


Abstract
Induction of drug enzyme activity in the intestine can strongly determine plasma levels of drugs. It is therefore important to predict drug-drug interactions in human intestine in vitro. We evaluated the applicability of human intestinal precision-cut slices for induction studies in vitro. Morphological examination and intracellular ATP levels indicated tissue integrity up to 24 h of incubation, whereas in proximal jejunum slices, the metabolic rate toward most substrates remained at 40 to 50% of initial values. In colon slices, the cytochrome P450 conversions were below the detection limit, but conjugation rates remained relatively constant during incubation. The inducibility of drug-metabolizing enzymes and P-glycoprotein was evaluated using prototypical inducers for five induction pathways. β-Naphthoflavone (aryl hydrocarbon receptor ligand) induced CYP1A1 (132-fold in colon and 362-fold in proximal jejunum) and UDP glucuronosyltransferase (UGT) 1A6 mRNA (9.8-fold in colon and 3.2-fold in proximal jejunum). In proximal jejunum, rifampicin (RIF) [pregnane X receptor (PXR) ligand] induced CYP3A4 (5.2-fold), CYP2B6 (2-fold), UGT1A6 (2.2-fold), and multidrug resistance-1 (MDR1)/ABCB1 mRNA (2.7-fold), whereas 6β-hydroxytestosterone formation (CYP3A4) increased 2-fold. In colon, RIF induced UGT1A6 32-fold and MDR1 2.2-fold. Dexamethasone (glucocorticoid receptor and PXR ligand) induced CYP3A4 mRNA (3.5-fold) and activity (5-fold) in proximal jejunum. Phenobarbital (constitutive androstane receptor activator) induced CYP3A4 (4.1-fold, only in jejunum), CYP2B6 (4.9-fold in colon and 2.3-fold in proximal jejunum), and MDR1/ABCB1 mRNA and CYP3A4 activity (2-fold only proximal jejunum). Quercetin (nuclear factor-E2-related factor 2 activator) induced UGT1A6 mRNA (6.7-fold in colon and 2.2-fold in proximal jejunum). In conclusion, this study shows that human intestinal precision-cut slices are useful to study induction of drug-metabolizing enzymes and transporters in the human intestine.
The modulation of the expression and activity of drug-metabolizing enzymes (DMEs) and drug transporters (DTs) by inducers is a major concern in the development of new drugs, because it potentially leads to changes in the bioavailability of drugs and may disturb the balance between toxification and detoxification (Lin and Lu, 1998). Like the liver, the intestine expresses a broad spectrum of DMEs (Pacifici et al., 1988; Kaminsky and Zhang, 2003) and DTs (Kunta and Sinko, 2004; Seithel et al., 2006). The latter are commonly referred to as phase III in drug metabolism. DMEs and DTs were shown to play a cooperative role in detoxifying and excreting xenobiotics (Benet and Cummins, 2001; Jeong et al., 2005).
Similarly to human liver (Madan et al., 2003; Vermeir et al., 2005), intestinal DMEs and DTs have been reported to be sensitive to induction (Cummins et al., 2001; Glaeser et al., 2004; Lampen et al., 2004). In vivo studies showed up-regulation of CYP3A4 mRNA and activity in human enterocytes after oral administration of rifampicin (Glaeser et al., 2004) and St. John's wort (Tannergren et al., 2004). However, unlike in the liver, the inducibility of DMEs and DTs in the intestine mediated via various nuclear receptor signaling pathways has not been investigated in detail.
To study the effect of inducers on DMEs and DTs, intact cell systems are required. Because the expression and characteristics of nuclear receptor ligands vary among species (Qatanani and Moore, 2005), it is crucial to test the induction profile of new compounds intended for human use on intact human tissue during drug development. The human-derived Caco-2 and LS180 cell lines (Hartley et al., 2006) have been proven to be useful for induction studies for some compounds (Galijatovic et al., 2000; Le Ferrec et al., 2002; Aiba et al., 2005). However, it is commonly known that these cell lines do not express all DMEs and DTs present in small intestine and colon (Prueksaritanont et al., 1996; Sun et al., 2002). In addition, Caco-2 cells are deficient in PXR, making them less suitable to study PXR-mediated induction (Hartley et al., 2006).
Recently, we validated the precision-cut slice technique using human intestinal tissue for metabolism studies and showed constant metabolic activity up to 4 h of incubation (van de Kerkhof et al., 2006); furthermore, this system was validated for studying drug-induced induction in rat small intestine and colon (van de Kerkhof et al., 2007). In the present study, we investigated the applicability of human intestinal precision-cut slices to study induction of DMEs (phase I and II) and DTs up to 24 h of incubation. Theodoropoulos et al. showed the inducibility of CYP3A4 in human fetal intestinal biopsies after incubation with calcitriol (Theodoropoulos et al., 2003), but to our knowledge, the present study is the first to test and prove the applicability of human, intact, nonfetal, intestinal tissue for induction studies. We first tested both the viability of the preparations and the stability of the metabolic rates in colon and proximal jejunum slices during 24-h incubations by measuring intracellular ATP levels, examining morphology, and determining stability of the metabolic rate using several substrates: testosterone (TT), substrate for CYP3A4, CYP2B6, and 17β-HSD (Farthing et al., 1981; Yamazaki and Shimada, 1997); 7-ethoxycoumarin (7EC), substrate for CYP1A1, CYP1A2, CYP2B6, and CYP2E1 (Shimada et al., 1999); 7-hydroxycoumarin (7HC), substrate for UGT1A6 and sulfotransferase; and midazolam, coumarin, diclofenac, and bufuralol, substrates for CYP3A4/5, CYP2A6, CYP2C9, and CYP2D6, respectively (Bjornsson et al., 2003).
