


Abstract
Substrates that are specific for certain UDP-glucuronosyltransferase (UGT) isoforms are usually used as specific inhibitors to identify UGT isoforms responsible for the glucuronidation of drugs. 1-Naphthol and 4-nitrophenol are probe substrates for human UGT1A6. In the present study, we found that UGT1A1-catalyzed estradiol 3-O-glucuronide formation and UGT1A4-catalyzed imipramine N-glucuronide formation in human liver microsomes were prominently decreased in the presence of 1-naphthol, but those by recombinant human UGT1A1 and UGT1A4, respectively, were not. Interestingly, when recombinant UGT1A6 was added in the reaction mixture, these activities by recombinant UGT1A1 and UGT1A4 were diminished in the presence of 1-naphthol. To interpret this phenomenon, the inhibitory effects of 1-naphthol O-glucuronide and UDP, products of the glucuronidation of 1-naphthol, were investigated. We found that UDP strongly inhibited the UGT1A1 (Ki = 7 μM) and UGT1A4 (Ki = 47 μM) activities in a competitive manner for the 5′-diphosphoglucuronic acid binding. These results suggest that UDP produced by UGT1A6-catalyzed 1-naphthol glucuronidation, but not 1-naphthol O-glucuronide and 1-naphthol per se, is the actual inhibition substance. Next, we examined the inhibitory effects of 15 compounds that are substrates of UGTs on estradiol 3-O-glucuronide formation in human liver microsomes compared with those by recombinant UGT1A1. Among them, 4 compounds (1-naphthol, 2-naphthol, 4-nitrophenol, and 4-methylumbelliferone) with high turnover rates (Vmax/Km value >200 μl/min/mg) showed more potent inhibition of the activity in human liver microsomes compared with that by the recombinant UGT1A1. Thus, we should pay attention to the inhibitory effects of UDP on UGT, which may cause erroneous evaluations in inhibition studies using human liver microsomes.
UDP-glucuronosyltransferases (UGTs) are a superfamily of enzymes that catalyze the formation of glucuronides by the transfer of glucuronic acid from a cofactor uridine 5′-diphosphoglucuronic acid (UDPGA) to hydroxyl, carboxyl, or amine groups of endogenous and exogenous substrates (Dutton, 1980). The hydrophilic glucuronides can readily be excreted from the body via bile and urine. Human UGTs are classified into two subfamilies, UGT1 and UGT2, based on similarities between their amino acid sequences and gene organization (Mackenzie et al., 2005). To date, 19 human UGT isoforms, UGT1A1, -1A3, -1A4, -1A5, -1A6, -1A7, -1A8, -1A9, -1A10, -2A1, -2A2, -2A3, -2B4, -2B7, -2B10, -2B11, -2B15, -2B17, and -2B28, have been identified (Mackenzie et al., 2005). Most UGT enzymes are expressed in liver, but some isoforms such as UGT1A7, -1A8, and -1A10 are exclusively expressed in intestine (Strassburg et al., 2000; Burchell et al., 2001).
Most pharmacokinetic drug-drug interactions occur at the metabolic level and usually involve changes in the activity of the major drug-metabolizing enzymes such as UGT. Identification of the UGT(s) involved in the metabolism of a given compound allows us to predict potential drug-drug interactions. Recombinant enzymes are useful tools to determine the isoform(s) in a qualitative manner. If multiple isoforms can catalyze the metabolism, inhibition studies using antibodies or chemicals for the activities in human liver microsomes can be employed to estimate the contribution of each isoform. Specific antibodies for individual human UGT isoforms are not available because of their high amino acid similarities. Alternatively, specific substrates for each UGT isoform can be used as inhibitors. The increasing availability of substrate and inhibitor “probes” for the individual UGTs could enable reliable identification of UGT responsible for glucuronidation in human liver microsomes.
For typical substrates for human UGT1A6, 4-nitrophenol (Hanioka et al., 2001a), and 1-naphthol (Uchaipichat et al., 2004), interesting phenomena have been reported when these substrates were used as inhibitors. 4-Nitrophenol inhibited UGT2B7-catalyzed 3′-azido-3′-deoxythymidine glucuronidation (Rajaonarison et al., 1991) and UGT1A4-catalyzed amitriptyline glucuronidation (Dahl-Puustinen and Bertilsson, 1987) in human liver microsomes. 1-Naphthol inhibited UGT2B7-catalyzed morphine glucuronidation (Bock et al., 1978) and UGT1A1-catalyzed SN-38 glucuronidation (Hanioka et al., 2001b) in human liver microsomes. It should be noted that the inhibition of SN-38 glucuronidation by 1-naphthol was not observed when recombinant UGT1A1 was used as an enzyme source (Hanioka et al., 2001b). The purpose of this study was to clarify the cause of the discrepancies between the inhibitory effects of such compounds on glucuronidation in human liver microsomes and those by recombinant UGTs. As for the mechanism, we found that UDP, which was produced by the glucuronidation of 1-naphthol added as an inhibitor, inhibited human UGT enzymes, obfuscating the inhibitory effects of 1-naphthol on glucuronidation. Next, we expanded the inhibition study using 15 compounds that are typical substrates of UGT to understand what kinds of substrates could have such effects in inhibition studies for glucuronidation.

