


Abstract
Excretion and metabolism of zoniporide were investigated in humans after intravenous infusion of [14C]zoniporide at an 80-mg dose. Bile was the primary route of excretion because 57% of dose was recovered in the feces after intravenous infusion. Zoniporide was primarily cleared via metabolism in humans. 2-Oxozoniporide (M1) was the major excretory and circulating metabolite in humans and was catalyzed by aldehyde oxidase (Km of 3.4 μM and Vmax of 74 pmol/min/mg protein). Metabolites M2 (17% of the dose) and M3 (6.4% of circulating radioactivity), in which the guanidine moiety was hydrolyzed to a carboxylic acid, were also detected in human feces and plasma, respectively, suggesting that hydrolysis was another route of metabolism of zoniporide in humans. The metabolism and excretion of [14C]zoniporide in rats and dogs were also evaluated. As in humans, bile was the primary route of excretion of the radiolabeled material in both species, and metabolism was the primary route of clearance. A comparison of plasma metabolites showed that for M3, rats had a higher concentration than human or dog. M1 was absent in dog and present in human and rat plasma at comparable levels, whereas comparison of excreta showed that the total body burden of M1 was greater in rat than that in human. No further evaluation of M2 was considered because it was detected only in the human fecal extracts. Hence, no further toxicological evaluation of the three human metabolites was undertaken.
Zoniporide [1-(quinolin-5-yl)-5-cyclopropyl-1H-pyrazole-4-carbonyl guanidine] (Fig. 1) was designed and synthesized as a highly selective inhibitor of the sodium/hydrogen exchanger (NHE-1) (Guzman-Perez et al., 2001; Knight et al., 2001; Marala et al., 2002; Tracey et al., 2003) and was being developed to reduce the perioperative myocardial ischemic injury in high-risk surgery patients. It inhibited 22Na+ uptake in fibroblasts expressing human NHE-1 in a concentration-dependent manner with an IC50 of 14 nM and had >150-fold selectivity over other NHE isoforms (Marala et al., 2002). Zoniporide also produced a reduction in infarct size and decreased the incidence and duration of potentially lethal arrhythmias after intravenous administration to animal models of ischemic-reperfusion injury (Knight et al., 2001; Tracey et al., 2003).

Structure of [14C]zoniporide. ∗, position of the radiolabel.
Preclinical pharmacokinetic studies of zoniporide in mice, rats, rabbits, dogs, and monkeys demonstrated that its clearance (Clp) was moderate to high in all species. The volume of distribution (Vd) was moderate, and the plasma t1/2 was 1.5 h or shorter in all species evaluated. There was a linear increase in systemic exposure of zoniporide in healthy male human volunteers, after a 1-h infusion of doses ranging from 0.003 to 1.0 mg/kg, as assessed by Cmax and the area under plasma concentration versus time curve (AUC0–tlast). The Cmax and AUC0–tlast ranged from 2 to 622 ng/ml and from 2 to 930 ng-h/ml, respectively, at these doses. The Clp and Vd were independent of dose, and the mean t1/2 was approximately 3 h. The most common adverse events reported by subjects receiving zoniporide were somnolence and postural hypotension. Other adverse events included nausea, vomiting, and headache.
In drug development, it is important to study the metabolism and the routes of excretion of a drug candidate in humans. Furthermore, assurance that all major circulating metabolites observed in humans are present in at least one animal species used for safety assessment provides greater confidence that animal toxicology studies are relevant for human safety (Baillie et al., 2002; Smith and Obach, 2005, 2006; Davis-Bruno and Atrakchi, 2006; Atrakchi, 2009). The recent Food and Drug Administration (2008) guidance on metabolites in safety testing recommends that human circulating metabolites exceeding 10% of the parent should be present in at least equal quantities in at least one of the preclinical species used in toxicological assessment (Davis-Bruno and Atrakchi 2006; Atrakchi 2009). Direct testing in animals is warranted for human metabolites that are absent or present in disproportionately lower amounts in preclinical species used for toxicology studies.
Conventionally, an assessment of major metabolites in humans and preclinical species is accomplished by definitive absorption, distribution, metabolism, and excretion studies in which radiolabeled drugs are administered and biological fluids are evaluated for a comprehensive and quantitative profile of metabolites. The current study was performed to compare the metabolism and excretion of zoniporide in humans and animal species used in toxicological testing using radiolabeled zoniporide. [14C]Zoniporide (Fig. 1) was administered to four young healthy male volunteers by intravenous infusion for 1 h, and the excreta and plasma were collected to assess the mass balance, routes of excretion, and circulating metabolites of zoniporide in humans. To assess the coverage of all human metabolites in toxicology species, [14C]zoniporide was also administered to Sprague-Dawley rats and Beagle dogs, and the metabolites in the excreta and in circulation were also identified to determine the major metabolic routes of zoniporide. In addition, the characterization of the enzymes responsible for the formation of M1 in humans and the enzyme kinetics for the formation of M1 were also evaluated in vitro in this study using human subcellular fractions.