Second, slices were incubated with several prototypical inducers that based on literature data (in vivo and/or in vitro) are reported to induce certain DMEs and DTs via the most relevant nuclear receptor pathways: β-naphthoflavone (BNF) via AhR (Lin and Lu, 1998); rifampicin (RIF) via PXR (Wang and LeCluyse, 2003); dexamethasone (DEX) via GR and PXR (Pascussi et al., 2001); phenobarbital (PB) via CAR (Wang and LeCluyse, 2003); and quercetin (Q), known to induce phase II metabolic reactions via Nrf2 (Bock et al., 2000). After exposure of intestinal slices to the model inducers, induction was tested by determining the relative mRNA expression of several genes involved in phase I (CYP1A1, CYP2B6, CYP3A4, and 17β-HSD) and phase II metabolism (UGT1A6) and phase III transport (MDR1/ABCB1). In addition, for a number of metabolic pathways the enzyme activity was determined after exposure to the inducing agents.
Materials and Methods
Chemicals. Dimethyl sulfoxide (DMSO), trifluoroacetic acid, low gelling temperature agarose (type VII-A), amphotericin B solution (250 μg/ml), d-glucose, 7HC, 7-hydroxycoumarin-glucuronide (7HC-GLUC), HEPES, testosterone, 11β-hydroxytestosterone (11β-TOH), 6β-TOH, RIF, DEX, BNF, Q, PB, and bovine serum albumin were purchased from Sigma-Aldrich (St. Louis, MO). Williams Medium E with Glutamax-I and gentamicin (50 mg/ml) solution were obtained from Invitrogen (Paisley, UK). Formaldehyde solution (3.8%) was purchased from Mallinckrodt Baker B.V. (Deventer, The Netherlands). Hydroxybufuralol maleate salt, bufuralol hydrochloride salt, 1-hydroxymidazolam, and 7-hydroxycoumarin-sulfate were purchased from Ultrafine (Manchester, UK). 4-Hydroxydiclofenac was obtained from BD Gentest (Woburn, MA). Midazolam was purchased from Lipomed AG (Arlesheim, Switzerland). Sodium azide was obtained from Kebo Laboratory (Spånga, Sweden). Sodium chloride EDTA, formic acid, Tris-HCl, dichloromethane, and sodium acetate were obtained from Merck (Darmstadt, Germany). Acetonitril (ACN) and methanol (MeOH) were purchased from Rathburn (Walkerburn, Scotland). Ethanol was obtained from Kemetyl (Haninge, Sweden). 2α-, 16α-, and 11α-TOH were obtained from Steraloids (Newport, RI).
Human Tissue. Human tissue originated from surgical resections, with approval from the regional ethical committee in Gothenburg (Sweden) and with consent from each of the individual patients; proximal jejunum was obtained from obese patients from Sahlgren's University Hospital (Gothenburg, Sweden), whereas colon samples were from patients with colon carcinomas from East Hospital (Gothenburg, Sweden). Donor characteristics are listed in Table 1. Pathological alterations were absent in the tissues used, as confirmed by a pathological examination of the tissue sample.
Donor characteristics
After surgical removal, tissue was directly stored in ice-cold constantly oxygenated (with carbogen, 95% O2 and 5% CO2) Krebs-bicarbonate-Ringer solution (pH 7.4) with the composition as reported earlier (Polentarutti et al., 1999), Generally, approximately 15 min elapsed between surgical removal of the tissue and arrival at the laboratory.
Preparation and Incubation of Human Tissue. Directly after the tissue arrived in the laboratory, the muscle layers and serosa were carefully removed. To remove the muscle and serosa, the intestinal tissue was pinned (with the mucosa downwards) on a fiber mat, whereafter the muscle and serosa were cut off with sharp scissors. The different layers are clearly visible by the naked eye. In all cases, preparation of mucosal tissue was finished within 1 to 2 h after excision.
Preparation of Precision-Cut Slices. Preparation of slices was described in an earlier study (van de Kerkhof et al., 2006). In brief, mucosal tissue was transferred to ice-cold oxygenated Krebs-Henseleit buffer (containing 10 mM HEPES and 25 mM d-glucose, pH 7.4). Tissue was cut in pieces of 7 to 9 mm width and variable length and were subsequently embedded in 37°C, 3% low-gelling temperature agarose in 0.9% NaCl solution, using a precooled (0°C) tissue embedding unit (Alabama Research and Development, Munford, AL), allowing the agarose solution to gel. After the agarose solution had gelled, precision-cut slices (thickness approximately 400 μm with wet weight of approximately 2–4 mg without agarose) were cut using a Krumdieck tissue slicer as described previously (de Kanter et al., 2005).
Incubation of Precision-Cut Slices. Slices were incubated individually in 12-well culture plates (Costar 3512; Corning Glassworks, Corning, NY) in 1.3 ml of Williams Medium E with Glutamax-I, supplemented with d-glucose (final concentration 25 mM), gentamicin (final concentration 50 μg/ml), and amphotericin B (final concentration 2.5 μg/ml). The culture plates were placed in a prewarmed cabinet (37°C) in plastic boxes. Slices were incubated under humidified carbogen and shaken using an orbital shaker at approximately 45 rpm.
