15 min, 3000g, 4°C), and the resulting supernatant was subjected to ultracentrifugation (60 min, 105,000g, 4°C) to give a clear supernatant representing the cytosolic fraction.
Cytosolic proteins were resolved by SDS-polyacrylamide (11%, w/w) gel electrophoresis. After electrophoresis, proteins were electrotransferred to Hybond enhanced chemiluminescence membrane (GE Healthcare, Freiburg, Germany). Bovine serum albumin (1%, w/v) dissolved in Tris-buffered saline (150 mM NaCl, 50 mM Tris-HCl, pH 7.6) containing 0.1% Tween 20 was used as blocking reagent for the membranes (for 1 h at 25°C) and for the dilution of antisera. For the detection of SULT, the following immunosera raised in sheep were used: anti-1A antiserum (raised against SULT1A3 and recognizing all the SULT1A forms) (final dilution, 1:10,000) (Richard et al., 2001), anti-1B1 antiserum (1:10,000), and anti-1C1 antiserum (1:5000) (Stanley et al., 2005). For the specific detection of SULT1A1 and SULT1A2—as opposed to all the other known human SULT forms, including SULT1A3—we used an antiserum (1:2000) raised in rabbits against a peptide sequence of SULT1A1 (Rubin et al., 1996). Because this peptide sequence only differs in a single amino acid from the respective sequence in SULT1A2, it also recognizes this form (Meinl et al., 2006). These antisera were a gift from M.W.H. Coughtrie (University of Dundee, Scotland). Further antisera were raised in rabbits and used in 1:2000 dilutions: anti-1E1 antiserum (Falany et al., 1995), anti-2A1 antiserum (Comer et al., 1993), and anti-2B1a antiserum (which also recognizes SULT2B1b) (Meloche and Falany, 2001). The anti-4A1 antiserum (1:2000) was raised in chicken (Falany et al., 2000). These antisera were provided by C.N. Falany (University of Alabama, Birmingham, AL). Because no antiserum against SULT1C2 was available, we expressed SULT1C2 cDNA in Escherichia coli BL-21 (DE3) using the pET28 vector (Merck Biosciences, Bad Schwalbach, Germany). Expression of SULT1C2 in this system led to the enclosure of the heterologous protein in bacterial inclusion bodies. As determined by SDS-polyacrylamide electrophoresis and staining with Coomassie blue, SULT1C2 made up nearly 95% of the total protein content of the inclusion bodies. The inclusion bodies were directly used for immunizing a rabbit. The resulting antiserum was finally diluted 1:10,000. The secondary goat anti-rabbit, donkey anti-sheep, and rabbit anti-chicken, coupled with horseradish peroxidase, were used in a dilution of 1:2000.
The specificity of some antisera was increased by preincubation with other SULT forms. Native anti-1C1 antiserum shows some cross-reactivity with SULT1B1, which comigrates with SULT1C1. Likewise, native anti-1C2 antiserum showed faint cross-reactivity with SULT1A3, a form abundant in Caco-2 and TC7 cells and migrating close to SULT1C2. These cross-reactivities were abolished by preincubating (1 h, 25°C) the anti-1C1 and anti-1C2 antisera with 1 ml of cytosolic fraction (nearly 5 mg of protein) from Salmonella strains expressing SULT1B1 and SULT1A3, respectively, diluted in 50 ml of blocking reagent.
Bands recognized by antisera were visualized using enhanced chemiluminescence detection kit according to the manufacturer's instructions (GE Healthcare, Braunschweig, Germany) together with the Fuji LAS-1000 imaging system (Raytest, Straubenhardt, Germany). To remove bound antibodies for reprobing with other antisera, membranes were incubated with 62.5 mM Tris-HCl, pH 6.7, containing 100 mM mercaptoethanol and 2% SDS at 50°C for 1 h. Afterward they were washed twice with Tris-buffered saline containing 0.1% Tween 20 for 15 min before new blocking reagent was applied. Membranes then were probed with the next antiserum as described. All the blots contained various controls (cDNA-expressed SULT) whose reactivity with the various antisera is known. Incomplete removal of an antibody or washing out of antigen before reprobing would have led to unspecific or insufficient signals, respectively, of these controls, which never was the case under the conditions used. Likewise, comparable results were obtained when we repeated the analysis with the antiserum used for the initial probing at the end of the reprobing series or when we changed the order of antisera in repeat experiments.