Inhibitory effects of 1-naphthol on estradiol 3-O(A), imipramine N(B), serotonin O(C), or propofol O(D) glucuronide formations in human liver microsomes (HLMs) and recombinant UGTs. Each point represents the mean of duplicate determinations. Control activities for estradiol 3-O-glucuronide formation in pooled HLMs and by recombinant UGT1A1 were 193 and 371 pmol/min/mg, respectively. Control activities for imipramine N-glucuronide formation in HLMs and by recombinant UGT1A4 were 67.2 and 25.2 pmol/min/mg, respectively. Control activities for serotonin O-glucuronide formation in HLMs and by recombinant UGT1A6 were 240 and 425 pmol/min/mg, respectively. Control activities for propofol O-glucuronide formation in HLMs and by recombinant UGT1A9 were 1.7 and 5.5 nmol/min/mg, respectively.
Materials and Methods
Chemicals and Enzyme Sources. UDPGA, UDP, alamethicin, estradiol, estradiol 3-O-glucuronide, 1-naphthol O-glucuronide, and 4-methylumbelliferone were purchased from Sigma-Aldrich (St. Louis, MO). Acetaminophen, ethynylestradiol, furosemide, ibuprofen, imipramine, ketoprofen, mycophenolic acid, 1-naphthol, 2-naphthol, 4-nitrophenol, propofol, serotonin, tranilast, and valproic acid were purchased from Wako Pure Chemicals Industries (Osaka, Japan). Pooled human liver microsomes were obtained from BD Gentest (Woburn, MA). Recombinant human UGT1A1, UGT1A4, UGT1A6, and UGT1A9 expressed in HEK293 cells and mock-transfected cells were prepared in our previous study (Fujiwara et al., 2007a,b). All other chemicals and solvents were of analytical grade or the highest grade commercially available.
Glucuronide Formations in Human Liver Microsomes and Recombinant UGT1A. Estradiol 3-O-, imipramine N-, serotonin O-, and propofol O-glucuronide formations were determined as described previously with slightly modifications (Fujiwara et al., 2007b). Briefly, a typical incubation mixture (200 μl of total volume) contained 50 mM Tris-HCl or potassium phosphate buffer (pH 7.4), 10 mM MgCl2, 2.5 mM UDPGA, 25 μg/ml alamethicin, 0.25 mg/ml pooled human liver microsomes or recombinant UGTs, and substrates (10 μM estradiol, 200 μM imipramine, 200 μM serotonin, and 50 μM propofol). In the inhibition study, inhibitors were also included. Estradiol, propofol, and inhibitors except tranilast were dissolved in methanol, and tranilast was dissolved in dimethyl sulfoxide. The final concentration of the organic solvents in the incubation mixture was 1% (v/v). The reaction was initiated by the addition of UDPGA after a 3-min preincubation at 37°C. After incubation at 37°C for 30 min (estradiol, serotonin, and propofol glucuronidations) or 60 min (imipramine glucuronidation), the reaction was terminated by the addition of 100 μl of ice-cold acetonitrile including 6% acetic acid (estradiol glucuronidation) or ice-cold acetonitrile (imipramine, serotonin, and propofol glucuronidations). After removal of the protein by centrifugation at 13,000g for 5 min, a 20-μl portion of the sample was subjected to HPLC. To estimate the inhibition type of UDP for substrate binding, analyses were conducted using a fixed concentration (2.5 mM) of UDPGA and varied concentrations of estradiol (2.5–40 μM), imipramine (0.25–1.5 mM), serotonin (1.25–10 mM), and propofol (25–200 μM). To estimate the inhibition type of UDP for UDPGA binding, analyses were conducted using a fixed concentration of estradiol (100 μM), imipramine (1.5 mM), serotonin (10 mM), or propofol (200 μM) and varied concentrations of UDPGA (0.25, 0.5, 1, and 2.5 mM). The type of inhibition and the Ki value were determined by a nonlinear regression analysis using the computer program k · cat (BioMetallics, Princeton, NJ).
2-Naphthol O-glucuronide formation was determined to obtain the kinetic parameters, according to the method of Terrier et al. (1999). The incubation mixture described above containing 2-naphthol (0.5–100 μM) was incubated at 37°C for 5 min after a 3-min preincubation. The reaction was terminated by the addition of 100 μl of ice-cold acetonitrile. After removal of the protein by centrifugation at 13,000g for 5 min, a 20-μl portion of the sample was subjected to HPLC. HPLC was performed using an l-7100 pump (Hitachi, Tokyo, Japan), an l-7485 FL detector (Hitachi), an l-7200 autosampler (Hitachi), a d-2500 integrator (Hitachi), and a CAPCELL PAK column (4.6 × 150 mm, 5 μm; Shiseido, Tokyo, Japan). The flow rate was 1.0 ml/min, and the column temperature was 35°C. Detection was accomplished with a fluorescence detector at 296-nm excitation and 336-nm emission. The mobile phases were 20% acetonitrile including 0.1% acetic acid. The 2-naphthol O-glucuronide formation was determined by the decrease of the peak area of the 2-naphthol. Kinetic parameters were estimated from the fitted curve using a computer program (KaleidaGraph; Synergy Software, Reading, PA) designed for nonlinear regression analysis. The following equation was used: V = Vmax · [S]/(Km + [S]), where V is the velocity of the reaction at substrate concentration S, Km is the Michaelis-Menten constant, and Vmax is the maximum velocity.

