


Abstract
The aim of this study was to evaluate drug metabolism in rat small intestinal and colon precision-cut slices during 24 h of incubation and the applicability of these slices for enzyme induction studies. Various parameters were evaluated: intracellular levels of ATP (general viability marker), alkaline phosphatase activity (specific epithelial marker), villin expression (specific epithelial marker), and metabolic rates of 7-ethoxycoumarin (CYP1A), testosterone (CYP3A and CYP2B), and 7-hydroxycoumarin (glucuronide and sulfate conjugation) conversions. ATP and villin remained constant up to, respectively, 5 and 8 h in small intestine and up to 24 h in colon. The metabolic rate remained constant in small intestinal slices up to 8 h and decreased afterward to 24 to 92%, depending on the substrate studied. The inducibility of metabolism in small intestinal and colon slices was tested with several inducers at various concentrations and incubation times. The following inducers were used: 3-methylcholanthrene, β-naphthoflavone, indirubin, and tert-butylhydroquinone (aryl hydrocarbon receptor ligands), dexamethasone (glucocorticoid receptor/pregnane X receptor ligand) and phenobarbital (constitutive androstane receptor ligand). After incubation with inducers, metabolic rates were evaluated with 7-ethoxycoumarin and testosterone (phase I) and 7-hydroxycoumarin (phase II) as substrate. All inducers elevated the metabolic rates consistent with the available published in vivo induction data. Induction of enzyme activity was already detectable after 5 h (small intestine) and after 8 h (colon) for 3-methylcholanthrene and β-naphthoflavone and was clearly detectable for all tested inducers after 24 h (up to 20-fold compared with noninduced controls). In conclusion, small intestinal and colon precision-cut slices are useful for metabolism and enzyme induction studies.
The intestine is highly sensitive for drug-drug and drug-diet interactions, influencing drug metabolism by inhibition and induction of drug-metabolizing enzymes (DMEs) (Pelkonen et al., 2001). This can lead to major changes in bioavailability of drugs and may also cause an imbalance between toxification and detoxification (Lin and Lu, 1998).
An unfortunate and serious example of adverse drug-drug interactions is the induction of CYP3A4 and P-glycoprotein by St. Johns Wort in transplantation patients, causing a serious decrease in cyclosporine plasma concentration, which can lead to organ rejection after transplantation (Barone et al., 2000; Ruschitzka et al., 2000). The importance of the induction of CYP3A4 by St. Johns Wort in the human intestine was demonstrated using the jejunal perfusion technique (Tannergren et al., 2004).
Pharmaceutical research would greatly benefit from an in vitro system to study drug-drug interactions in the human and animal intestine and, thus, to be able to predict potential substrate interactions and undesirable side effects/toxicity of drugs. Preferably, this should be a system that could also make use of human intestinal material, as many drug interactions are highly species-specific. Such a system should consist of intact cells that remain viable, expressing genes and proteins and capable of metabolizing drugs for at least 24 h of incubation. Only a few in vitro systems have been reported to meet these criteria. Caco-2 cells have been used for induction studies at the enzyme activity level (Galijatovic et al., 2000) and LS180 cells for studies at the mRNA level (Aiba et al., 2005). However, these cell cultures clearly differ from the complex structure and metabolic function of normal intestinal tissue. The mouse intestinal explant technique is an intact tissue system that has been reported to be useful for studying the induction of fatty acid-binding proteins (Mallordy et al., 1995). Important in this respect is the precision-cut slice system, an intact tissue system in which induction at the mRNA level has been described using rat small intestinal tissue (Martignoni et al., 2004). At the activity level, a first indication of the inducibility of DMEs in intestinal slices with β-naphthoflavone (bNF) was reported by our laboratory (van de Kerkhof et al., 2005).
In the present study, we further assessed 1) the viability and metabolic capacity of rat small intestinal and colon precision-cut slices after long-term incubation (e.g., up to 24 h) and 2) their applicability for DME induction studies. Viability was assessed after incubation by measuring the intracellular ATP levels in both small intestinal and colon slices. ATP levels measured in slices are indicative for the viability of all cell types in the tissue, but for drug metabolism studies the viability of the enterocytes is of particular interest. As a viability marker for enterocytes, alkaline phosphatase activity was determined in small intestinal slices. In addition, gene expression of villin (epithelial marker) and GAPDH (housekeeping gene for all cell types) was monitored up to 24 h of incubation. Finally, enzyme activity levels were evaluated using 7-ethoxycoumarin O-deethylation (7EC) (CYP1A, phase I), testosterone hydroxylation (TT) (CYP2B and CYP3A, phase I), and 7-hydroxycoumarin conjugation (7HC) (glucuronidation and sulfation, phase II).
To study DME induction, we focused on three major induction pathways: induction via CAR (regulating CYP2B and CYP3A) (Wang and LeCluyse, 2003), PXR (mainly regulating CYP3A, but also CYP2B) (Wang and LeCluyse, 2003), and AhR (CYP1A1) (Lin and Lu, 1998). We chose bNF (Lin and Lu, 1998), 3-methylcholanthrene (3MC) (Abdelrahim et al., 2006), indirubin (IR) (Guengerich et al., 2004), and tert-butylhydroquinone (tBHQ) (Schreiber et al., 2006) as AhR ligands. Dexamethasone (DEX) was used as a GR/PXR ligand (Pascussi et al., 2001) and phenobarbital (PB) as a CAR mediator (Wang and LeCluyse, 2003). Slices were incubated with several inducers at various concentrations and incubation periods (0, 5, 8, and 24 h), followed by 3 h of incubation with a substrate (7EC, 7HC, or TT). For PB studies, induction was also monitored at the mRNA level (CYP2B15, CYP3A9, CYP1A1, and CAR).
Materials and Methods
Chemicals.para-Nitrophenylphosphate, para-nitrophenol, 6β- and 11β-hydroxytestosterone (TOH), testosterone, androstenedione, 7HC, 7HC-glucuronide (7HC-GLUC), low gelling temperature agarose (type VII-A), DMSO, and tBHQ were purchased from Sigma-Aldrich (St. Louis, MO). Gentamicin, Williams' medium E with Glutamax-I, and amphotericin B (Fungizone) solution were obtained from Invitrogen (Paisley, UK). 7EC and bNF were obtained from Fluka (Buchs, Switzerland). 2-Amino-2-methyl-1,3-propanediol (ammediol) was purchased from Aldrich (Steinheim, Germany). 16α-TOH was obtained from Steraloids (Newport, RI). Acetic acid, sodium azide, sodium chloride, and calcium chloride were obtained from Merck (Darmstadt, Germany). HEPES was obtained from ICN Biomedicals, Inc (Illkirch, France). 7HC-sulfate (7HC-SULF) was a kind gift from Mr. P. Mutch, GlaxoWellcome (Herts, UK). PB was obtained from Bufa B.V. (Uitgeest, Holland). DEX was purchased from Genfarma B.V. (Maarssen, Holland). 3MC was obtained from Supelco (Bellefonte, PA). Indirubin was obtained from Tebu-bio (Heerhugowaard, The Netherlands). All reagents and materials were of the highest purity that is commercially available.