Viability Testing of Precision-Cut Slices. Morphology. The condition of precision-cut slices was evaluated after 24 h of incubation by examining their morphology microscopically. After incubation, slices (three per experiment) were stored in 3.8% formaldehyde solution at 4°C. In addition, directly after the tissue arrived, a small piece of tissue was put in ice-cold 3.8% formaldehyde as a control. Samples were sent to HistoCenter AB (Frölunda, Sweden) for further processing and hematoxylin and eosin staining. Colon slice incubations for morphology were performed in two experiments with tissue from two subjects in triplicate; proximal jejunum slice incubations were performed in five experiments with tissue from five subjects in triplicate.
Intracellular ATP levels. Intracellular ATP levels were evaluated in proximal jejunum slices to judge the overall metabolic condition of the tissue after 4 and 24 h of incubation. Directly after tissue preparation, three pieces of tissue were snap-frozen as controls. Intracellular ATP levels were determined as described earlier (van de Kerkhof et al., 2006). ATP was determined in four experiments using four different donor tissues in triplicate.
Stability of metabolic rate in time. The stability of the metabolic rate was determined between 0 to 3 h and 24 to 27 h of incubation. To evaluate phase I metabolism, slices were incubated with TT (final concentration 250 μM, 1% MeOH), 7EC (final concentration 500 μM, 1% MeOH), or a cocktail of four compounds: midazolam (3 μM), diclofenac (5 μM), bufuralol (10 μM), and coumarin (3 μM) with 0.35% MeOH as a final solvent concentration. For cocktail incubations, no metabolic interactions were detected between the substrates at these concentrations (T. Andersson, AstraZeneca, Molndal, Sweden, personal communication). To evaluate phase II metabolism, slices were incubated with 7HC (final concentration 500 μM, 1% MeOH). As a control, medium was incubated with substrate, but without the slice. After incubations with 7EC or 7HC, medium was harvested for analysis of the metabolites. The slice was harvested for protein determination. After incubations with TT and the cocktail, slice and medium were harvested together for metabolite analysis. All samples were stored at -20°C until further use. Metabolism experiments with proximal jejunum and colon were performed on tissues from five donors in triplicate.
Induction Studies. To test the induction of DMEs and DTs, slices prepared from proximal jejunum (up to six individual donors) and colon tissue (up to three individual donors) were preincubated with BNF (final concentration 50 μM, 0.5% DMSO), DEX (final concentration 100 μM, 0.5% DMSO), RIF (final concentration 30 μM, 0.5% DMSO), PB (500 μM), Q (final concentration 10 μM, 0.5% DMSO), 0.5% DMSO (control for BNF, Q, DEX, and RIF), or 0% DMSO (control for PB). Subsequently, slices were either harvested for RNA isolation by snap-freezing in liquid N2 or for proximal jejunum only were further incubated for 3 h with a substrate to assess induction of metabolic activity. After 3 h of incubation with the substrate for metabolism, the reactions were stopped as described above. The following combinations of inducers and substrates were tested: BNF with 7EC and 7HC, RIF with TT, DEX with TT, PB with TT, and Q with 7HC.
Gene Expression Levels. After thawing, total RNA was isolated using the RNeasy Mini Kit (QIAGEN, Hilden, Germany); 2 μg of total RNA was used to synthesize 50 μl of cDNA using the Promega Reverse Transcription System (Promega, Madison WI). Subsequently, the genes of interest were analyzed by two different detection systems that were dependent on the available primers. Villin, CYP1A1, and CYP3A4 (Table 2) were analyzed with the SYBR Green method. cDNA (1.25 μl) was used in real-time PCR reactions using SYBR Green reaction mixture (Applied Biosystems, Warrington, UK) to assess expression of the target genes. The cycling conditions comprised 10 min at 95°C and 40 cycles at 95°C for 15 s, at 56°C for 15 s, and at 72°C for 40 s followed by a dissociation stage (at 95°C for 15 s, at 60°C for 15 s, and at 95°C for 15 s).
Primers used for SYBR green method
For TaqMan analysis, Assay-on-Demand Gene Expression Products were purchased for villin, MDR1/ABCB1, and UGT1A6 (Table 3). Primers and probe for the UGT1A6 gene were custom-designed and had the following sequences: probe sequence (5′ 6-carboxyfluorescein → 3′ nonfluorescent quencher): CCACATGACTTTTTCCC, forward primer sequence 5′-CCAGGTGCTACACAAAGTTTTCAGA-3′ and reverse primer sequence 5′-AAGGAAGTTGGCCACTCGTT-3′. cDNA (1.0 μl) was used in real-time PCR reactions using a TaqMan reaction mixture (Applied Biosystems, Warrington, UK), and the appropriate primers and probes. The cycling conditions comprised 10 min at 95°C and 40 cycles at 95°C for 15 s and at 60°C for 1 min.