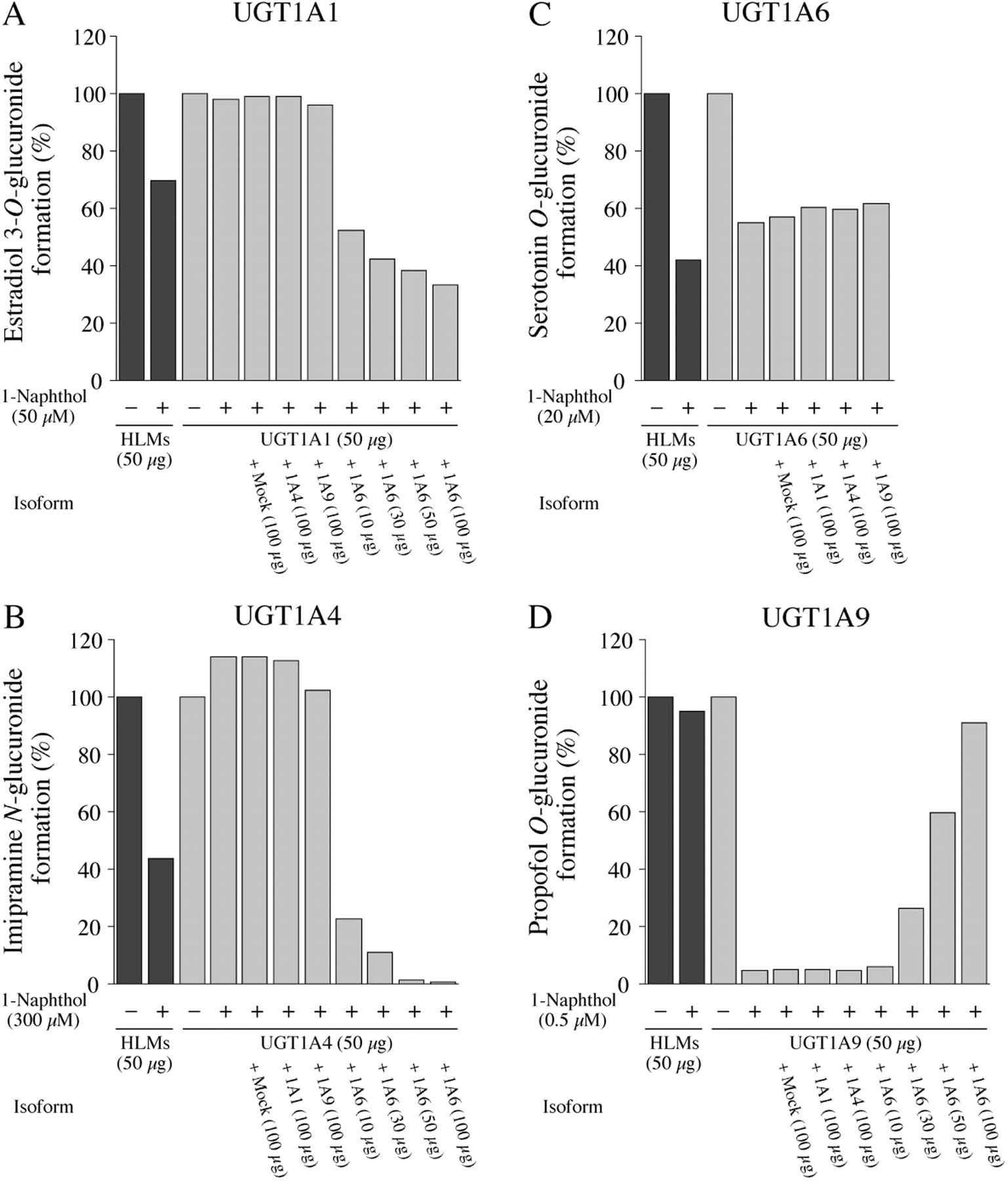
Effects of 1-naphthol on estradiol 3-O(A), imipramine N(B), serotonin O(C), or propofol O(D) glucuronide formations in HLMs and by recombinant UGTs in the presence of other UGT1A isoforms. Each column represents the mean of duplicate determinations. The concentrations of 1-naphthol for the inhibition of estradiol 3-O-, imipramine N-, serotonin O-, or propofol O-glucuronide formations were 50, 300, 20, and 0.5 μM, respectively. Control activities for estradiol 3-O-, imipramine N-, serotonin O-, and propofol O-glucuronide formations in HLMs and by recombinant UGTs are described in the legend for Fig. 1.
Statistical Analyses. Statistical analyses were performed using the two-tailed Student's t test. A value of P < 0.05 was considered statistically significant.