Animals. Male Wistar (HsdCpb:WU) rats weighing ∼350 g were purchased from Harlan (Horst, The Netherlands). Rats were housed in a temperature- and humidity-controlled room on a 12-h light/dark cycle with food (Harlan chow no. 2018; Harlan) and tap water ad libitum. The animal ethical committee of the University of Groningen approved the use of animals for these experiments.
Preparation of Precision-Cut Slices. Under isoflurane-N2O-O2 anesthesia, the small intestine and colon were excised from the rat and put in ice-cold, oxygenated Krebs-Henseleit buffer (containing 10 mM HEPES and 25 mM d-glucose, pH 7.4). Segments of 3 cm were excised from the colon or small intestine (between 25 and 40 cm from the stomach) and subsequently flushed with ice-cold Krebs-Henseleit buffer. One side of the segment was tightly closed, filled with 3% (w/v) agarose solution in 0.9% NaCl (37°C) and then cooled in ice-cold Krebs-Henseleit buffer, allowing the agarose solution to gel. Subsequently, the filled segment was embedded in 37°C agarose solution using a precooled (0°C) tissue embedding unit (Alabama R&D, Munford, AL). After the agarose solution had gelled, precision-cut slices (thickness of ∼400 μm and slice wet weight of ∼2 mg) were cut using a Krumdieck tissue slicer as described earlier (de Kanter et al., 2005).
Incubation of Precision-Cut Slices. The slices were incubated individually in a 12-well culture plate (Greiner Bio-One GmbH, Frickenhausen, Austria) in 1.3 ml of Williams' medium E (with Glutamax-I), supplemented with d-glucose (final concentration 25 mM), gentamicin (final concentration 50 μg/ml), and amphotericin B (final concentration 2.5 μg/ml). The culture plates were placed in a prewarmed cabinet (37°C) in plastic boxes under humidified carbogen (95% O2 and 5% CO2) and shaken back and forth 90 times/min.
Viability Testing.Alkaline phosphatase activity. The precision-cut slices were incubated in triplicate for 0, 5, 8, and 24 h. After incubation, the slices were taken out of the wells, placed in 1 ml of 0.05 M ammediol buffer (pH 9.8 at 4°C), and medium and slices were stored separately at 4°C until further analysis. AP activity was determined as described before (van de Kerkhof et al., 2005).
Intracellular ATP levels. Intracellular ATP levels in slices were evaluated up to 24 h of incubation. Directly after tissue excision, three pieces of tissue were snap-frozen as “in vivo” controls. Intracellular ATP levels were determined according to the method described earlier (van de Kerkhof et al., 2006). ATP content was determined in two to eight experiments in triplicate.
Gene expression levels. Two micrograms of total RNA, isolated from six snap-frozen slices using the RNeasy Mini Kit (QIAGEN, Hilden, Germany), was used to synthesize 50 μl of cDNA using the Promega Reverse Transcription System (Promega, Madison, WI). cDNA (1.25 μl) was used in real-time PCR reactions using an SYBRGreen reaction mixture (Applied Biosystems, Warrington, UK) and the appropriate primers listed in Table 1. Agarose gel electrophoresis and dissociation curves confirmed homogeneity of the PCR products. Only for CAR, a minor second product was formed in colon slices. The cycle threshold value (Ct value) is inversely related to the abundance of mRNA transcripts in the initial sample. Mean Ct value of duplicate measurements was used to calculate the difference of Ct value for the target gene and the reference villin gene (ΔCt), which was compared with the corresponding ΔCt value of the control experiment (ΔΔCt). Data are expressed as fold induction of the gene of interest according to the formula 2-ΔΔCt.
Primer information of the rat genes under study
Induction Studies. Precision-cut slices were prepared from small intestine (25-40 cm from the stomach) and colon and incubated with the selected inducers for several incubation times at various concentrations: bNF (0, 5, 8, and 24 h of preincubation at 50 μM), 3MC (0, 5, 8, and 24 h of preincubation at 5 μM), IR (0 and 24 h of preincubation at 10, 100, and 1000 nM), tBHQ (0, 5 and 24 h of preincubation at 50 μM), DEX (0, 5, and 24 h of preincubation at 100 μM), and PB (0, 8, and 24 h of preincubation at 2, 2.5, 4, and 8 mM). Subsequently, slices were transferred to fresh medium and incubated for 3 h with model substrates: TT (250 μM), 7EC (500 μM), or 7HC (500 μM). The substrates were added as 100× concentrated stock dissolved in MeOH (final medium concentration: 1% MeOH). As controls, slices were incubated without substrate and medium was incubated with substrate without slices.

Viability characterization of small intestinal and colon slices up to 24 h of incubation. Several parameters were monitored: intracellular ATP (A), alkaline phosphatase (only small intestine) (B), GAPDH mRNA expression (C), and villin mRNA expression (D). Results are means ± S.E.M. of slices of two to eight rats. In each experiment at least three slices were incubated per time point. *, p < 0.05; **, p < 0.01 compared with t = 0 h.
The model inducers were added as a 200× concentrated stock solution in DMSO (100 times for tBHQ) with a final concentration of 0.5 or 1% DMSO, with the exception of PB, which was directly added to the medium (0% DMSO). Control slices were incubated in medium supplemented with the same DMSO concentration (0, 0.5, or 1%).
After 24 h of PB incubation (4 mM), slices were also harvested for RNA isolation. Expressions of several genes as a ratio to villin expression were tested: CYP2B15 (same primers are coding for CYP2B1 according to others) (Caron et al., 2005), CYP3A9, CYP1A1, and CAR. Furthermore, slices were incubated with a combination of PB (4 mM) and DEX (1 μM) during 24 h; slice incubations with only DEX (1 μM) for 24 h were then used as controls.
Metabolite Analysis.Testosterone analysis. After TT incubation, slice and medium were collected together and stored at -20°C until further use. Sample extraction and high-performance liquid chromatography analysis was performed as described earlier (van de Kerkhof et al., 2006).
7EC and 7HC analysis. As it was previously shown that 7EC and 7HC and their metabolites are not significantly retained in the tissue, analysis was performed on medium samples only (de Kanter et al., 205). Medium samples were stored at -20°C and analyzed for 7HC-GLUC and 7HC-SULF using high-performance liquid chromatography analysis as described earlier (van de Kerkhof et al., 2005). Phase I metabolism of 7EC was calculated by adding up the amount of formed 7HC, 7HC-GLUC, and 7HC-SULF.
Protein Determination. After incubation with 7EC or 7HC, slices were stored at -20°C until further use. After thawing, 20 μl of 5 N NaOH was added to the slice followed by 40 min of incubation at 37°C to dissolve the tissue; 980 μl of water was added to dilute the NaOH concentration to 0.1 M, after which the mixture was homogenized by 5 s of sonication. Samples were diluted and the protein content was determined using Bio-Rad protein assay dye reagent (Bio-Rad, Munich, Germany) with bovine serum albumin as standard. After testosterone incubation, the protein of the slices was not determined. For these slices, the average protein contents of slices that were incubated with 7EC and 7HC within the same experiment were used.
Statistics. Statistical significance was determined using Student's t test; p < 0.05 is considered significant. For gene expression the ΔΔCt values were used to determine the statistical significance of differences.
Results
Slice Characterization during 24 h of Incubation. The viability of the slices was monitored by several parameters, i.e., intracellular ATP levels, AP activity, gene expression levels, and activity during incubation.