Assay-on-Demand gene specific probes used for TaqMan analysis
The following combinations of genes and inducers were tested: CYP1A1, UGT1A6 after BNF exposure, CYP3A4, MDR1/ABCB1 after exposure to DEX, CYP3A4, CYP2B6, MDR1/ABCB1 after exposure to RIF, CYP3A4, CYP2B6, MDR1/ABCB1 after exposure to PB, and UGT1A6 after exposure to Q. The cycle number at the threshold (Ct) is inversely related to the abundance of mRNA transcripts in the initial sample. The mean Ct of duplicate measurements was used to calculate the difference of Ct for the target gene and the reference villin gene (ΔCt), which was compared with the corresponding ΔCt of the control experiment (ΔΔCt). Data are expressed as fold induction of the gene of interest according to the formula 2-ΔΔCt.
Metabolite Analyses.Testosterone sample analysis. After thawing (at 4°C), samples were homogenized using a sonicator. Then, 11β-TOH (internal standard) and 6 ml of dichloromethane were added. The mixture was vortexed and then centrifuged using a Beckman-Coulter CS-6KR centrifuge (Beckman Coulter, Fullerton, CA) (10 min, 4°C, 800g). The water layer was removed, and the remaining dichloromethane phase was evaporated overnight. The residue was dissolved in 130 μl of 50% MeOH and then analyzed by HPLC as described previously (van 't Klooster et al., 1993), using a Guard RP C18 (10 mm) precolumn and two Chromosphere RP C18 (100 × 3 mm, particle size 5 μm) columns in series. Detection occurred at 254 nm wavelength.
Cocktail sample analysis. Slices were homogenized in the medium by sonication, and 2 volumes of ACN were added to precipitate the proteins. Then, samples were centrifuged (10 min, 20°C, 4000g). The supernatant, used for metabolite analysis, was diluted with 2 volumes of water (final concentration of ACN 30%) and subsequently pipetted into a 96-well plate together with standards (1-hydroxymidazolam, 4-hydroxydiclofenac, 1-hydroxybufuralol, and 7-hydroxycoumarin) dissolved in 30% ACN. The plate was centrifuged for 20 min (4000g, 4°C) before injection occurred. For liquid chromatography separation, a reversed-phase HyPurity C18 analytical column (50 mm × 2.1 mm i.d., 5 μm; ThermoQuest, Runcorn, UK) with a HyPurity C18 guard column (10 mm × 2.1 mm id, 5 μm) at 40°C was used. The mass spectrometric analyses were performed with an API4000 quadrupole mass spectrometer equipped with an electrospray interface (Applied Biosystems/MDS Sciex, Concord, ON, Canada). This method was described in more detail by van de Kerkhof et al. (2006). The precipitate that was left after removal of the supernatant was stored at -20°C for protein determination to be able to relate the metabolite formation to the amount of protein in the slice.
7EC and 7HC sample analyses. After thawing, sodium azide (final concentration 0.1 mg/ml) was added to the samples to prevent bacterial growth. Subsequently, the samples were centrifuged (3 min, 4°C, 10,000g), and metabolites were determined by HPLC. 7EC, 7HC, 7HC-GLUC, and 7-hydroxycoumarin-sulfate were used as references. Chromatography was performed by HPLC as described before (van de Kerkhof et al., 2006).
Protein Determination. To correct for the differences in slice volume, the protein content was determined on all slice samples after ATP analysis and 7EC, 7HC, or cocktail incubations to be able to relate metabolic rate to protein content. For testosterone experiments, the protein content could not be determined. Therefore, the average of the protein values of 7HC incubations was used. After incubation, samples were stored at -20°C until further use. Subsequently, samples were thawed, and 5 N NaOH was added: 20 μl of 5 N NaOH for slice samples was used for ATP determination and 7HC metabolism in Eppendorf cups and 200 μl of 5 N NaOH for cocktail slice samples. Forty minutes of incubation at 37°C followed to dissolve the tissue. Then, water was added to obtain a final concentration of 0.1 N NaOH needed for the analysis. Tissue was homogenized using sonication and further diluted to determine the protein content using Bio-Rad protein assay dye reagent (Bio-Rad, Munich, Germany) with bovine serum albumin as standard. For testosterone experiments, the protein content could not be determined, as the proteins were removed together with the water layer during extraction. Therefore, the average of the protein values of 7HC incubations was used.
Statistics. Statistical significance of the elevation of gene expression or activity by the inducing compound compared with control incubations was evaluated using a paired Student's t test.
Results
Viability Testing of Small Intestinal and Colon Slices. Three different parameters, morphology, ATP levels, and metabolic stability, were assessed to evaluate the viability and functionality of proximal jejunum and colon slices after 24 h of incubation. Small pieces of nonincubated tissue served as controls for morphology and ATP levels.