Materials and Methods
Reference Compounds, Radiolabel Zoniporide, and Chemicals.
All synthetic standards for the metabolites were synthesized at Pfizer Global Research and Development (Groton, CT) using standard procedures. [14C]Zoniporide mesylate was synthesized by the radiochemistry group at Pfizer Global Research in Groton under good manufacturing practice conditions. The label was located in the pyrazole ring of the molecule (Fig. 1). The purity of the radiolabeled material was >99%. For animal studies, [14C]zoniporide (radiochemical purity 99.4%, specific activity was 26.6 mCi/mmol) was synthesized by the radiochemistry group at Pfizer Global Research in Groton. All other reagents and solvents used in the studies were of the highest grade available and were obtained from commercial sources. EcoLite(+) scintillation cocktail was obtained from ICN Pharmaceuticals (Irvine, CA). Carbo-Sorb and Permafluor E+ scintillation cocktails were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA).
Study Design, Dosing, and Sample Collection in Humans.
This was an open-label, single-dose inpatient study conducted with four nonsmoking healthy male human volunteers aged between 18 and 55 years and weighing between 74 and 79 kg. Before the study started, an institutional review committee approved the protocol and the informed consent document. All study participants gave written informed consent before initiation of the study.
All volunteers were administered a single 80-mg dose of [14C]zoniporide mesylate (100 μCi) by intravenous infusion over 1 h in water at a 10 mg/ml concentration. The specific activity of the dose was 1.25 μCi/mg. Urine was collected into containers surrounded by dry ice at predose (−8 to 0), 0 to 12, and 12 to 24 h and at 24-h intervals during the study through 144 h postdose (8 days). Feces were collected before dosing and then over 24-h intervals up to 144 h postdose (8 days). The total weight of the urine and feces was recorded after each collection. Blood samples (sufficient to provide 6 ml of plasma) were collected for pharmacokinetic evaluation of total radioactivity and zoniporide at times 0 (just before dosing) and at 0.5, 1, 2, 4, 8, 12, 24, 36, 48, and 24 h after the start of the infusion. Subsequent samples were taken at 24-h intervals through 144 h. Additional blood sufficient to provide a minimum of 20 ml of plasma was collected at 1, 4, 8, and 12 h postdose from the start of the infusion for characterization of metabolites. All samples were collected in heparinized tubes and centrifuged at approximately 4°C. Samples were stored frozen until the day of analysis.
Animal Studies.
All studies were conducted in a research facility accredited by the American Association for the Accreditation of Laboratory Animal Care. All study animals were acclimated to standard housing and environmental conditions in metabolism cages and rooms where light cycles, temperature, and humidity were documented daily for 2 days before the experiments.
Rats.
Three male and three female Sprague-Dawley rats were housed individually in Nalgene metabolism cages and fasted overnight. A single dose of [14C]zoniporide was administered to all animals intravenously at a dose of 10 mg/kg containing 50 μCi of radioactivity. The dose was prepared by dissolving [14C]zoniporide in 20% SBE-β-cyclodextrin at a concentration of 2.1 mg/ml. The specific activity of the dose was 28.8 μCi/mg. Urine was collected into containers surrounded by dry ice at predose (−24 to 0), 0 to 8, and 8 to 24 h and at 24-h intervals during the study and feces over 24-h intervals, up to 168 h postdose. After the last urine and fecal collection, the cages were washed with 1:1 ethanol-water (v/v), and the wash was collected. The weight of each fecal sample, cage wash, and cage debris was determined. All animals were euthanized by CO2 asphyxiation at the end of the study. For pharmacokinetic studies, six Sprague-Dawley rats (n = 3/sex) were dosed intravenously with 10 mg/kg [14C]zoniporide. The dose was prepared by mixing unlabeled and radiolabeled zoniporide in 20% w/v SBE-β-cyclodextrin. The concentration and specific activity of the dose was 3.34 mg/ml and 7.46 μCi/mg, respectively. Blood samples were collected in heparinized tubes at times 0 (just before dosing) 3, 15, 30, 60, 120, 240, 360, and 480 min. After sampling, whole blood was centrifuged at 14,000 rpm for 3 min, and plasma was transferred to Eppendorf snap-capped tubes and stored at −20°C until assay. For identification of circulating metabolites, a separate group (two animals, one per sex, per each time point) of animals were dosed intravenously at a dose of 10 mg/kg [14C]zoniporide (containing 75 μCi of radioactivity). The specific activity of the dose was 30 μCi/mg. Blood was collected at 1, 2, 4, 8, and 24 h by sacrificing the rats at each sampling time. Samples were collected in heparinized tubes and centrifuged at approximately 4°C. All samples were stored frozen at −20°C until the day of analysis.