Results
Inhibitory Effects of 1-Naphthol on Estradiol 3-O-, ImipramineN-, SerotoninO-, and PropofolO-Glucuronide Formations in Human Liver Microsomes and Recombinant UGT1As. We previously confirmed that the estradiol 3-O-, imipramine N-, serotonin O-, and propofol O-glucuronide formations were specifically catalyzed by UGT1A1, UGT1A4, UGT1A6, and UGT1A9, respectively (Fujiwara et al., 2007b). As shown in Fig. 1, the inhibitory effects of 1-naphthol on the estradiol 3-O-glucuronide formation in human liver microsomes (IC50 was 141 μM) were more potent than those by recombinant UGT1A1 (IC50 was 294 μM). 1-Naphthol moderately inhibited the imipramine N-glucuronide formation in human liver microsomes (IC50 was 229 μM) but not the activity by recombinant UGT1A4 (Fig. 1B). 1-Naphthol showed inhibition of serotonin O-glucuronide formation in human liver microsomes and recombinant UGT1A6 with similar potencies (IC50 values were 18 and 21 μM, respectively) (Fig. 1C). For propofol O-glucuronide formation, 1-naphthol showed more potent inhibition for the activity by recombinant UGT1A9 (IC50 was 0.08 μM) than that in human liver microsomes (IC50 was 7.9 μM) (Fig. 1D). Thus, it was clearly demonstrated that the inhibitory effects of 1-naphthol on the UGT1A1, UGT1A4, and UGT1A9 activities were different between human liver microsomes and recombinant UGTs.
Effects of the Presence of UGT1A6 on the Inhibitory Effects of 1-Naphthol. The substantial difference between these enzyme sources is that human liver microsomes express multiple UGT isoforms, whereas the recombinant system expresses only one targeted isoform. Because 1-naphthol is a specific substrate of UGT1A6, it was surmised that the differences between the inhibitory effects of 1-naphthol on the activities in human liver microsomes and those by recombinant UGT might be due to the presence of UGT1A6 in human liver microsomes. We investigated the inhibitory effects of 1-naphthol on the activities of each of the recombinant UGTs in the presence of recombinant UGT1A6. The estradiol 3-O-glucuonide formation catalyzed by recombinant UGT1A1 was not inhibited by 1-naphthol but was inhibited in the presence of recombinant UGT1A6 in a dose-dependent manner (Fig. 2A). In contrast, the presence of recombinant UGT1A4, UGT1A9, and mock cell lysate showed no effect (Fig. 2A). Likewise, the imipramine N-glucuronide formation by recombinant UGT1A4 was inhibited by 1-naphthol in the presence of recombinant UGT1A6, but not with recombinant UGT1A1 and UGT1A9 (Fig. 2B). The inhibitory effect of 1-naphthol on the serotonin