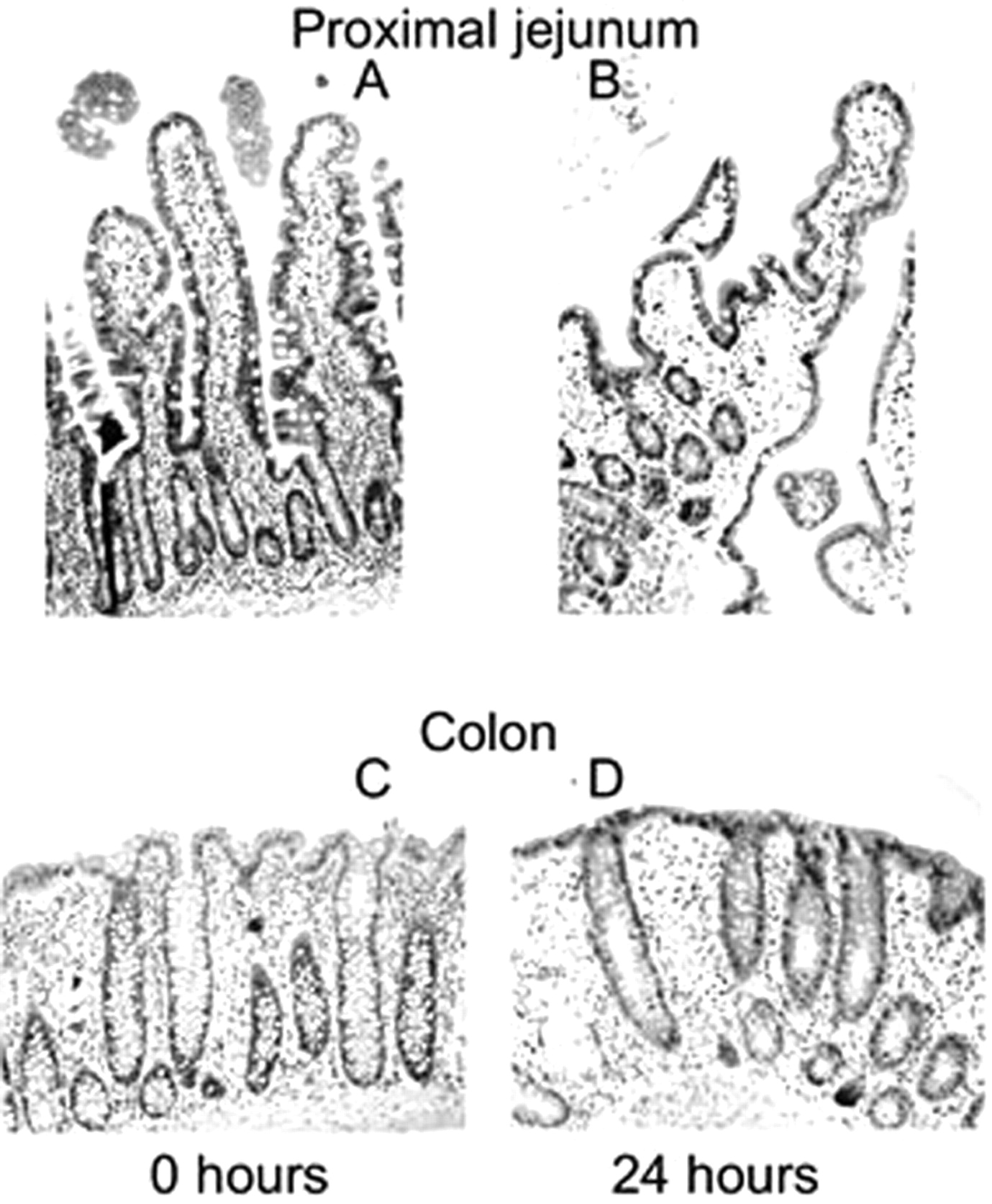
Morphology. The viability of colon and proximal jejunum slices was assessed by morphological evaluation. In general, flattening of the villi was observed after 24 h of incubation. The cells of interest for metabolism, the epithelial cells, however, still showed their columnar shape and no cell swelling or necrosis was observed. In addition, goblet cells remained intact as well. In the colon slices, the initial tissue morphology remained intact up to 24 h of incubation (Fig. 1).
Intracellular ATP levels. General viability of proximal jejunum slices was assessed in four experiments by measurement of intracellular ATP levels. The level of ATP in mucosal sheets harvested directly after preparation was 1.5 ± 0.3 nmol/mg of protein (average ± S.E.M.) and increased significantly during the first 4 h of incubation to 2.6 ± 0.5 nmol/mg of protein (p < 0.02) after which it nonsignificantly decreased to 2.0 ± 0.3 nmol/mg of protein after 24 h of incubation.

Representative micrographs of small intestinal tissue as control (A) and slices after 24 h of incubation (B) and colon tissue as control (C) and colon slices after 24 h of incubation (D). Hematoxylin and eosin (original magnifications, 100× for proximal jejunum and 200× for colon).
Stability of metabolic rate in time. To assess the metabolic stability, slices were incubated from 0 to 3 h or from 24 to 27 h with several substrates. In small intestinal slices (Table 4), the metabolic rates remained at 40 to 50% of the initial rates for most substrates (phase I and II conversions) after 24 h of preincubation. The rate of 7HC glucuronidation (mediated by UGT1A6) and androstenedione formation (mediated by several P450s and 17β-HSD) (Farthing et al., 1981) after 24 h of incubation was maintained at 76% of the initial metabolic rate. Formation of 4-hydroxydiclofenac (CYP2C9), however, was retained at only 9% of the 0- to 3-h control incubation. CYP3A4 activity assessed with either 1-hydroxymidazolam, 6β-TOH, or 2β-TOH was maintained at 22 to 42% of 0- to 3-h levels.
Enzyme activity in proximal jejunum slices after 0 and 24 h of incubation Activities are expressed as mean ± S.E.M.
In colon slices (Table 5), the phase I activities were under the detection limit al all incubation times. Glucuronidation and sulfation rates were comparable with those in proximal jejunum and remained at 100% up to 24 h of incubation.