Formation of Phase 2 Metabolites of B[a]P in Caco-2 and TC7 Cells. Caco-2 and TC7 cells were grown on gelatin-coated (1%, w/v, in deionized water) glass Petri dishes (28 cm2) and provided with fresh medium every 2nd day. After reaching confluence, the cells were cultured for 15 days (TC7) or 17 days (Caco-2) and subsequently treated for 48 h with potential enzyme inducers (indole-3-carbinol, oltipraz, flavone, or β-NF) or the delivering solvent only (0.1% dimethylsulfoxide). After removal of the inducers by a change of the medium, the substrate B[a]P (10 μM) was added to each Petri dish. After 12 h of incubation, media were collected and stored at –20°C until analysis for B[a]P metabolites.
Sample preparation and high-performance liquid chromatographic analysis were performed as described by Buesen et al. (2002). In brief, samples were subjected to a solid-phase extraction using Bakerbond ODS C18 (J. T. Baker, Griesheim, Germany) glass cartridges. The solvent was removed by evaporation, and the sample was reconstituted with 60 μl of methanol. The separation of the B[a]P metabolites was performed using high-performance liquid chromatography (HP 1100 series, Agilent Technologies, Waldbronn, Germany) on a reversed-phase C18 column (PAH 16 plus, 5-μm particle size, J. T. Baker). The detection was carried out using a diode-array detector (HP 1100 series). B[a]P metabolites were quantified using an internal standard, indeno[1,2,3-cd]fluoranthene.
Results
Genotyping of SULT. Functional polymorphisms have been described for SULT1A1 and SULT1A2 leading to an Arg213His or Asn235Thr exchange in the respective protein. Because these polymorphisms are associated with altered stability and intrinsic activity of the enzymes, we examined the SULT1A1/1A2 genotype of Caco-2 and TC7 cells. Using PCR, we obtained specific amplicons of the SULT1A1 and SULT1A2 genes from the cell lines. The SULT1A1 amplicons of both cell lines were completely digested by Bsp143II, whereas the respective SULT1A2 amplicons were resistant to digestion with BpiI, indicating that both cell lines are homozygous for the reference-type (high-activity) forms of SULT1A1 and SULT1A2.
Expression of SULT Proteins in Caco-2 and TC7 Cells. We used nine antisera raised against various SULT forms and a SULT peptide to study expression of these proteins in Caco-2 and TC7 cells. Standards of all 11 human forms were available as proteins expressed in Salmonella typhimurium. In general, the same blot was probed with the different antisera after removal of the preceding antibody. We changed the order of the antisera in repeat experiments. Because this did not affect the result, we conclude that the stripping procedure removed the preceding antibody quantitatively but preserved the SULT proteins on the nitrocellulose membrane.
Highest expression of all the SULT was detected in TC7 cells (right half of Fig. 1) at the latest point of analysis (14 days after the cells had reached confluence). The anti-1A antiserum detected the SULT1A1, SULT1A2, and SULT1A3 standards and faintly the SULT1B1 standard (which comigrated with SULT1A2). TC7 cells showed signals comigrating with SULT1A1, SULT1A2/1B1, and SULT1A3 (Fig. 1A). The SULT1A3 signal was at least 10-fold stronger than the other two signals. The anti-1B1 antiserum detected the SULT1B1 standard with high sensitivity and showed some cross-reactivity with SULT1A3. Both proteins were detected in TC7 cells by the anti-1B1 antiserum (Fig. 1B). The anti-1C1 antiserum (after increasing its specificity by pretreatment with SULT1B1) recognized the SULT1C1 standard with high selectivity. In