Intracellular ATP levels. Intracellular ATP levels were measured in small intestinal and colon slices at various time points during 24 h of incubation to evaluate viability (Fig. 1A). In small intestinal and colon tissue, the in vivo intracellular ATP was, respectively, 0.8 and 0.9 nmol/mg protein and increased during the slicing procedure to, respectively, 3.9 and 2.4 nmol/mg protein (0 h), after which it decreased to, respectively, 1.0 and 1.7 nmol/mg protein after 24 h of incubation. In small intestinal slices, ATP levels remained constant up to 5 h and decreased significantly afterward to 75% (8 h) and 25% (24 h), but this value at 24 h was not lower than the in vivo value, in contrast with colon slices in which no significant decrease was observed for 24 h.
Alkaline phosphatase activity. AP activity was measured in small intestinal slice tissue to assess the AP activity of epithelial cells during incubation, as well as in the incubation medium (Fig. 1B). The AP activity within the slice remained constant (approximately 100 U/min/slice) during incubation, whereas it increased in the incubation medium.
GAPDH and villin expression. GAPDH and villin expressions were measured at various time points in both small intestine and colon slices. GAPDH (generally applied as a housekeeping gene) remained constant up to 24 h of incubation in slices of both organs (Fig. 1C). In colon slices, villin expression (Fig. 1D) also remained constant up to 24 h of incubation. In contrast, the villin expression in small intestinal slices was constant up to 8 h of incubation and decreased afterward, possibly indicating that small intestinal slices lost epithelial cells after 8 h of incubation.
Metabolic rates during incubation. Slices were preincubated for 0, 5, 8, and 24 h after which the metabolic conversion of several model compounds was monitored. In general, in both small intestinal and colon slices (Tables 2 and 3), the metabolic activity (per milligram of protein) was constant up to 8 to 11 h and decreased during further incubation, but the rate of decline of activity varied between substrates. For colon slices, the protein content per slice was constant up to 27 h. For small intestinal slices, however, the protein content per slice decreased during incubation to 71% (5 h), 64% (8 h), and 49% (24 h) compared with 0- to 3-h incubated slices (data not shown).
In small intestinal slices, 7EC metabolism and androstenedione, 7HC-GLUC, and 7HC-SULF formation remained constant up to 11 h of incubation (Table 2). In contrast, the 6β-TOH formation rate had already decreased, nonsignificantly, after 5 h of preincubation. Then, after 24 h of preincubation, 7HC and 6β-TOH formation significantly decreased to 38 and 24%, respectively, compared with 0 to 3 h of incubation. Androstenedione, 7HC-GLUC, and 7HC-SULF formation tended to decrease, although not significantly, to, respectively, 43 and 77% of the 0- to 3-h rates.
In colon slices, considerable activity (32-196%) was retained up to 27 h of incubation (Table 3). The metabolic rates for all tested reactions remained high (>81%) after 8 h of preincubation. 7HC-GLUC, 7HC-SULF, and androstenedione formation remained constant up to 27 h of incubation, but 7EC metabolism and 7HC sulfation decreased nonsignificantly to 41 and 54%, respectively. Formation of 6β-TOH increased nonsignificantly to 196% after 24 to 27 h of incubation compared with 0 to 3 h.
Induction Studies.bNF and 3MC. Although the rate of 7EC conversion declined to 38 and 32% during incubation up to 27 h in, respectively, small intestine and colon, a very strong induction of phase I metabolism of 7EC was found when slices were preincubated with either bNF or 3MC (Fig. 2). In small intestinal slices (Fig. 2A) preincubated with bNF (50 μM) or 3MC (5 μM) for 5, 8, or 24 h, the metabolic rate increased strongly and was already prominently induced after only 5 h of preincubation (>16-fold compared with DMSO control). The fold induction was the highest after 8 h of preincubation (20-fold for bNF and 19-fold for 3MC) and induction remained prominent after 24 h of preincubation with a metabolic rate of 113 pmol/min/mg protein (12-fold for both bNF and 3MC; p < 0.05, compared with the DMSO control for each time point).
In colon slices (Fig. 2B), metabolic conversion of 7EC was also induced by bNF and 3MC. When slices were preincubated for 8 or 24 h with bNF or 3MC, the metabolic rate increased from 7.2 to 146 and 62 pmol/min/mg protein, respectively (p < 0.05) but was not significantly elevated after 5 h. Furthermore, the induction (compared with DMSO controls) was the highest after 24 h of preincubation (21-fold with bNF and 10-fold with 3MC). After 8 h of preincubation, induction was 5-fold for bNF and 4-fold with 3MC.
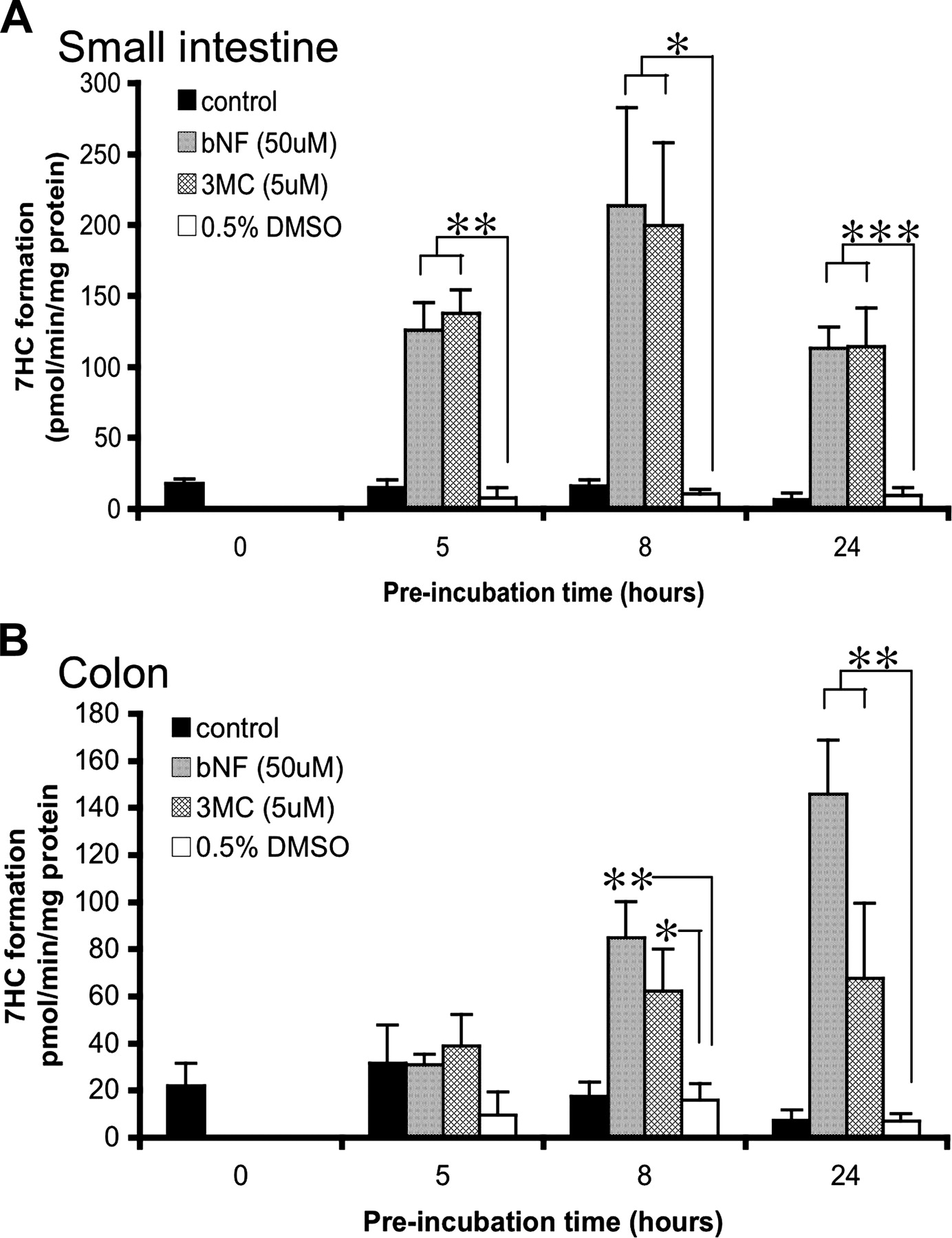
Induction of 7EC activity in precision-cut slices prepared from small intestine (25-40 cm from stomach) (A) and colon (B). Slices were preincubated for 0, 5, 8, and 24 h with control medium, 50 μM bNF, 5 μM 3MC, or 0.5% DMSO (solvent of bNF and 3MC stock solutions). Subsequently, slices were transferred to fresh medium containing 7EC (500 μM) for 3 h of incubation. Results are means ± S.E.M. of slices of three to seven rats. In each experiment three slices were incubated per treatment. Significant differences toward the activities of slices incubated with 0.5% DMSO per incubation time are indicated: *, p < 0.05; **, p < 0.01, and ***, p < 0.001.