Enzyme activity in colon slices after 0 and 24 h of incubation Activities are expressed as mean ± S.E.M.
Induction Studies. To investigate the induction of DMEs and DTs in human intestinal tissue, slices were incubated with BNF, Q, DEX, RIF, or PB and subsequently harvested for mRNA isolation or further incubated with substrates to analyze enzyme activity. To calculate the average fold induction and to determine statistical significance for each inducer, responses of all tissue samples (responsive and nonresponsive) were taken into account. In the following section, for each inducer the number of responsive (induction of >1.5 fold) and nonresponsive tissue samples is indicated.
After incubation with BNF (Fig. 2), CYP1A1 mRNA was clearly induced in all proximal jejunum (362-fold, p < 0.05) and colon (132-fold, p < 0.05) samples. Furthermore, mRNA of UGT1A6 was induced in all three tissue samples that were tested (proximal jejunum: 3.2-fold, p < 0.05, n = 3; colon: 9.8-fold, n = 3). In colon, however, induction was not statistically significant because of the high variation between the three individual samples (2-, 4- and 23-fold induction, respectively). Besides CYP1A1 mRNA, the rate of 7EC O-deethylation also was increased in slices after preincubation with BNF and was significantly higher than that in fresh slices or slices incubated for 24 h without BNF or DMSO. However, there was no significant difference between the rate of 7EC O-deethylation in BNF-treated slices and the 0.5% DMSO controls in which EC metabolism rates also were increased in three of five experiments.
After incubation with DEX (Fig. 3), CYP3A4 mRNA expression was induced in proximal jejunum (3.5-fold, n = 5, induction in four of five experiments, p < 0.05). In colon, CYP3A4 mRNA was only induced (10-fold) in slices of one of the two individual samples tested. MDR1/ABCB1 mRNA expression showed a tendency to increase in proximal jejunum (1.8-fold, n = 5; in tissue of three of the five donors induction was >1.5-fold), but no change was observed in colon slices. Hydroxylation of testosterone was also tested in proximal jejunum slices after incubation with DEX. Formation of 6β-TOH increased in slices of five of six donors. The average increase in activity was 1.5-fold (n = 6), but this increase was not statistically significant. Induction of 2β-TOH could not be measured, because DEX disturbed the HPLC measurement by eluating at the same retention time as 2β-TOH.
After incubation with RIF (Fig. 4), CYP3A4 mRNA expression was prominently induced in proximal jejunum (5-fold, p < 0.05, n = 5, all tissues responsive), but no significant induction was observed in colon slices (average induction 1.3-fold, n = 2, one tissue responsive). Furthermore, CYP2B6 (proximal jejunum: 2.2-fold, n = 5, four tissues responsive, p < 0.05; colon: 2.0-fold, n = 2, one tissue responsive), UGT1A6 (proximal jejunum: 2.2-fold, n = 5, p < 0.05, all tissues responsive; colon: 32-fold, n = 2, one tissue responsive), and MDR1/ABCB1 (proximal jejunum: 2.7-fold, n = 5, p < 0.05, all tissues responsive; colon: 2.9-fold, n = 2, both tissues responsive) were induced. In proximal jejunum slices, the metabolic activity also was increased by RIF: 6β-TOH and 2β-TOH formation increased 2-fold (n = 6, p < 0.05, all tested samples responsive). Formation of 16α-TOH increased nonsignificantly (p = 0.06) by 1.5-fold compared with the DMSO controls (n = 6, five of six tissues responsive).
After incubation with PB (Fig. 5), all genes that were tested were significantly induced in proximal jejunum. CYP3A4 was induced 4.1-fold (p < 0.05, n = 5, all tissues responsive), CYP2B6 was induced 2.3-fold (p < 0.05, n = 5, four of five tissues responsive), and MDR1/ABCB1 was induced 2.2-fold (p < 0.05, n = 5, all tissues responsive). Only colon tissue was available for two donors (which impeded calculation of statistical significancy), and in slices of both tissues, CYP3A4 was not influenced, but CYP2B6 and MDR1/ABCB1 tended to increase (4.9- and 2-fold, respectively).
Besides CYP3A4 mRNA expression, the metabolic rate toward both CYP3A4-mediated formation of 6β-TOH and 2β-TOH also increased in all proximal jejunum samples (n = 6) (p < 0.05). In contrast with CYP2B6 mRNA expression, the formation of 16α-TOH (CYP2B-mediated) in proximal jejunum slices was not induced after 24 h of incubation.
After incubation with Q (Fig. 6), UGT1A6 was induced in proximal jejunum (2.2-fold, n = 3, p < 0.05, all tissues responsive) and colon slices (6.7-fold, tested in one experiment only). Formation of 7HC-GLUC, mediated by UGT1A6, was not influenced by Q after 24 h of incubation (n = 4).
Discussion
In the present study, we investigated the applicability of human intestinal precision-cut slices for drug induction studies. Therefore, we assessed the viability and functionality of intestinal slices during 24 h of incubation. In addition, we tested the induction of several phase I, II, and III genes by five prototypical inducers, covering the most relevant (nuclear) receptor pathways (AhR, Nrf2, PXR, GR, and CAR).