particular, it did not react with any other SULT form comigrating with SULT1C1. The presence of SULT1C1 protein could be clearly identified in TC7 cells (Fig. 1C). The same was the case for SULT1C2 (Fig. 1D). However, the anti-1C2 antiserum additionally recognized a second, faster migrating band (designated X in Fig. 1D). When we preincubated the anti-1C2 antiserum with bacterially expressed SULT1C2, both bands disappeared, indicating high structural similarity of both antigens. Moreover, the intensity of band X correlated with that of the SULT1C2 band in the various TC7 and Caco-2 cultures. Thus, band X may represent a degradation product of SULT1C2. SULT1E1 was detectable with a relatively low expression level using anti-1E1 antiserum (Fig. 1E). SULT1E1 migrates between SULT1B1 and SULT1A3, other forms detected by anti-1E1 antiserum in TC7 cells. Anti-1E1 antiserum gave rise to a further band (designated Y in Fig. 1E) migrating more slowly than SULT1A3 and SULT1C2, the SULT forms detected with the highest apparent molecular weight. This additional band was stained with similar intensity in each lane containing Caco-2 or TC7 cytosol and weakly in some lanes loaded with standards. The nature of this band is unknown. Using anti-2A1 anti-serum, SULT2A1 was detected in TC7 cells (Fig. 1F). A similarly strong, but faster, migrating band (designated Z in Fig. 1F) was also detected with this antiserum. The intensity of band Z correlated with that of the SULT2A1 band in the various TC7 and Caco-2 cultures. Thus, band Z may represent a degradation product of SULT2A1. SULT2B1a, SULT2B1b, and SULT4A1 were not detected in any TC7 and Caco-2 cultures (data not shown).

Immunoblot analysis of the expression of SULT in Caco-2 and TC7 cells cultured for varying time periods. Cells were harvested after reaching confluence, defined as day 0 (d0) or on a later day as indicated. Cytosolic fraction (100 μg of total protein) was electrophoresed, blotted, and stained using the antiserum indicated on the left side of each panel. After removing antibodies, the same blot was probed with another antiserum. Cytosols from S. typhimurium strains engineered for expression of human SULT were used as standards: SULT1A3 (2 μg of total cytosolic protein for left lane, 25 μg for right lane), SULT1B1 (1 μg), SULT2A1 (2 μg), SULT1A1 (2 μg), SULT1E1 (8 μg), SULT1A2 (2 μg), SULT1C1 (0.5 μg), and SULT1C2 (1 μg). The electrophoretic mobility of the standards is indicated by arrows on the right side of the panels. X, Y, and Z indicate unknown bands discussed in the main text.
SULT levels were much lower in young TC7 cultures than in differentiated cells. On day 0, when cells just had reached confluence, only SULT1A3 and SULT1C2 were detected (Fig. 1, A and D). The SULT1A3 level was slightly increased on day 4 and strongly increased on days 9 and 14. SULT1C2 was weakly expressed at the initial two times of analysis but strongly expressed at the later times. The other SULT forms only became detectable on day 9 (Fig. 1, B, C, E, and F). At this stage, expression levels were similar to those observed on day 14.
SULT expression in parental Caco-2 cells (Fig. 1, left) was similar to that in TC7 cells with the following modifications: expression of several SULT forms started earlier in Caco-2 cells than in TC7 cells. SULT1B1 (Fig. 1B) and SULT2A1 (Fig. 1F) levels were much lower in Caco-2 cells than in TC7 cells. SULT1E1 was the only form that was detected in TC7 cells but was absent in Caco-2 cells (Fig. 1E). Even when we increased the amount of Caco-2 protein to 200 μg/lane, no SULT1E1 signal was detected (data not shown).