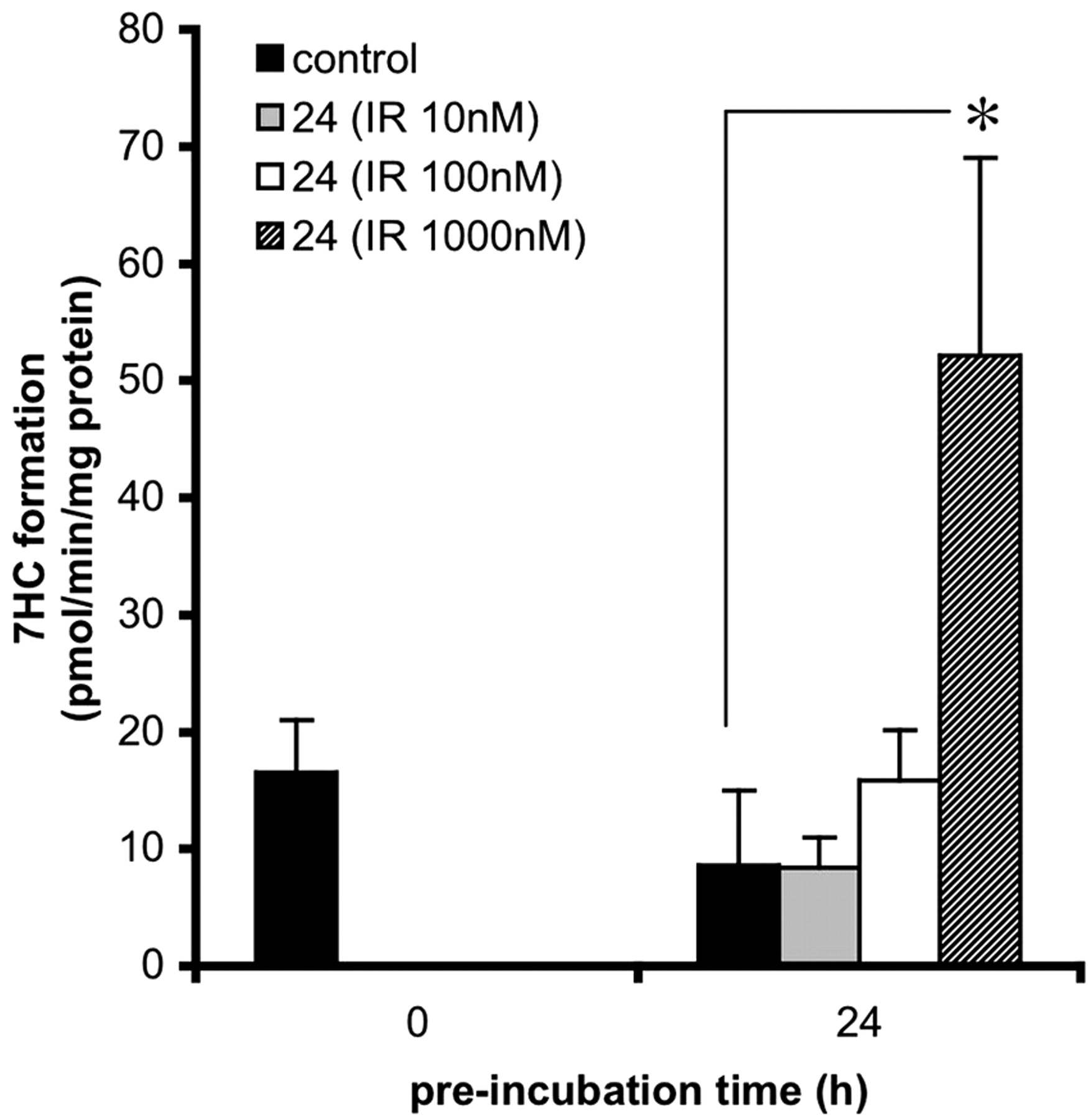
Induction of 7EC activity in precision-cut slices prepared from small intestine (25-40 cm from stomach). Slices were incubated for 0 and 24 h with control medium, 0.5% DMSO (solvent of IR stock solutions), and IR at different concentrations: 10, 100, or 1000 nM. Subsequently, slices were transferred to fresh medium for 3 h of substrate incubation with 500 μM 7EC. Results are means ± S.E.M. of slices of four rats. In each experiment three slices were incubated per treatment. Significant differences toward the activities of slices incubated with 0.5% DMSO are indicated: *, p < 0.05.
Slices of small intestine and colon were also incubated with 7HC after 0, 5, 8, and 24 h of preincubation with bNF and 3MC. Neither bNF nor 3MC influenced the conjugation rates of 7HC (data not shown).
IR. IR is reported to be an endogenous ligand for the AhR (Guengerich et al., 2004). Therefore, small intestinal slices were preincubated with indirubin at various concentrations (10, 100, and 1000 nM) for 24 h after which the metabolic rate of 7EC toward 7HC was determined (Fig. 3). A significant induction (6.1-fold compared with the DMSO control) was observed after 24 h of preincubation with 1000 nM IR (p < 0.05).