O-glucuronide formation catalyzed by UGT1A6 was not affected by UGT1A1, UGT1A4, and UGT1A9 (Fig. 2C). In contrast, the inhibitory effects of 1-naphthol on the propofol O-glucuronide formation catalyzed by recombinant UGT1A9 were restored by the addition of recombinant UGT1A6 (Fig. 2D). These results suggest that the UGT1A6-dependent glucuronidation of 1-naphthol would affect the inhibitory effects of 1-naphthol on the UGT1A1, UGT1A4, and UGT1A9 activities in human liver microsomes.

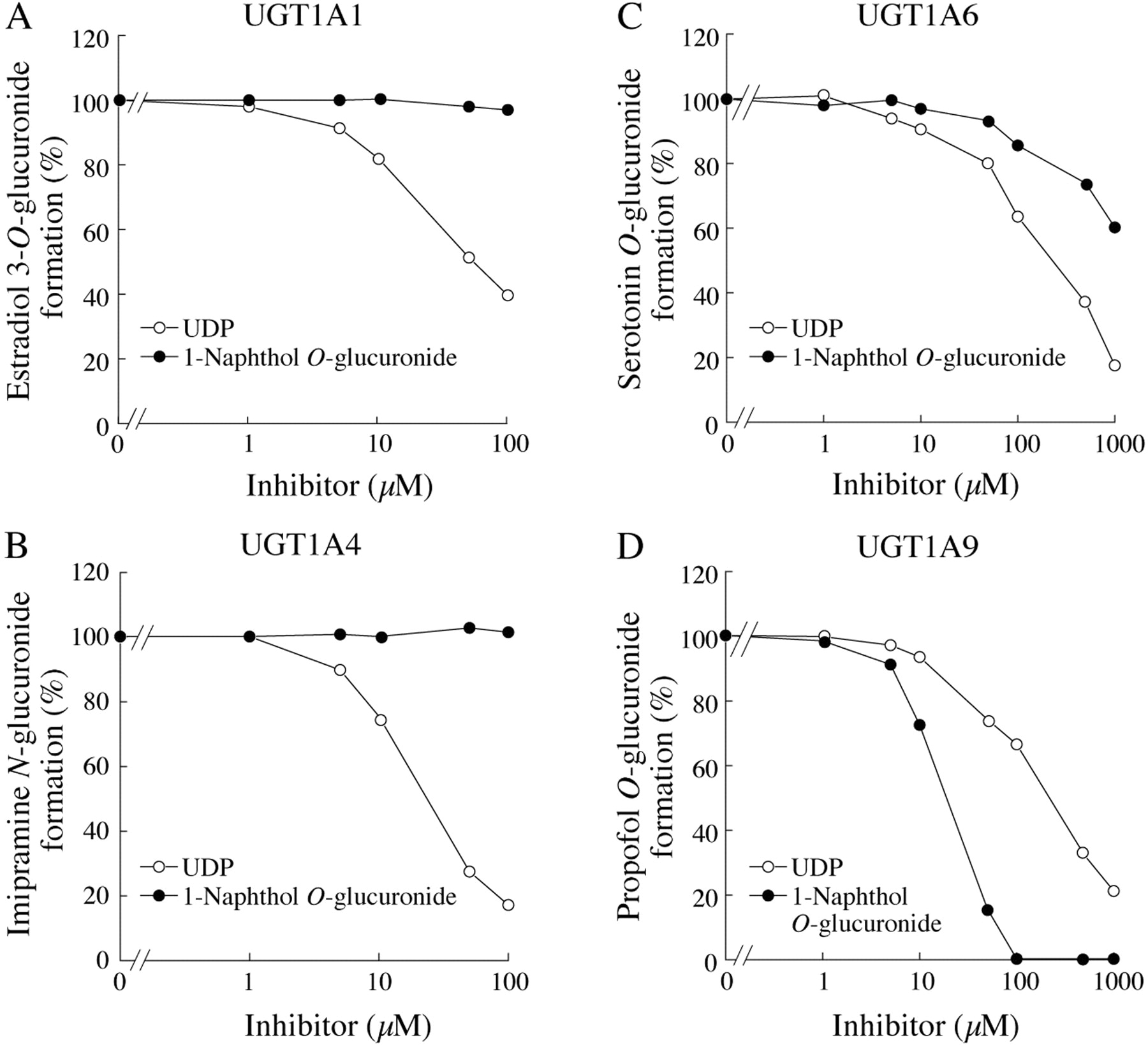
Inhibitory effects of UDP and 1-naphthol O-glucuronide on estradiol 3-O(A), imipramine N(B), serotonin O(C), or propofol O(D) glucuronide formations by recombinant UGTs. Each point represents the mean of duplicate determinations. Control activities for estradiol 3-O-, imipramine N-, serotonin O-, and propofol Oglucuronide formations in HLMs and by recombinant UGTs are described in the legend for Fig. 1.
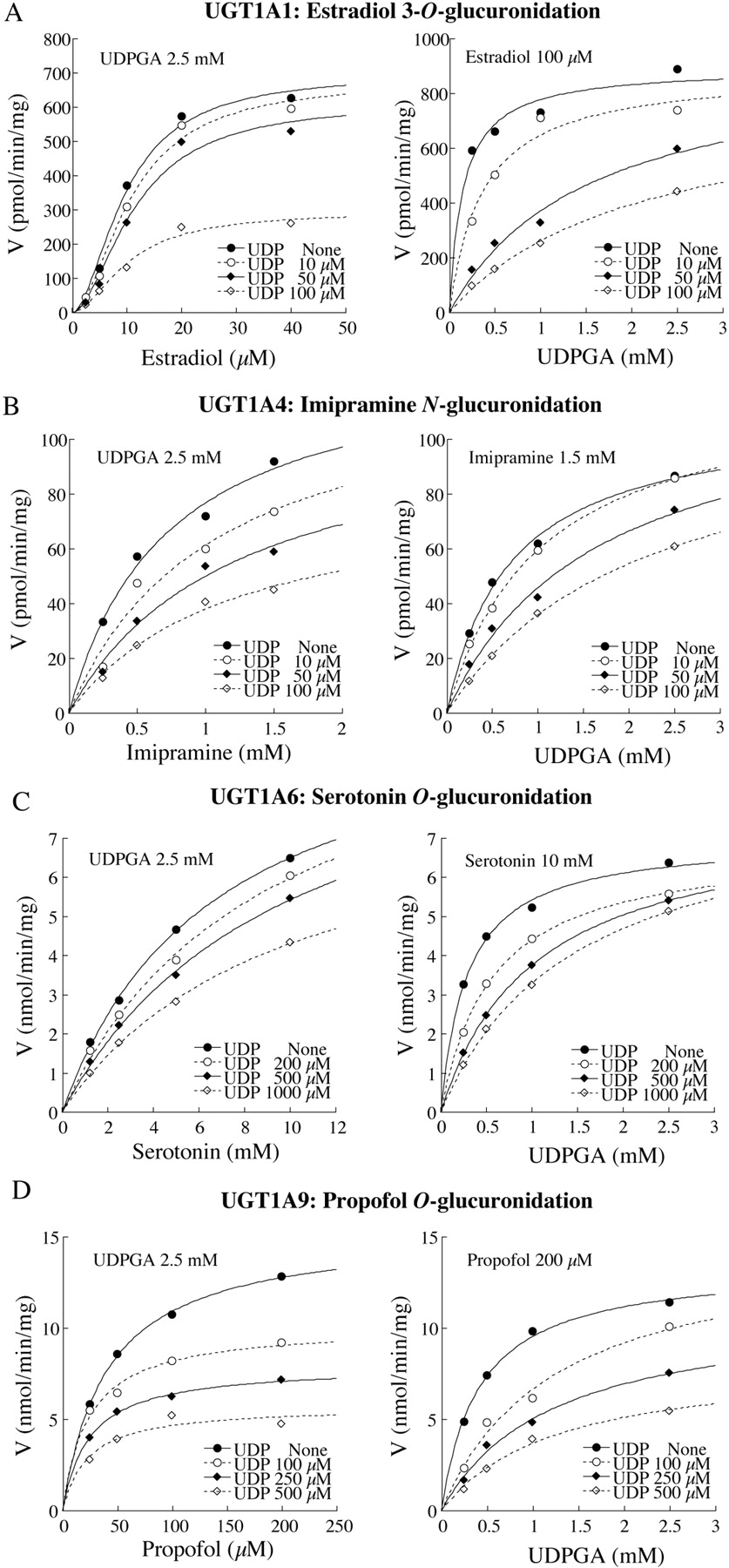
Inhibitory Effects of 1-NaphtholO-Glucuronide and UDP on Recombinant UGT1A1, UGT1A4, UGT1A6, and UGT1A9 Activities. The effects of 1-naphthol O-glucuronide and UDP, which are products of 1-naphthol glucuronidation, on the UGT activities were investigated using recombinant UGTs as the enzyme sources (Fig. 3). Although 1-napthol O-glucuronide prominently inhibited the UGT1A9 activity (IC50 = 26 μM), it did not inhibit the UGT1A1 and UGT1A4 activities. Its inhibitory effects on the UGT1A6 activity were trivial. In contrast, UDP strongly inhibited the UGT1A1 (IC50 = 56 μM) and UGT1A4 (IC50 = 31 μM) activities and moderately inhibited the UGT1A6 (IC50 = 307 μM) and UGT1A9 (IC50 = 290 μM) activities. These results suggest that UDP formed from 1-naphthol glucuronidation by UGT1A6 would cause more potent inhibition toward activities in human liver microsomes than that by recombinant UGT1A1 and UGT1A4. We performed the kinetic analyses to determine the inhibition type of UDP (Fig. 4). UDP noncompetitively inhibited the binding of estradiol, imipramine, and propofol to UGT. The inhibition type for the serotonin binding to UGT1A6 was a mixed type of competitive and noncompetitive. In contrast, UDP competitively inhibited the UDPGA binding to all UGT1A1 (Ki = 7 μM), UGT1A4 (Ki = 47 μM), UGT1A6 (Ki = 259 μM), and UGT1A9 (Ki = 143 μM).