We already mentioned that the anti-1A1 antiserum recognized a SULT1A2/1B1 band in Caco-2 and TC7 cells (Fig. 1A). This band was also recognized by an antiserum that selectively detects SULT1B1 but not SULT1A2, implying that SULT1B1 was present. Whether SULT1A2 was also present required further clarification. To this end we used an anti-SULT1A1 peptide antiserum that only recognizes SULT1A1 and SULT1A2 but not any other human SULT form known. Because this antiserum is less sensitive than the anti-SULT1A antiserum (raised against a full-length protein), we had to load larger amounts of the cytosolic preparations. As a control we used the SULT1B1 standard at a level equal or higher than present in test samples (Fig. 2A). The anti-SULT1A1 peptide antiserum recognized two bands in TC7 and Caco-2 cells. They comigrated with the SULT1A1 and SULT1A2 standards (Fig. 2B). The SULT1B1 standard was not recognized by the anti-SULT1A1 peptide antiserum, indicating that the SULT1A2 band contained SULT1A2 protein (rather than the signal reflected a cross-reactivity of the antiserum with SULT1B1). In this analysis, we included four human intestinal samples from our recent study (Teubner et al., 2007), two from ileal mucosa and two from colonic mucosa. Levels of SULT1A1 and SULT1B1 were clearly higher in these samples than in TC7 and Caco-2 cells (Fig. 2). One ileal sample and one colonic sample also showed faint signals at the height of the band of the SULT1A2 standard. It may be possible to increase the intensity of this band by loading larger amounts of the tissue preparations on the gel; however, this would also increase the signal of the neighboring, very strong SULT1A1 band. In any case, it is clear that SULT1A1 levels strongly exceeded those of SULT1A2 in the intestinal samples, whereas these forms were expressed at nearly equal levels in TC7 cells; parental Caco-2 cells showed a 1.7-fold higher level of SULT1A1 protein than that of SULT1A2.
Table 1 contains a summary of the levels of the SULT proteins in differentiated Caco-2 and TC7 cells, as estimated from the immunoblots. For comparison we have added data for human ileum and colon mucosa from another study in which we had used similar methods (Teubner et al., 2007). Moreover, the table contains data published by Tamura et al. (2001) on the expression of various SULT mRNA in Caco-2 cells. These findings will be discussed under Discussion.
SULT expression levels in Caco-2 and TC7 cells compared with colonic and ileal mucosa

Verification of the expression of SULT1A2 in Caco-2 and TC7 cells, taking into account its comigration and possible immuno cross-reactivity with SULT1B1. Based on results from preceding experiments, we estimated the level of SULT1B1 in the various enzyme sources. To give an equal SULT1B1 background signal, the total protein amount in each lane was adjusted to contain 15 ng of SULT1B1 protein, except for the SULT1A2 standard (no SULT1B1) and Caco-2 cells (maximum load, 300 μg of total cytosolic protein, containing 3.7 ng of SULT1B1 protein) (A). The same blot then was reprobed with an anti-SULT1A1 peptide antiserum that recognizes SULT1A1 and SULT1A2 but no other human SULT forms; in particular, note that the SULT1B1 standard was not recognized by this antiserum (B). In addition to the cytosolic preparations from Caco-2 cells (19 days after reaching confluence, 300 μg of protein) and TC7 cells (14 days after reaching confluence, 190 μg of protein) and from Salmonella strains expressing SULT1B1 and SULT1A2 (1.5 and 0.5 μg of total protein equivalent to 15 ng of the corresponding SULT), we analyzed two ileal samples (50 μg of protein each) and two colonic samples (65 and 75 μg). Because of the strongly differing total amounts of protein loaded, migration varied somewhat between the lanes; in particular, migration was enhanced when very much protein was loaded (i.e., with the Caco-2 and TC7 samples). This was true for SULT1A1 and SULT1A2 (bottom), as well as the SULT1B1 reference band (top), which may be used as a curve template.
Effects of Caco-2 Cell Differentiation on Sulfation Capacity of Intact Cells. To see whether the increasing level of SULT proteins with the stage of differentiation enhances the sulfation capacity, we exposed Caco-2 cultures to the model substrate 3-hydroxybenzo-[a]pyrene (3-OH-B[a]P) at various times (2–20 days) after reaching confluence. During the observation period, the sulfation rate (measured as B[a]P-3-sulfate appearing in the medium) increased nearly 3-fold (Fig. 3). Similar results were obtained when 1-hydroxybenzo-[a]pyrene was used as the substrate (data not shown).