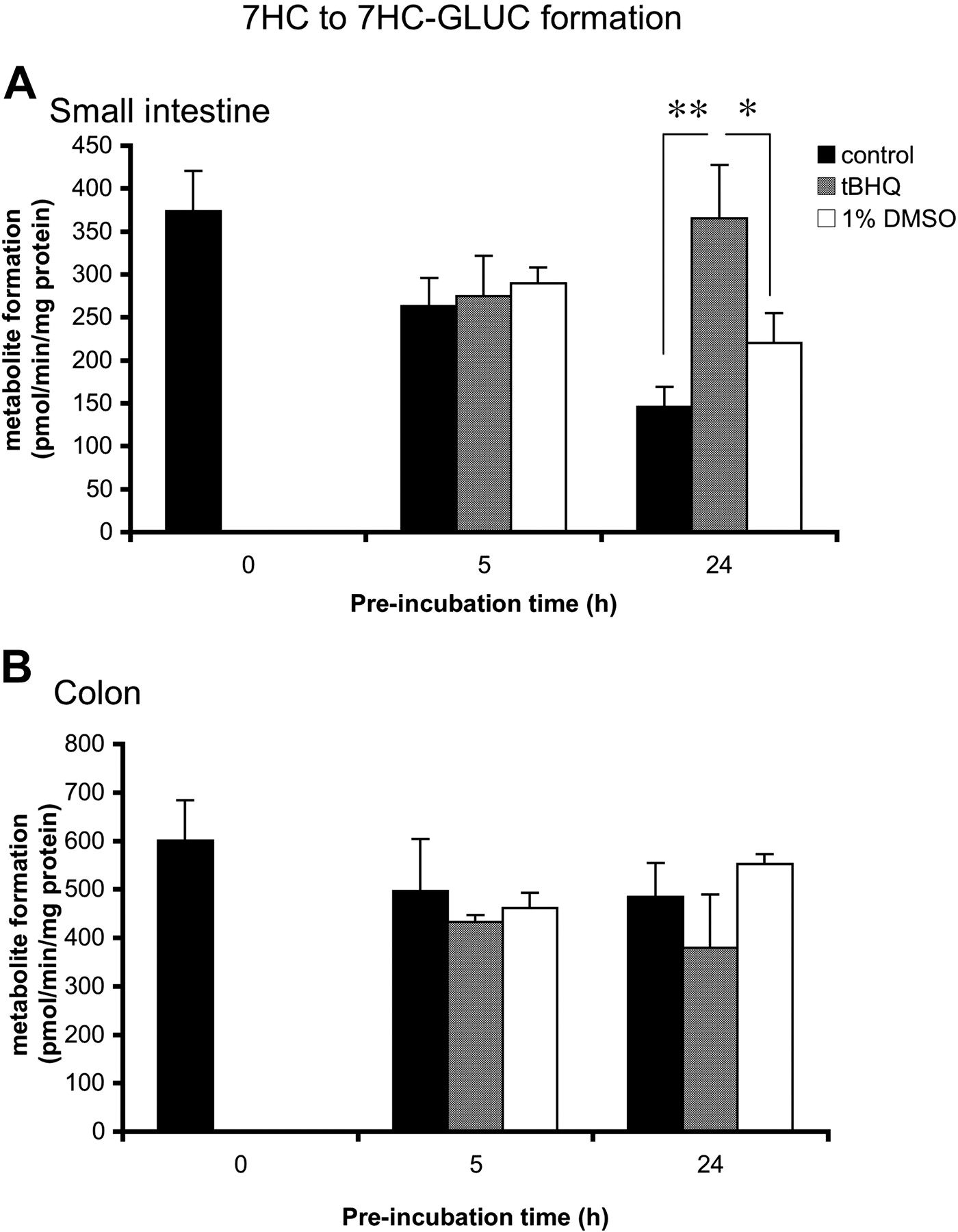
tBHQ. Some phase II enzymes are reported to be induced by tBHQ via the AhR pathway (Schreiber et al., 2006). To test the induction potential of tBHQ in rat intestine, we incubated slices (small intestine and colon) with tBHQ (50 μM) for 0, 5, and 24 h, after which slices were incubated for 3 h with 7HC. In small intestinal slices (Fig. 4A), a 1.7-fold induction was significant (p < 0.05) after 24 h of incubation, (24-h DMSO control, 220 ± 35 pmol/min/mg protein; 24-h induced levels, 365 ± 62 pmol/min/mg protein). No effect could be detected by tBHQ on 7HC glucuronidation after 5 h of incubation. In colon slices (Fig. 4B), no effect could be observed after preincubation with tBHQ. In addition, in both small intestinal and colon slices no effect was observed on 7HC sulfation (data not shown).
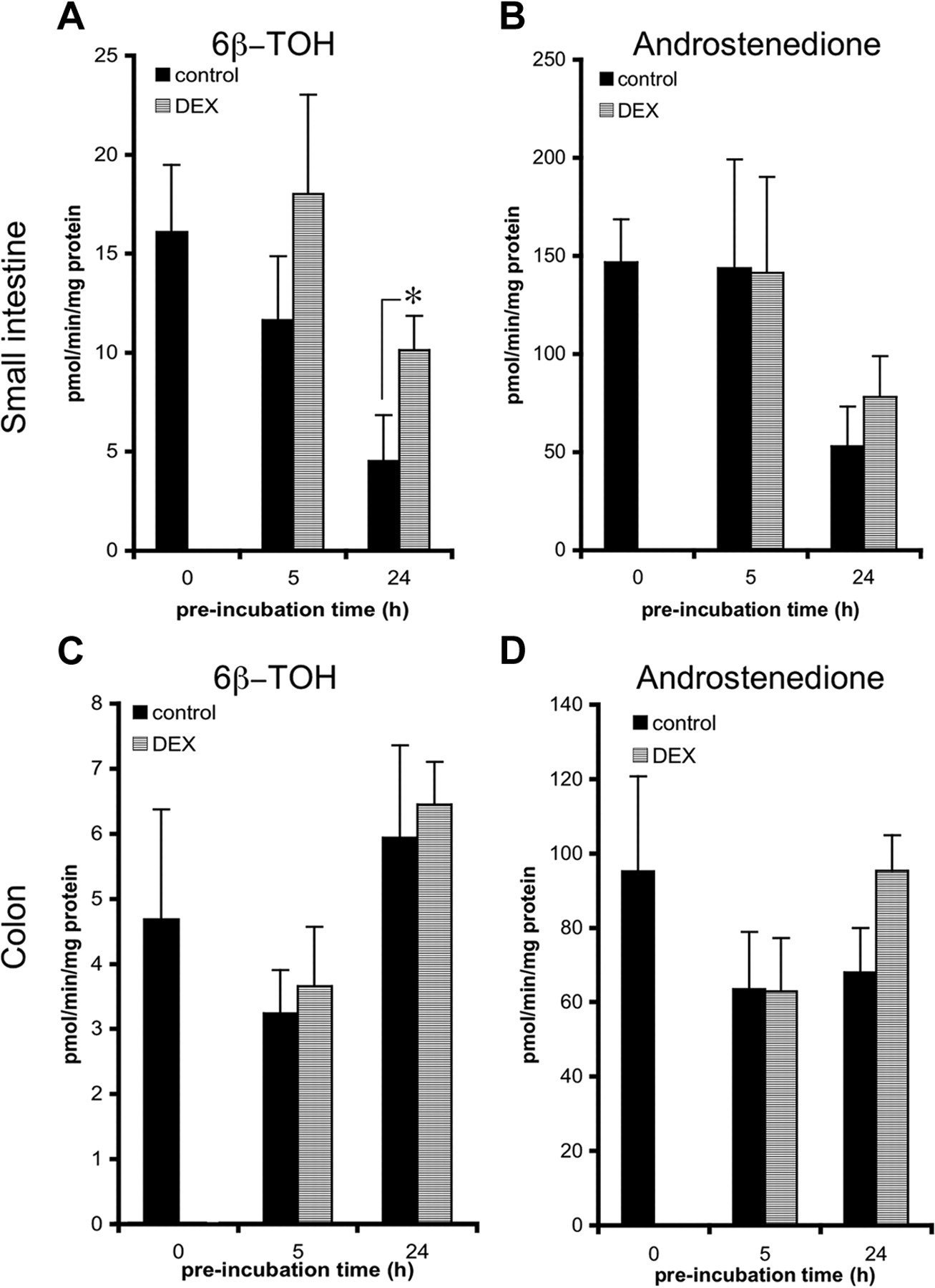
Dexamethasone. Small intestinal and colon slices were incubated with dexamethasone (100 μM) to investigate the ability of this compound to induce metabolic enzymes in precision-cut slices (Fig. 5). When control slices were incubated with TT, two TT metabolites, namely, 6β-TOH and androstenedione, were detected. When small intestinal slices were preincubated with DEX, the rate of 6β-TOH formation (Fig. 5A) increased 1.7-fold, from 11.6 (control) to 18 pmol/min/mg protein (p < 0.08, compared with the DMSO control) after 5 h and 2.3-fold, from 4.5 (control) to 10.1 pmol/min/mg protein (p < 0.05, compared with the DMSO control) after 24 h. For androstenedione formation (Fig. 5B), only a slight, nonsignificant increase was observed after 24 h of preincubation (from 53 to 78 pmol/min/mg protein). In colon slices (Fig. 5C/D), 6β-TOH formation was not significantly increased by DEX. However, in five of six experiments, a slight, but nonsignificant, increase of 6β-TOH formation was observed after 5 h of incubation. After a 24-h preincubation of colon slices with DEX, a small, nonsignificant, elevation of androstenedione formation from 68 to 95 pmol/min/mg protein was observed.