Comparison of the Inhibitory Effects of a Variety of Compounds on Estradiol 3-O-Glucuronide Formation in Human Liver Microsomes and Recombinant UGT1A1. The inhibitory effects of the 15 UGT substrates on the estradiol 3-O-glucuronide formation in human liver microsomes were compared with those by recombinant UGT1A1 (Table 1). The 15 compounds were classified into three groups, A, B, and C, as follows. Group A included 8 compounds of which the inhibitory effects on the activities of both human liver microsomes and recombinant UGT1A1 were weak (residual activities were >75%). Because these compounds are not substrates of UGT1A1, weak inhibitory effects were feasible. Group B included 3 compounds of which the inhibitory effects on the activities of both human liver microsomes and recombinant UGT1A1 were potent (residual activities were <40%). Ethynylestradiol and tranilast are the substrates of UGT1A1 (McGinnity et al., 2004; Katoh et al., 2007). Propofol, a substrate of UGT1A9, has been reported to inhibit UGT1A1 activities (Williams et al., 2004; Kaji and Kume, 2005). Therefore, they could potently inhibit the estradiol 3-O-glucuronide formation in both human liver microsomes and recombinant UGT1A1. Group C included 4 compounds of which the inhibitory effects on the activities in human liver microsomes were more potent than those by recombinant UGT1A1. They are all substrates of UGT1A6 (Hanioka et al., 2001b). As an index of UDP formation, the intrinsic clearances (Vmax /Km values) of glucuronidation of the 15 compounds in human liver microsomes were evaluated (Table 1). When the residual activities in human liver microsomes and recombinant UGT1A1 were plotted against the intrinsic clearances, the 15 compounds could be divided into three parts concurring with the groups A, B, and C (Fig. 5). All four compounds in group C with large differences in the inhibitory effects on the activities between human liver microsomes and recombinant UGT1A1 showed high intrinsic clearances (>200 μl/min/mg). These results suggest that UDP produced by the UGT1A6-catalyzed glucuronidation of compounds caused potent inhibitory effects on the UGT1A1 activity in human liver microsomes.
Residual activities of estradiol 3-O-glucuronide formation in human liver microsomes and recombinant UGT1A1 in the presence of 15 compounds that are substrates of UGTs, the kinetic parameters of the 15 compounds in human liver microsomes, and the main isoforms involved in the glucuronidation
Residual activities of estradiol 3-O-glucuronide formation in human liver microsomes and recombinant UGT1A1 were determined in the presence of 15 kinds of compounds (100 μM). Control activities for estradiol 3-O-glucuronide formations in HLMs and by recombinant UGT1A1 are described in Fig. 1 legend. The data are the means ± S.D. of three independent determinations.