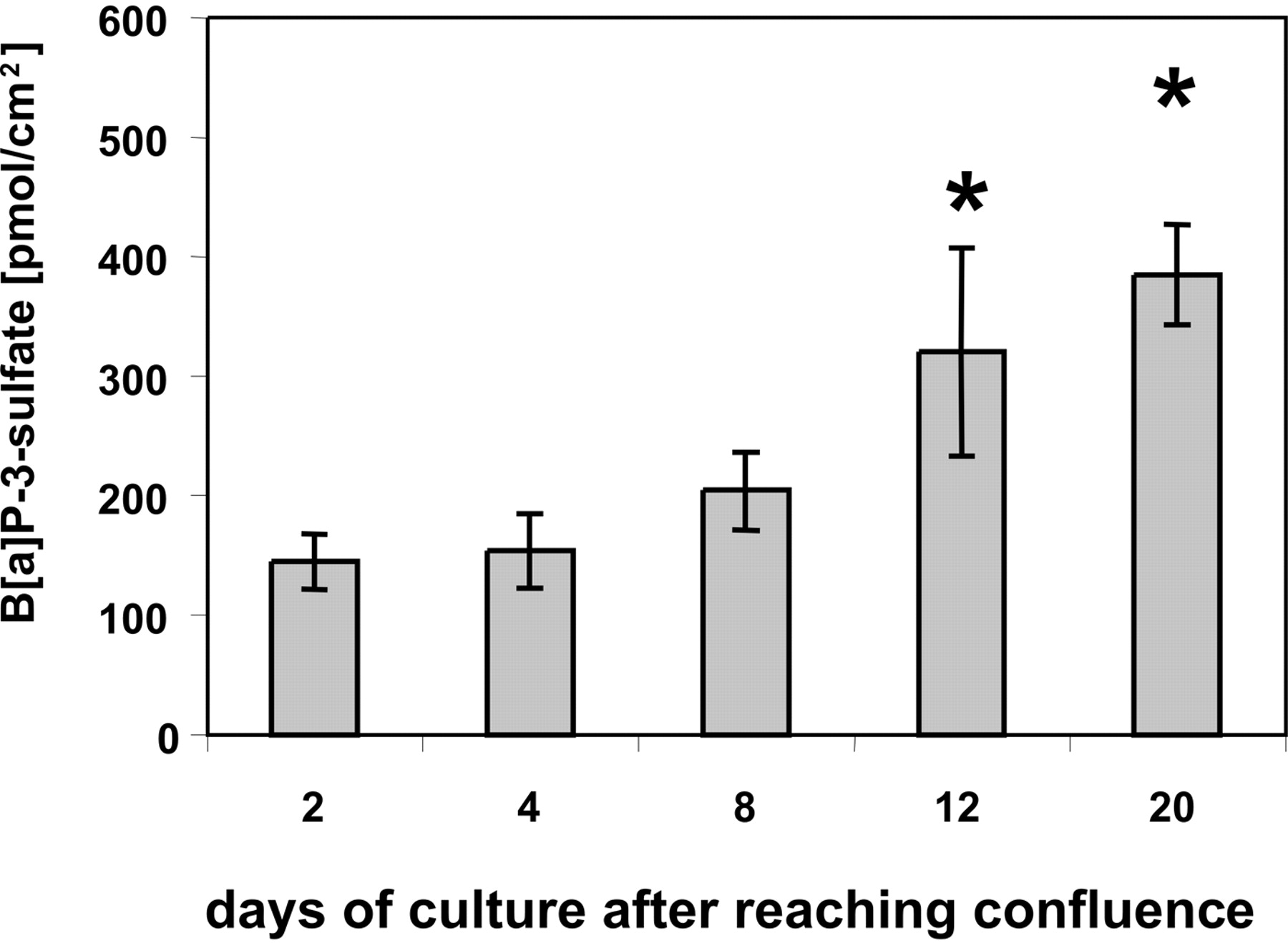
Effect of differentiation stage of Caco-2 cells on the conversion of 3-OH-B[a]P to B[a]P-3-sulfate. After reaching confluence (day 0), cells were grown for different periods as indicated and subsequently incubated with medium containing 5 μM 3-OH-B[a]P for 12 h. The medium then was analyzed for the formation of B[a]P-3-sulfate. Values are mean ± S.D. from triplicate determinations. *, p < 0.05 (one-way analysis of variance, Tukey-Kramer post-test): 12-day cultures compared with 2- or 4-day cultures, and 24-day cultures compared with 2-, 4-, or 8-day cultures.