PB. Small intestinal and colon slices were incubated with PB (0, 2, 2.5, 4, and 8 mM) to study the influence of this compound on the metabolic conversion of TT, 7EC, and 7HC (Fig. 6). In small intestinal and colon slices, an effect of PB at 4 mM, but not at 2 mM, on 7HC glucuronidation was observed [p < 0.02, compared with control (0 mM after 24 h) and p < 0.04] (Fig. 6, A and F, respectively). Sulfation, however, was not affected (Fig. 6, B and G).
Doubling the concentration of PB to 8 mM drastically decreased the glucuronidation rate in small intestine and colon to 0.2 and 21%, respectively, and the sulfation rate to 60 and 7%, respectively. To investigate whether PB affected the viability of small intestinal slices, control incubations with 2, 4, and 8 mM PB were performed, and the intracellular ATP levels were measured afterward. Concentrations of 0, 2, and 4 mM PB had no effect on the ATP content (0.9 nmol ATP/mg protein), but ATP levels were drastically decreased when slices were incubated with 8 mM (0.2 nmol ATP/mg protein).

Induction of 7HC glucuronidation in precision-cut slices prepared from (A) small intestine (25-40 cm from stomach) and (B) colon. Slices were preincubated for 0, 5, and 24 h with control medium, 1% DMSO, or 50 μM tBHQ (1% DMSO) and subsequently transferred to fresh medium and incubated with 500 μM 7HC for 3 h. Results are means ± S.E.M. of slices of three to four rats. In each experiment three slices were incubated per treatment. Significant differences are indicated: *, p < 0.05; **, p < 0.01.
In small intestine and colon, PB (2.5 mM) induced the metabolic rates of 7HC formation (Fig. 6, C and H). Induction was detectable after 8 h (1.8-fold in small intestine and 2.9-fold in colon compared with control) and 24 h of preincubation (3.1-fold in small intestine and 4.1-fold in colon compared with control). Induction was only significant in colon (p < 0.05) after 8 h of preincubation. However, the induction in small intestine was clear, as prominent increases of metabolic rates were found in three of four experiments after 8 h and in three of three experiments after 24 h. PB (2.5 mM) did not influence the formation of 6β-TOH (Fig. 6, D and I) or androstenedione (Fig. 6, E and J).
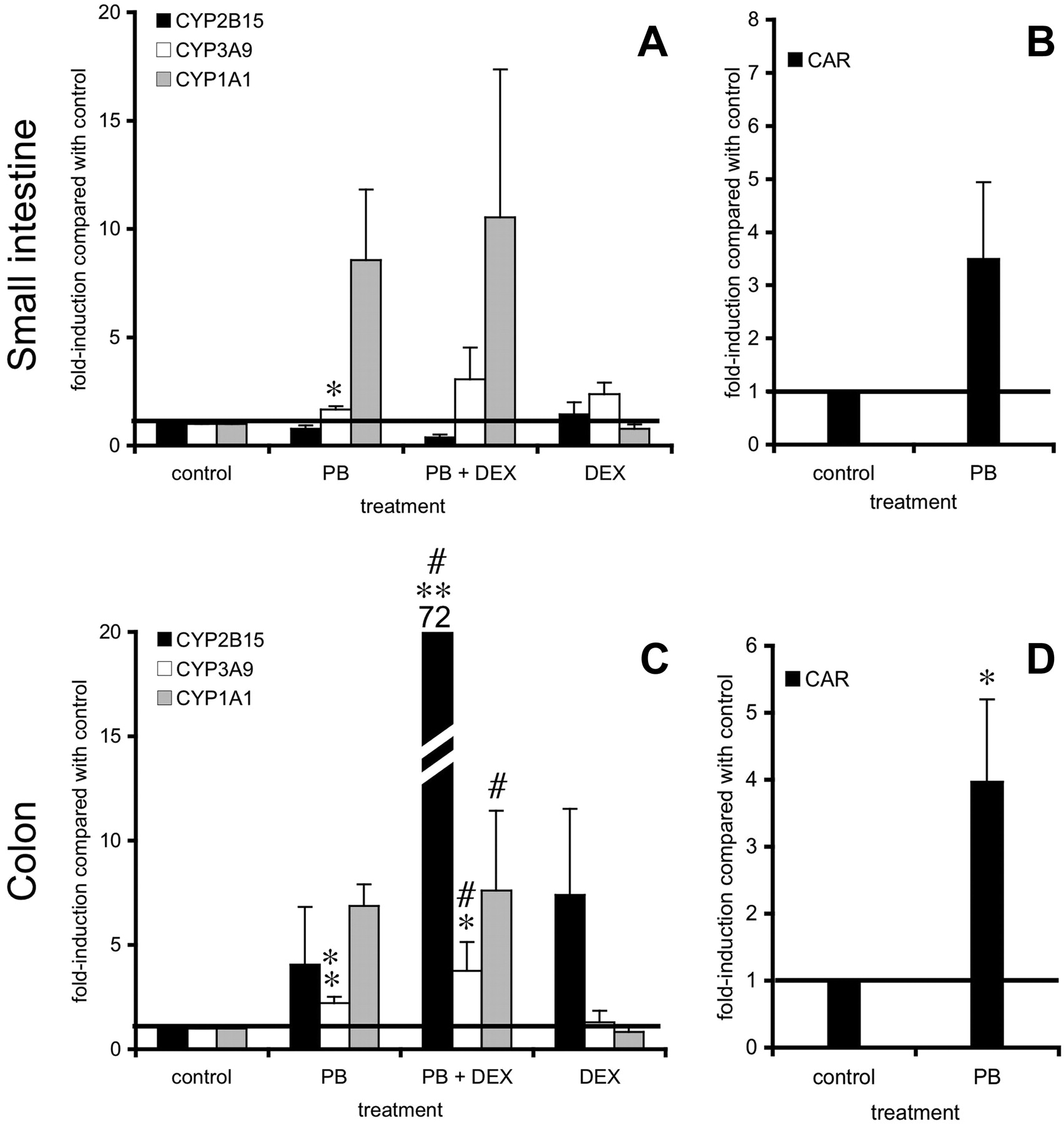
Gene expression during incubation. PB was expected to induce CYP2B. However, the formation of 16α- and 16β-TOH was below the detection limit in intestinal slices and remained undetectable after 24 h of preincubation with PB. Therefore, the presence of CAR and CYP2B15 mRNA was investigated during incubation. Slices were harvested after several incubation periods: 0, 5, 8, and 24 h in control medium. Slice preparation (small intestine and colon) did not influence the expression of CAR and CYP2B15 (data not shown). Incubation of small intestinal and colon slices for 24 h did not change CAR expression either. CYP2B15 mRNA expression, however, had already decreased in both tissues significantly after 5 h of incubation (small intestine, 12 ± 0.05% of control value with p < 0.05; colon, 28 ± 14% of control value with p < 0.05) and remained low up to 24 h of incubation (small intestine, 5 ± 0.1% of control value; colon, 41 ± 21% of control value).