Inhibitory effects of UDP on the kinetics of estradiol 3-O(A), imipramine N(B), serotonin O(C), or propofol O(D) glucuronide formations by recombinant UGTs. UDP noncompetitively inhibited the binding of estradiol, imipramine, and propofol to UGT. The inhibition type for the serotonin binding to UGT1A6 was a mixed type of competitive and noncompetitive. UDP was a competitive inhibitor for the UDPGA binding. Each point represents the mean of duplicate determinations.
Discussion
Substantial progress has been made in identifying typical substrates for individual UGT isoforms. Although UGTs display broad and overlapping substrate specificities, some compounds are specific for certain UGT isoforms. Such substrates can be used as specific inhibitors to identify the UGT isoform(s) contributing to the glucuronidation of drugs (Court, 2005). However, previous reports have shown confusing results, namely that 4-nitrophenol and 1-naphthol, typical substrates for human UGT1A6, inhibited UGT1A1, UGT1A4, or UGT2B7-catalyzed glucuronidations in human liver microsomes but did not inhibit the activities by recombinant UGTs (Bock et al., 1978; Dahl-Puustinen and Bertilsson, 1987; Rajaonarison et al., 1991; Hanioka et al., 2001b). In the present study, we found that 1-naphthol showed more potent inhibition of the UGT1A1- and UGT1A4-catalyzing activities in human liver microsomes than that by recombinant enzymes (Fig. 1, A and B), whereas 1-naphthol more potently inhibited the UGT1A9-catalyzing activity by recombinant enzymes than that in human liver microsomes (Fig. 1D). As for the mechanism responsible for this discrepancy between human liver microsomes and recombinant enzymes, we found that the prominent inhibition of UGT1A1 and UGT1A4 activities could be attributed to the inhibitory effects of UDP produced by UGT1A6-catalyzed 1-naphthol glucuronidation in human liver microsomes. In contrast, it was considered that the UGT1A6-catalyzed 1-naphthol glucuronidation in human liver microsomes attenuated the inhibitory effects of 1-naphthol on the UGT1A9 activity, because 1-naphthol has the most potent inhibitory effects toward UGT1A9 within 1-naphthol, 1-naphthol O-glucuronide, and UDP. Thus, when human liver microsomes are used as an enzyme source for UGT inhibition studies, we should carefully interpret the results.
Luukkanen et al. (2005) have reported that UDP competitively inhibits the binding of UDPGA to human UGT1A9. We found that UDP competitively inhibited not only UGT1A9 but also UGT1A1, UGT1A4, and UGT1A6 for the UDPGA binding, although the Ki values varied. The carboxyl-terminal domain of UGT proteins is highly conserved and is responsible for the UDPGA binding (Pillot et al., 1993; Tukey and Strassburg, 2000). Therefore, it is conceivable that UDP commonly inhibits UGTs of any isoform, tissue, or species, as supported by previous reports (Gotze et al., 1971; Winsnes, 1972; Hallinan et al., 1979; Koster and Noordhoek, 1983; Yokota et al., 1998). A particular interesting finding in our study was that UDP produced by glucuronidation of the substrate (or inhibitor) of certain UGT isoforms exerts inhibitory effects on other UGT isoforms, raising the question of what kinds of UGT substrates can cause inhibition by UDP. To answer this question, we compared the inhibitory effects of 15 kinds of typical substrates on the activities between human liver microsomes and recombinant enzymes by calculating the Vmax/Km values of the glucuronidations of the substrates (Fig. 5; Table 1). We found that the substrates with high Vmax/Km values (>200 μl/min/mg) showed prominent inhibitory effects toward the glucuronidation in human liver microsomes compared with those when using recombinant enzymes. This would be important information for the selection of appropriate compounds for inhibition studies using human liver microsomes.