The viability studies revealed that in proximal jejunum slices 1) ATP levels were preserved during 24 h of incubation, 2) epithelial cells remained intact, as judged by morphological evaluation, although some flattening of the villi occurred, and 3) metabolic phase I and II reactions were retained at an appreciable level. The morphology of colon slices remained intact, and the conjugation rates remained constant during 24 h of incubation. P450-mediated metabolism, however, was below the detection level in colon at all time points. In proximal jejunum slices, the rates of P450-mediated metabolism were retained to different extents (9–51%), depending on the substrate. Because all P450 reactions use the same cofactor NADPH, the differential maintenance of P450 isoenzyme activity may be explained either by the differences in half-lives of the isoenzymes, which is in line with reported findings in human liver slices (Renwick et al., 2000) and/or in the lack of natural inducers, which are normally present in the intestinal lumen or the blood, in the incubation medium. Further studies are required to investigate these hypotheses.
To investigate the applicability of human intestinal slices for induction studies, both proximal jejunum and colon slices were incubated with different prototypical inducers: BNF (AhR ligand), RIF (PXR ligand), DEX (GR and PXR ligand), PB (CAR ligand), and Q [Nrf2, described to induce UGT expression (Galijatovic et al., 2000)].
CYP1A1 mRNA and the activity level of 7EC O-deethylation were prominently induced after BNF incubation, which confirms published findings in LS180 cells (Hartley et al., 2006). The activity level of 7EC O-deethylation after DMSO (control) incubation, however, was also increased in three of five experiments. DMSO, however, did not up-regulate CYP1A1 mRNA levels. It is possible that DMSO regulates CYP1A1 expression at post-transcriptional levels, perhaps by protecting the enzyme from degradation. DMSO (2%) was found earlier to selectively maintain levels of CYP1A1 in rat hepatocyte cultures. However, it was concluded that DMSO stimulated de novo synthesis of CYP isoenzymes (Lindsay et al., 1991). It is not clear, therefore, to what extent DMSO or BNF is responsible for the effect on CYP1A1 activity in the present study. Interestingly, 7EC O-deethylation was up-regulated by BNF in all five experiments, i.e., also in the two experiments in which DMSO did not show any effect, which indicates that AhR-mediated induction can be detected in human intestinal slices. This finding is further strengthened by the observation that BNF induced CYP1A1 and UGT1A6 mRNA expression, which is in agreement with published findings with Caco-2 cells (Abid et al., 1995; Münzel et al., 2003).

Slices were exposed to BNF (50 μM) for 24 h after which mRNA expressions of CYP1A1 and UGT1A6 (A, proximal jejunum and colon) and 7EC O-deethylation (B, proximal jejunum) were evaluated. For mRNA induction studies, the fold induction was compared with control slices that were incubated for 24 h with 0.5% DMSO (represented by the line at 1-fold). Results show mean ± S.D. for proximal jejunum (three donors for mRNA expression and five donors for activity analysis) and colon (three donors); in each experiment three slices were incubated per treatment. Significant differences toward the control incubations: *, p < 0.05.

Slices were exposed to DEX (100 μM) for 24 h after which mRNA expression of both CYP3A4 and MDR1/ABCB1 (A, proximal jejunum and colon) and testosterone hydroxylation (B, proximal jejunum) were evaluated. For mRNA induction studies, the fold induction was compared with control slices that were incubated for 24 h with 0.5% DMSO (represented by the line at 1-fold). Results show mean ± S.D. for proximal jejunum (five donors for mRNA analysis and six donors for activity measurement) and colon (two donors); in each experiment three slices were incubated per treatment. Significant differences toward the control incubations: *, p < 0.05.

Slices were exposed to RIF (30 μM) for 24 h after which mRNA expression of CYP3A4, CYP2B6, UGT1A6, and MDR1/ABCB1 (A, proximal jejunum and colon) and testosterone hydroxylation (B–D, proximal jejunum) were evaluated. For mRNA induction studies, the fold induction was compared with that of control slices incubated for 24 h with 0.5% DMSO (represented by the line at 1-fold). Results show mean ± S.D. for proximal jejunum (five donors for mRNA analysis and six donors for activity measurement) and colon (two donors); in each experiment three slices were incubated per treatment. Significant differences toward the control incubations: *, p < 0.05; **, p < 0.01.
DEX is a known agonist for GR (Savas et al., 1999) as well as an activator and ligand for human PXR, in particular at micromolar concentrations (>10 μM) (Pascussi et al., 2001). CYP3A4 mRNA and activity were induced in our studies with DEX using concentrations of 100 μM. In addition, induction of MDR1/ABCB1 mRNA was detected in our study and is in agreement with published findings in LS174T cells (Geick et al., 2001) and in LS180 cells (Schuetz et al., 1996).

Slices were exposed to PB (500 μM) for 24 h after which mRNA expression of CYP3A4, CYP2B6, and MDR1/ABCB1 (A, proximal jejunum and colon) and testosterone hydroxylation (B–D, proximal jejunum) were evaluated. For mRNA induction studies, the fold induction was compared with that of control slices incubated for 24 h (represented by the line at 1-fold). Results show mean ± S.D. for proximal jejunum (five donors for mRNA analysis and six donors for activity measurement) and colon (two donors); in each experiment three slices were incubated per treatment. Significant differences toward the control incubations: *, p < 0.05; **, p < 0.01.