In small intestine, PB did not induce the expression of CYP2B15 mRNA (Fig. 7A). In contrast, CYP3A9 (1.7-fold, p < 0.05) was significantly increased. CYP1A1 (8.6-fold) and CAR (3.5-fold) mRNA expressions were also induced in all individual experiments (Fig. 7, A and B). Addition of DEX to the incubation medium did not affect the inducing ability of PB for the cytochrome P450s tested. Furthermore, several pilot experiments were performed by incubating proximal jejunum slices for 5 h with either PB (4 mM) alone or with PB (4 mM), DEX (10 μM), insulin (1 μM), and 5% fetal calf serum as described by others (Martignoni et al., 2004) using proper controls without PB. All of the above-mentioned incubation conditions did not induce CYP2B15 mRNA levels (data not shown).
In colon slices (Fig. 7, C and D), however, CYP2B15 mRNA expression tended to increase after PB incubation in three of six experiments. The induction of CYP1A1 (7-fold), CYP3A9 (2-fold, p < 0.05), and CAR (4-fold, p < 0.05) expression was found in all experiments performed (n = 5-6). Medium supplementation with DEX (1 μM) significantly increased the induction of CYP2B15 from 4-up to 72-fold in colon. Furthermore, CYP3A9 and CYP1A1 induction remained significant after incubation with both PB (4 mM) and DEX (1 μM) compared with only DEX (1 μM) supplementation as a control.
Discussion
Recently, we presented rat intestinal precision-cut slices as a tool to study drug metabolism up to 3 h of incubation (de Kanter et al., 2005; van de Kerkhof et al., 2005). In the present study, we further investigated the applicability of intestinal slices up to 24 h of incubation for both metabolism and induction studies. Therefore, we evaluated the viability up to 24 h of incubation and treated slices up to 24 h with five prototypical inducers after which drug metabolism was evaluated.
Intracellular ATP levels are considered to be a proper measure for the overall viability of the tissue. The intracellular ATP levels of small intestinal and colon tissue directly after excision (in vivo levels) were 0.9 nmol/mg protein, but increased during slicing, which is similar to what was reported for rat liver and lung slices (De Kanter et al., 2002). This result suggests that ATP is also synthesized during the slicing procedure at 4°C in the presence of oxygen, which is in agreement with findings by others (Minor and Isselhard, 1996). The ATP levels in small intestine remained constant up to 8 h, after which it declined to 25% of control values after 24 h, but it was never lower than the tissue value.
Furthermore, the viability of the enterocytes (metabolizing cells) was studied in more detail. The AP activity in slices remained constant up to 24 h. In addition, the increase of enzyme activity in medium indicates that the cells are capable of de novo synthesis of AP during incubation. In addition, villin expression was evaluated during incubation, as it is commonly considered the housekeeping gene for enterocytes (Engman et al., 2001) in which it is exclusively expressed (West et al., 1988). Assuming that 1) the protein content is a measure for the amount of tissue in the slice, 2) the expression of villin is directly correlated to the amount of enterocytes in the slice, and taking into account the fact that 3) GAPDH remained constant in both small intestinal and colon slices up to 24 h, our results indicate that in colon slices no cells are lost up to 24 h, because all parameters that were tested remained constant (villin and GAPDH expression, the amount of protein per slice, and ATP and activity levels). In small intestinal slices, on the basis of the decreased protein content, it seemed that some cells are lost during the first 8 h. The constant levels of villin, GAPDH, and metabolic rates expressed per milligram of protein indicate that this occurs to the same extent for all cell types present in the slice. After 8 h of incubation, however, small intestinal slices have lost more enterocytes in relation to other tissue cells, as judged by the decreased villin expression per total RNA. The protein content of slices decreased to ∼50% after 24 h. The activity per milligram of protein decreased after 8 h of incubation, but remained clearly detectable after 24 h. The decline was different for the various metabolic reactions tested, and this might be explained by either a lack of endogenous stimuli present in the incubation medium (“de-induction”) and/or differences in half-lives of various isoenzymes, being in agreement with earlier findings by others in rat liver slices (Renwick et al., 2000) and cultured rat hepatocytes (McMillan et al., 1991). The decrease in villin can at least be partly explained by the normal apoptosis of enterocytes, as the in vivo lifespan of these cells is ∼2 days (Kaminsky and Zhang, 2003). Apparently, in the slices proliferation cannot compensate for the loss of cells by apoptosis. However, further research is needed to support this hypothesis.

Induction of TT conversion to 6β-TOH (A and C) and androstenedione (B and D) in precision-cut slices prepared from small intestine (25-40 cm from stomach) (A and B) and colon (C and D). Slices were incubated for 0, 5, and 24 h with control medium, 0.5% DMSO, or 100 μM DEX and subsequently transferred to fresh medium and incubated with 250 μM TT for 3 h. Results are means ± S.E.M. of slices of three to six rats. In each experiment three slices were incubated per treatment. Significant differences toward the activities of slices are indicated: *, p < 0.05.
Summarizing these data, colon slices remain viable up to 24 h of incubation; in small intestinal slices, the ATP content and the amount of enterocytes per slice decrease after 8 h. In addition, some metabolic activity is lost after 8 h but remains clearly detectable after 24 h of incubation. Therefore, small intestinal slices can be used for quantitative metabolism studies up to 8 to 11 h and for qualitative studies up to 24 to 27 h. The observed decrease in metabolic rate is in accordance with those found in hepatocytes, but, as in hepatocyte studies, this decrease does not necessarily impede detection of induction by inducing drugs (Wortelboer et al., 1991).