Comparison of the Metabolism of B[a]P in Caco-2 and TC7 Cells. We previously showed that Caco-2 cells metabolize B[a]P to B[a]P-3-sulfate and B[a]P-1-sulfate (Ebert et al., 2005). Pretreatment of the cells with various compounds (indole-3-carbinol, oltipraz, flavone, and β-NF) enhanced the expression CYP1A1 and CYP1B1 and the conversion of B[a]P to its sulfated metabolites. We have repeated this experiment and extended it to TC7 cells. Cells were exposed to the inducers for 48 h. After removing the inducers by a change of the medium, the substrate, B[a]P, was added for 12 h, followed by determination of metabolites (B[a]P-1-sulfate, B[a]P-3-sulfate, and 3-OH-B[a]P-glucuronide) present in the medium.
Nonpretreated Caco-2 cells produced similar amounts of both sulfates. All the pretreatments (with indole-3-carbinol, oltipraz, flavone, or β-NF) enhanced the formation of both sulfated metabolites by a factor of 1.5 to 2.2 (Fig. 4A).

Formation of major phase 2 metabolites of B[a]P by Caco-2 (A) and TC7 (B) cells after pretreatment for 48 h with 50 μM each indole-3-carbinol, oltipraz, flavone, or β-NF as determined by high-performance liquid chromatography from cell culture supernatants. Data are expressed as mean ± S.D. from three individual cultures. *, p < 0.05, ***, p < 0.001 compared with cultures treated with the delivering solvent (0.1% dimethylsulfoxide) only (one-way analysis of variance, Tukey-Kramer post-test).
The total level of B[a]P metabolites detected in nonpretreated TC7 cells was nearly 2-fold higher than in nonpretreated Caco-2 cells. Moreover, the metabolite profile differed between both cell lines. TC7 cells formed nearly twice as much B[a]P-3-sulfate as B[a]P-1-sulfate, and in addition they formed 3-OH-B[a]P-glucuronide, a metabolite not seen in parental Caco-2 cells under any conditions (pretreatments) used in the present study. Pretreatment of TC7 cells with indole-3-carbinol, oltipraz, and β-NF (but not with flavone) increased the rate of formation of the various metabolites (Fig. 4B). Most of these increases were statistically significant. The increases in the levels of sulfated metabolites in TC7 cells (1.3–2.4-fold) were similar to the increases observed in Caco-2 cells. However, stronger induction factors were found for the formation of 3-OH-B[a]P-glucuronide, in particular in cells treated with β-NF (increase by a factor of 7). This differential induction led to an increase in the ratio of 3-OH-B[a]P-glucuronide and B[a]P-3-sulfate from 0.12 in control cultures to 0.57 in β-NF-pretreated cultures.
Effects of PAH on SULT mRNA Levels in Caco-2 Cells. Various PAH induce P450 and phase 2 enzymes involved in the biotransformation of planar molecules. We determined levels of SULT1A1, SULT1A3, and SULT1B1 using semiquantitative reverse transcription-PCR in differentiated Caco-2 cells (18 days after confluence) after a 24-h treatment with a series of PAH [B[a]P (10 μM), pyrene (10, 25 μM), phenanthrene (10, 20 μM), chrysene (10, 20 μM), benzo[k]fluoranthene (10, 20 μM), and dibenzo[a,l]pyrene (10, 20 μM)]. None of the treatments affected the SULT mRNA levels (data not shown).
Discussion
We recently studied the expression of SULT in the human gastrointestinal tract (Teubner et al., 2007). We showed that it is a rich source of various SULT forms, the enzymes are primarily expressed in differentiated cells of the mucosa, and the patterns of SULT forms varied between different sections along the gastrointestinal tract. Several SULT forms showed their highest expression in the ileum (see also Table 1). However, the presence of SULT1A2 and SULT1C1 proteins could be clearly shown in cecum and stomach, respectively, whereas corresponding signals in immunoblots with ileal samples were absent or faint, near the limit of detection.
Caco-2 and TC7 cells are widely used as cell culture models for intestinal functions. For this reason we have explored the expression of SULT proteins in these cells. The presence of seven SULT forms could be unambiguously shown in differentiated Caco-2 cells (SULT1A1, SULT1A2, SULT1A3, SULT1B1, SULT1C1, SULT1C2, and SULT2A1). A further form, SULT1E1, was selectively detected in differentiated TC7 cells. This diversity is larger than found in any other cell lines or human tissues that have been investigated for SULT expression on the protein level.