Rifampicin is a known activator and ligand of human PXR (Savas et al., 1999). Rifampicin induced the mRNA expression levels of CYP3A4, CYP2B6, MDR1/ABCB1, and UGT1A6. In addition, the activities of CYP2B6 (16α-TOH) and CYP3A4 (6β-TOH/2β-TOH) enzymes were induced. These observations are in line with published findings. Rifampicin, for example, was reported to induce CYP3A4 (Glaeser et al., 2005) and MDR1/ABCB1 (Greiner et al., 1999) in human enterocytes after oral administration of RIF to healthy volunteers. In vitro, RIF induced CYP3A4, UGT1A6, and MDR1/ABCB1 (Schuetz et al., 1996; Hartley et al., 2006) in LS180 cells. In colon slices, the results obtained with RIF are difficult to interpret, because only one of the two donor tissues responded.
Phenobarbital induces the nuclear translocation of CAR, eventually inducing the transcription of several genes (Qatanani and Moore, 2005). Furthermore, PB seems to be a ligand for both CAR and PXR (Masahiko and Honkakoski, 2000). After incubation with PB, mRNA expressions of CYP2B6 (proximal jejunum and colon), CYP3A4, and MDR1/ABCB1 (only proximal jejunum) were induced in our slice system. This finding is in line with published findings showing induction of CYP3A4 and MDR1/ABCB1 mRNA in LS180 cells by PB (Schuetz et al., 1996). Furthermore, CYP3A4 activity (6β-TOH and 2β-TOH formation) was induced after 24 h of slice incubation, but CYP2B6 activity (16α-TOH formation) was not induced. A possible explanation might be that 16α-TOH formation, being a marker of rodent CYP2B activity, is not mediated by human CYP2B6. No report could be found in which the induction of CYP2B enzyme activity or RNA expression in human intestinal cells/tissue was measured.

Slices were exposed to Q (10 μM) for 24 h after which mRNA expression of UGT1A6 (A, proximal jejunum and colon) and 7HC glucuronidation (B, proximal jejunum) were evaluated. For mRNA induction studies, the fold induction was compared with that of control slices incubated for 24 h with 0.5% DMSO (represented by the line at 1-fold). Results show mean ± S.D. proximal jejunum (three donors for mRNA analysis and four donors for activity measurement) and colon (one donor); in each experiment three slices were incubated per treatment. Significant differences toward the control incubations: *, p < 0.05.
Quercetin mediates induction via Nrf2 and has been described to induce UGT1A6 in Caco-2 cells after 72 h of incubation (Bock et al., 2000). Induction of UGT1A6 at the mRNA level was clearly detected, but induction at the activity level (7HC glucuronidation mediated by UGT1A6) was not yet observed. This result could be due to the relatively short incubation time chosen in our studies (24 h). Induction of DMEs has been shown to be dependent on the concentration of the inducer and incubation period (Jigorel et al., 2006), on the presence of supplements in the incubation medium (Ringel et al., 2002), and also on the type of cell line used (Hartley et al., 2006). Therefore, “false-negative” results could be explained by these factors. The limited amount of tissue did not allow testing of concentration or time dependence in these studies. Future studies are required to study the kinetics of enzyme induction in human intestinal slices in more detail.
To summarize, because the morphological evaluation and ATP content indicated tissue integrity and the metabolic rates of all compounds that were tested were still detectable after 24 h of preincubation, we conclude that slices are useful for qualitative drug metabolism studies up to 24 h of incubation. Furthermore, mRNA of all selected “model” genes was induced as expected after slice exposure to prototypical agents for five putative induction mechanisms (AhR, Nrf2, GR, PXR, and CAR). This increase was in most cases accompanied by an increase of activity of corresponding metabolic enzymes. Therefore, we conclude that human intestinal precision-cut slices provide a powerful tool to study induction in human intestinal tissue, thereby providing the opportunity to characterize and study these mechanisms in more detail.
Acknowledgments
The authors thank Prof. Dr. D. K. F. Meijer for valuable advice and Dr. H. J. Koster and Dr. M. Rooseboom for critical reading of the manuscript, Sara Leandersson and Xueqing Li for excellent assistance with the liquid chromatography/mass spectrometry analysis, and Åsa Sjöberg, Constanze Hilgendorf, and Marina de Jager for excellent technical assistance.
Footnotes
-
This study was supported by the Technology Foundation STW, the applied science division of NWO and the technology programme of the Ministry of Economic Affairs, and Yamanouchi Europe.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.018820.
-
ABBREVIATIONS: DME, drug-metabolizing enzyme; DTs, drug transporters; PXR, pregnane X receptor; TT, testosterone; 17β-HSD, 17β-hydroxysteroid dehydrogenase activity; 7EC, 7-ethoxycoumarin; 7HC, 7-hydroxycoumarin; UGT, UDP glucuronosyltransferase; BNF, β-naphthoflavone; AhR, aryl hydrocarbon receptor; RIF, rifampicin; DEX, dexamethasone; GR, glucocorticoid receptor; PB, phenobarbital; CAR, constitutive androstane receptor; Q, quercetin; Nrf2, nuclear factor-E2-related factor 2; MDR1, multidrug resistance-1; DMSO, dimethyl sulfoxide; 7HC-GLUC, 7-hydroxycoumarin glucuronide; TOH, hydroxytestosterone; ACN, acetonitrile; MeOH, methanol; PCR, polymerase chain reaction; HPLC, high-performance liquid chromatography; P450, cytochrome P450.
- Received September 11, 2007.
- Accepted December 18, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}