Slices were incubated with model inducers to test the applicability of slices for induction studies. In a recent study, we showed the induction of 7EC O-deethylation after 24 h of bNF exposure in intestinal slices (van de Kerkhof et al., 2005). In the current study, induction of 7EC O-deethylation (CYP1A) was observed with bNF, 3MC (small intestine and colon), and IR (small intestine), well known AhR ligands (Lin and Lu, 1998; Guengerich et al., 2004; Abdelrahim et al., 2006). This finding is in line with published in vivo studies, in which orally administered bNF or 3MC induced 7EC O-deethylation 2- to 100-fold in small intestine (McDanell and McLean, 1984; Rosenberg, 1991; Spatzenegger et al., 2000).
In our in vitro studies, induction (at activity level) was already clearly detectable after 5 h in small intestinal slices and is in line with in vivo studies showing CYP1A1 induction in rat small intestine readily at 3 h (Zhang et al., 1997) and 12 h after bNF administration (Zhang et al., 1996). Unfortunately, no such data are available for colon tissue, which impedes comparison with results of the present study.
The AhR pathway is known to be involved in the induction of both phase I (Abdelrahim et al., 2006) and II metabolism (Auyeung et al., 2003). tBHQ (AhR ligand) (Schreiber et al., 2006) has been reported to induce UGT activity in Caco-2 cells (Munzel et al., 1999), and, indeed, it induced glucuronidation in small intestine but not in colon slices. However, two other AhR ligands, 3MC and bNF, did not induce 7HC conjugation via UGT1A6 (Ikushiro et al., 2004) or UGT1A7/8 (Webb et al., 2005), which is in line with reported findings that UGT1A6-8 mRNA expression was not induced in rat duodenum after in vivo administration of bNF (Shelby and Klaassen, 2006).

Influence of PB on 7HC-glucuronidation and 7EC and TT metabolism in precision-cut slices prepared from small intestine (25-40 cm from stomach) (A-E) and colon (F-J). Slices were preincubated for 0 and 24 h with control medium, 2 or 4 mM PB and transferred to fresh medium with 7HC (500 μM) (A and B and F and G) for 3 h of substrate incubation. Slices were preincubated for 0, 5, 8, and 24 h with 2.5 mM PB and then transferred to fresh medium containing either 7EC (500 μM) (C and H) or TT (250 μM) (C and E and I and J) for 3 h of additional incubation. Results are means ± S.E.M. of slices of three to six rats. In each experiment three slices were incubated per treatment. Significant differences toward 24 h control are indicated: *, p < 0.05.
Dexamethasone, a known rodent PXR agonist at the concentration used, induces CYP3A in rat (Savas et al., 1999). In the present study, induction of 6β-TOH formation by DEX was observed readily after 5 h (although not significant) and 24 h, which is in agreement with in vivo studies showing the induction of the CYP3A1/2 protein (Zhang et al., 1996) and mRNA (Martignoni et al., 2004). In colon tissue, 6β-TOH formation was slightly but nonsignificantly induced after 5 and 24 h in five of six experiments. Possibly it may take longer than 24 h to induce CYP3A activity in colon as was reported in Caco-2 cells (Cummins et al., 2001).
PB induces DMEs via CAR (Wang and LeCluyse, 2003). In the present study, 7HC glucuronidation was induced by PB in both organs. In contrast to our findings, in vivo administered PB was reported not to induce UGT1A6-8 in rat duodenum (Shelby and Klaassen, 2006). 7EC O-deethylation (CYP1A) was induced by PB (2.5 mM) in both small intestine and colon, which is in line with findings in hepatocytes, showing CYP1A1 induction by PB (Sadar et al., 1996).
Testosterone appeared not to be a good substrate for CYP2B metabolism in intestinal slices, and, therefore, mRNA expression was examined. However, no induction of CYP2B15 expression was observed, despite the continuous expression of CAR during 24 h of incubation. In contrast, induction of CYP1A1 (confirming our activity data), CYP3A9, and CAR mRNA was observed in both organs.
In rat hepatocytes, it was shown that addition of DEX (100 nM) to the medium increased the inducibility of CYP2B (Ringel et al., 2002). In the present study in colon but not in small intestinal slices, DEX appeared to enormously induce CYP2B15 mRNA. This suggests that PB-mediated induction is differentially regulated in rat small intestine and colon, but this hypothesis should be confirmed in further research.
In this study, induction was detected after 5, 8, and/or 24 h of exposure dependent on the inducer. Induction after 3MC and bNF was readily detectable after 5 h and the highest after 8 h, but induction after tBHQ and DEX was only detectable after 24 h of incubation. These data suggest that for proper detection of the inducing capacity of drugs, intestinal precision-cut slices should be incubated with these drugs for several incubation times.

Gene expression of small intestinal (25-40 cm from stomach) (A and B) and colon (C and D) slices after 24 h of incubation with control medium, PB (4 mM), PB (4 mM) + DEX (1 μM), or DEX (1 μM). Several genes were studied: CYP2B15, CYP3A9, CYP1A1 (A and C) and CAR (B and D). Slice incubation expression is corrected for villin and the control values were set at 1. The horizontal line indicates control levels (fold induction = 1). Results are means ± S.E.M. of four to six experiments. In each experiment six slices were incubated per treatment and harvested together. Significant differences toward 24-h control medium are indicated: *, p < 0.05; **, p < 0.01. Significant differences between PB + DEX and DEX incubations are indicated: #, p < 0.05.
To conclude, colon precision-cut slices are a useful tool for quantitative drug metabolism studies up to 24 to 27 h, whereas slices of small intestine can be used to measure drug metabolism quantitatively up to 8 to 11 h and qualitatively up to 24 to 27 h. The current study demonstrates that this model is also very suitable to study drug-induced induction of metabolism in vitro in the intestine, as the AhR, PXR, and CAR pathways are functional in both small intestinal and colon slices. This model provides an opportunity to further investigate mechanisms of induction in various regions of the intestine. Moreover, when applied also to human tissue, it may predict and increase our understanding of drug-induced changes in intestinal metabolism and bioavailability in patients.
Acknowledgments
We thank Prof. Dr. D. K. F. Meijer for excellent advice and critical reading of the manuscript.
Footnotes
-
doi:10.1124/dmd.106.014563.
-
This study was supported by the Technology Foundation STW, the Applied Science Division of NWO (the Dutch organization for scientific research), and the technology programme of the Ministry of Economic Affairs and by Yamanouchi Europe.
-
ABBREVIATIONS: DME, drug-metabolizing enzyme; bNF, β-naphthoflavone; GADPH, glyceraldehyde 3-phosphate dehydrogenase; 7EC, 7-ethoxycoumarin; TT, testosterone; 7HC, 7-hydroxycoumarin; CAR, constitutive androstane receptor; PXR, pregnene X receptor; AhR, aryl hydrocarbon receptor; 3MC, 3-methylcholanthrene; IR, indirubin; tBHQ, tert-butylhydroquinone; DEX, dexamethasone; GR, glucocorticoid receptor; PB, phenobarbital; TOH, hydroxytestosterone; 7HC-GLUC, 7-hydroxycoumarin glucuronide; DMSO, dimethyl sulfoxide; 7HC-SULF, 7-hydroxycoumarin sulfate; AP, alkaline phosphatase; PCR, polymerase chain reaction; Ct value, cycle threshold value; UGT, UDP glucuronosyltransferase.
- Received December 22, 2006.
- Accepted March 1, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}