SULT1A3 was the most abundant form in Caco-2 and TC7 cells (Table 1). Its level in TC7 cells exceeded that in ileal mucosa, the tissue with the highest SULT1A3 expression studied to date. SULT1A1 protein levels in colon and ileum are nearly as high as those of SULT1A3. In some subjects they even surmounted the SULT1A3 levels (Teubner et al., 2007). On the contrary, levels of SULT1A1 only amounted to 6 and 3% of those of SULT1A3 in Caco-2 and TC7 cells, respectively (Table 1). This low level of SULT1A1 facilitated the unambiguous demonstration of the presence of SULT1A2 in the cell lines. Normally, it is difficult to study expression of the SULT1A2 protein in human tissues because this form is 96% identical to SULT1A1. Thus, differences in electrophoretic mobility are small (Fig. 1A), and most antisera are cross-reactive for these forms. Nowell et al. (2005) recently raised an antiserum against a characteristic peptide fragment of SULT1A2 protein; this antiserum was specific for SULT1A2 as compared with SULT1A1 and SULT1A3. Nevertheless, the authors failed to detect SULT1A2 in any human tissue samples studied, including large numbers of samples from liver, colon, and small intestine. Possibly this anti-SULT1A2 peptide antibody was not sufficiently sensitive. However, using conventional antisera, we succeeded to unambiguously show the presence of SULT1A2 protein in liver and cecum samples; marginal signals were also seen in individual samples from kidney, colon, and ileum (Meinl et al., 2006; Teubner et al., 2007). In the latter tissues, the high level of SULT1A1 disturbed the analysis of SULT1A2. The detection limit for SULT1A2 in ileum is above the levels that could be shown readily in Caco-2 and TC7 cells (Fig. 2A; Table 1).
Gut is a major site of expression of SULT1B1 (Wang et al., 1998; Teubner et al., 2007). This protein was also detected in Caco-2 and TC7 cells. Levels were much lower in the parental Caco-2 line than in the TC7 subline. Levels in this subline amounted to 64 and 18% of those found in colon and ileum, respectively (Table 1).
Antisera recognizing SULT1C1 (Stanley et al., 2005) and SULT1C2 (present study) were only raised recently. Thus, most data on their tissue distribution are based on analyses on the mRNA level. SULT1C1 mRNA was detected in kidney, stomach, thyroid gland, and fetal liver (Her et al., 1997), as well as in ovary and some regions of the brain (Dooley et al., 2000). Stanley et al. (2005) detected SULT1C1 protein (termed SULT1C2 in that study) in various fetal tissues; the highest levels were found in fetal small intestine. To our knowledge, stomach is the only adult tissue in which SULT1C1 has been unambiguously detected to date (Teubner et al., 2007); low levels, at the limit of detection, may also be present in small and large bowel (Teubner et al., 2007). Here we showed expression of SULT1C1 in Caco-2 and TC7 cells. The highest levels of SULT1C2 mRNA were found in fetal lung and kidney; lower levels were detected in fetal heart and in adult kidney, ovary, and spinal cord (Sakakibara et al., 1998). The present study is the first showing natural expression of SULT1C2 protein. The protein was found in parental Caco-2 and TC7 cells. Data on the expression in human gut are not yet available.
SULT1E1 plays a key role in the regulation of estrogen hormones (Falany, 1997; Glatt, 2002). It is expressed in various estrogen-dependent tissues, such as endometrium and mammary gland, as well as in small intestine, where it may prevent exposure of the organism to active estrogen hormones from food. SULT1E1 was detected in TC7 cells at levels similar to those found in ileum but was not detectable in parental Caco-2 cells and colon mucosa (Table 1). SULT2A1 is another enzyme involved in the regulation of steroid hormones; unlike SULT1E1, it primarily acts on alcoholic hydroxyl groups. It was highly expressed in ileum, Caco-2 cells, and TC7 cells but was not detected in colon (Table 1).