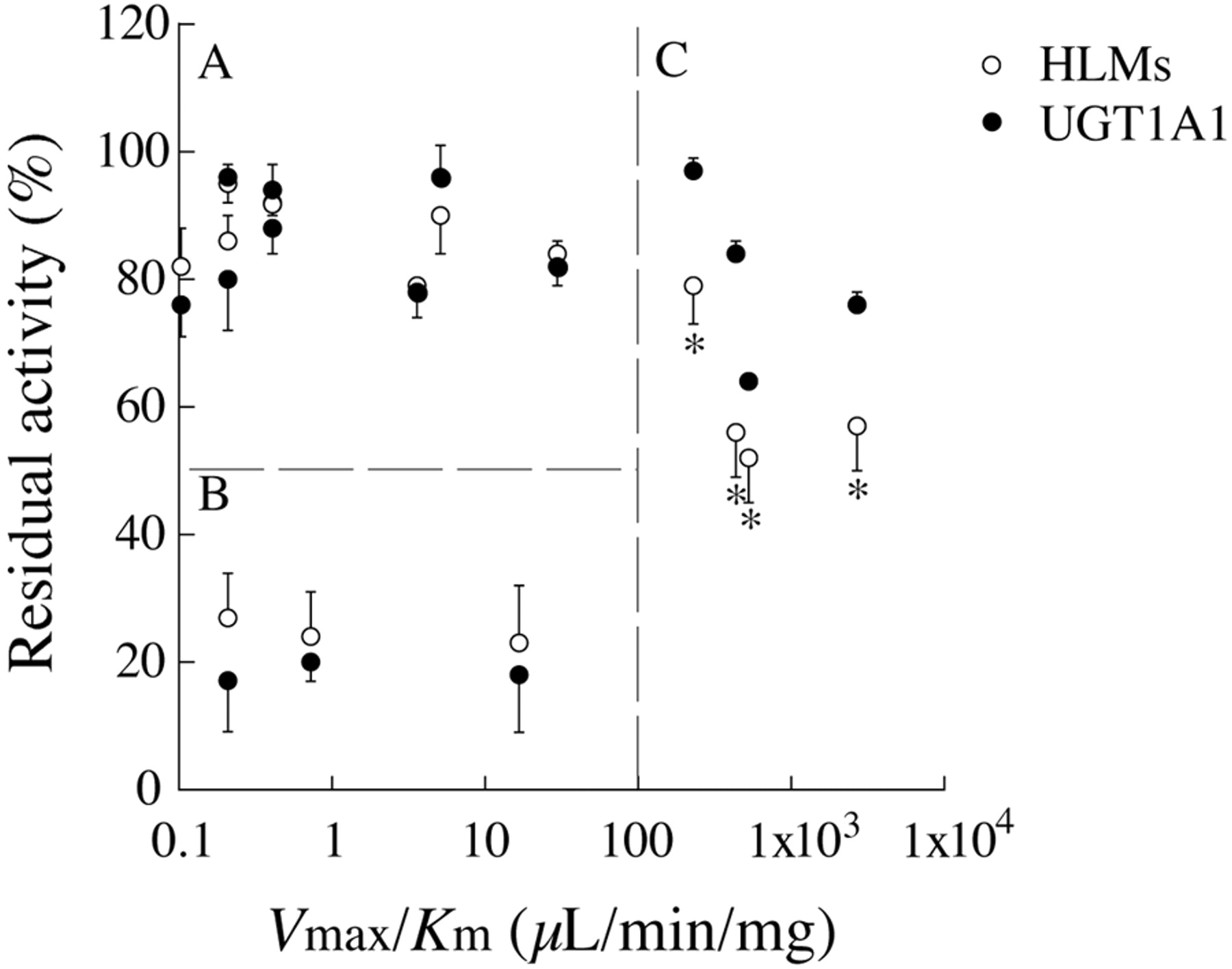
Plot of the residual activities of UGT1A1 and the Vmax /Km values of glucuronidations of the 15 inhibitors. Residual activities of estradiol 3-O-glucuronide formations in human liver microsomes and by recombinant UGT1A1 are shown in Table 1. The Vmax/Km values of the glucuronidations in HLMs of the 15 inhibitors are also shown in Table 1. The data are the means ± S.D. of three independent determinations. *, P < 0.05 compared with that by recombinant UGT1A1.
The active site of UGT is in the luminal side of the endoplasmic reticulum. Therefore, UDP, a by-product of glucuronidation, is generated at the inside of the endoplasmic reticulum lumen. In intact cells, nucleoside diphosphatase in lumen immediately hydrolyzes UDP to inorganic phosphate and uridine monophosphate (Finch et al., 1979), which is exported to the cytoplasm by a nucleotide sugar transporter (Hirschberg et al., 1998). In contrast, UGT in vitro can freely access UDP because the nucleoside diphosphatase in the microsomal membrane, of which activity is calcium-dependent (Wang and Guidotti, 1998), cannot hydrolyze UDP in the usual calcium-free incubation mixture. Therefore, preferential inhibition by UDP might be feasible in microsomal preparations compared with intact cells. This may at least partly explain why the extrapolation from in vitro clearance using human liver microsomes, but not hepatocytes, to in vivo clearance usually results in underestimation (Lin and Wong, 2002; Soars et al., 2002; Miners et al., 2006).
In conclusion, we demonstrated that UGT substrates with high turnover rates might confuse the identification of the UGT isoform responsible for the glucuronidation of drugs, when such compounds were used as inhibitors for the activities in human liver microsomes, owing to the production of UDP and/or glucuronide or reduction of the inhibitor per se. This finding is important for avoiding misinterpretations in the identification of UGT isoforms.
Acknowledgments
We acknowledge Brent Bell for reviewing the manuscript.
Footnotes
-
H.Y. was supported as a Research Fellow of the Japan Society for the Promotion of Science.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.018705.
-
ABBREVIATIONS: UGT, UDP-glucuronosyltransferase; HPLC, high performance liquid chromatography; UDPGA, UDP-glucuronic acid; SN-38, 7-ethyl-10-hydroxycamptothecin; HLM, human liver microsome.
- Received September 2, 2007.
- Accepted November 9, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}