Tamura et al. (2001) studied the expression of various SULT forms in Caco-2 cells on the mRNA level and for selected forms also on the activity level. Their mRNA data agree, semiquantitatively, with our protein data for SULT1A1, SULT1A3, SULT1C1, SULT1C2, SULT2A1, and SULT2B1a/b (Table 1). However, there were discrepancies with respect to SULT1E1 and SULT1B1. Tamura et al. (2001) detected SULT1E1 mRNA but no estrogen sulfation and concluded that the protein might be instable or degraded. Indeed, we did not detect the protein in parental Caco-2 cells, but we found moderate levels in TC7 cells. The discrepancy with the other enzyme, SULT1B1, is more difficult to explain. We found the protein, but Tamura et al. (2001) missed the mRNA in Caco-2 cells, although they detected it in other cell lines tested concurrently.
We detected a strong increase in SULT protein levels when cells were kept in confluent stage for an extended period. We have not studied whether these increases are associated with increased levels of mRNA.
The parental Caco-2 cell line and the TC7 subline differ in phase 2 metabolism (Münzel et al., 1999; Sabolovic et al., 2000); often the ratio of glucuronidated to sulfo-conjugated metabolites is enhanced in TC7 cells. This difference was confirmed—with an extreme result—in the present study. A glucuronidated metabolite of B[a]P, 3-OH-B[a]P-glucuronide, was just detected in TC7 cells but not in Caco-2 cells, in which only the corresponding sulfate, B[a]P-3-sulfate, was found. In principle, this shift in the metabolite pattern in TC7 cells could be caused by increased activity of UGT or decreased activity of SULT. The latter was not the case, as the expression of all the SULT forms studied was equal or higher in TC7 cells compared with parental Caco-2 cells (Table 1). Pretreatment of Caco-2 and TC7 cells with various aromatic agents enhanced the biotransformation of B[a]P to sulfated metabolites (in both cell lines) and glucuronidated metabolites (only detected in TC7 cells). However, mRNA levels of major phenol SULT were unchanged, suggesting that the enhanced formation of sulfates was only caused by induction of P450 (shown previously in Caco-2 cells) (Ebert et al., 2005). The situation may be different for UGT, at least in β-NF-treated TC7 cells. This treatment enhanced the biotransformation of B[a]P to its 3-hydroxy-glucuronide much more than the formation of its 3-sulfate (7-versus 1.3-fold). Indeed, in a previous study we observed an induction of UGT mRNA in Caco-2 cells by various PAH, which are agonists of the aryl hydrocarbon receptor, as is β-NF (Lampen et al., 2004).
In summary, we have shown that Caco-2 and TC7 cells are rich in various SULT forms, as is intestinal mucosa. Both cell lines express SULT2A1; TC7 cells also express SULT1E1 protein, enzymes that are present in ileal but not colonic mucosa. Thus, the expression profile of the cell lines is more similar to that in ileum than in colon, although the cell lines are derived from a colon tumor. This supports the current use of well differentiated Caco-2 and TC7 cells as a model for the small intestine. However, the expression patterns in the cell lines do not exactly match those found in ileum: levels of SULT1A1 and SULT1B1 are lower, and those of SULT1C1 are higher in the cell lines than in ileal mucosa. The expression of SULT in the cell lines was strongly dependent on the differentiation state. High expression was only seen after the cells had been kept in a confluent differentiated state for an extended period. Similarly, expression of SULT in the gut in vivo is strongly focused to the differentiated enterocytes (Teubner et al., 2007).
Acknowledgments
We thank Sabine Braune and Birgit Kuehlein for excellent technical assistance.
Footnotes
-
This work was supported by Bundesministerium für Bildung und Forschung (Grant BIO/0313053A). In addition, the study was supported by the Deutsche Forschungsgemeinschaft (La1177/4–2) and the Federal Institute for Risk Assessment (BfR).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.018036.
-
ABBREVIATIONS: P450, cytochrome(s) P450; UGT, UDP-glucuronosyltransferase(s); PAH, polycyclic aromatic hydrocarbon(s); B[a]P, benzo-[a]pyrene; SULT, soluble sulfotransferase(s); β-NF, β-naphthoflavone; PCR, polymerase chain reaction(s); nt, nucleotide(s); 3-OH-B[a]P, 3-hydroxybenzo[a]pyrene.
- Received August 24, 2007.
- Accepted October 25, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}