Dogs.
Four noncannulated (two per gender) Beagle dogs were housed individually in stainless steel metabolism cages and administered [14C]zoniporide by intravenous administration at a dose of 3 mg/kg (containing 150 μCi of radioactivity). The dose was prepared by dissolving the salt of unlabeled and labeled zoniporide (specific activity 26.6 mCi/mmol) into 20 ml of 20% SBE-β-cyclodextrin. The specific activity of the dose was 5 μCi/mg. Urine was collected in containers surrounded by dry ice at predose (−12 to 0), 0 to 8, and 8 to 24 h and at 24-h intervals during the study through 168 h postdose. Feces were collected before dosing and then over 24-h intervals, up to 168 h postdose. The total weight of urine and feces was recorded after each collection. Blood samples were collected for pharmacokinetic evaluation of zoniporide and total radioactivity at times 0 (just before dosing) 0.5, 1, 2, 4, 8, 12, and 24 and at 24-h intervals through 168 h postdose. For characterization of circulating metabolites, additional blood was collected at 0.5, 1, 2, 4, 8, and 12 h after an intravenous dose. Samples were collected in heparinized tubes and centrifuged at approximately 4°C. All samples were stored frozen at −20°C until the day of analysis.
Quantitation of Radioactivity.
Radioactivity in the plasma, urine, and feces was determined by liquid scintillation counting. Aliquots of plasma (50–100 μl) and urine (100–500 μl) were mixed with EcoLite(+) scintillation cocktail (6 ml) and counted in triplicate in a model L 1409 DSA liquid scintillation counter (PerkinElmer Life and Analytical Sciences–Wallac Oy, Turku, Finland). For determination of radioactivity in feces, the weight of each fecal sample was determined, and the samples were homogenized in 2 parts of deionized water using a Stomacher blender 400. After homogenization, triplicate aliquots (250–500 μl) of each sample were transferred into tared cones and pads, weighed, and combusted in an automatic sample PerkinElmer 308 oxidizer (PerkinElmer Life and Analytical Sciences). The resulting 14CO2 was trapped in Carbo-Sorb and mixed with Permafluor E+ scintillation fluid, and the radioactivity was quantified by liquid scintillation counting. Radioactivity less than twice the background value was considered to be below the limit of determination. Samples collected before dosing were used as controls and counted to obtain a background count rate.
The radioactivity in the plasma was expressed as nanogram-equivalents of zoniporide per milliliter. The compound equivalents were determined by dividing the microcuries per milligram of sample by the specific activity of the compound (1.25 μCi/mg). Samples containing radioactivity (disintegrations per minute) less than or equal to twice the background were considered to be zero in the calculation of the means. Radioactivity in the urine and feces was expressed as a percentage of the administered dose per time interval.
Quantitation of Zoniporide in Human, Rat, and Dog Plasma.
Plasma samples were analyzed for zoniporide using a validated liquid chromatography-tandem mass spectrometry method. A 200-μl aliquot of each human plasma sample or a 50-μl sample from rat and dog plasma was treated with 100 μl of a solution of an internal standard (100 μg/ml [15N3]zoniporide). The samples were basified with sodium carbonate solution, and the analytes were extracted with methyl tert-butyl ether. The organic layer was separated and evaporated to dryness, and the residue was reconstituted with 200 μl of a mixture of acetonitrile and 2 mM ammonium acetate (15:85), the extracts were analyzed using a PE Sciex API 3000 mass spectrometer with a TurboIonSpray source (PerkinElmerSciex Instruments, Waltham, MA). The analytes were separated chromatographically using a Betabasic C18 column (100 × 2 mm, 5 μm) and detected in a positive ion mode using the protonated molecular ion as the precursor ion monitored at m/z 321.0 > 262.0 for zoniporide and at m/z 324.0 > 262.0 for the internal standard. Data collection and integration were performed using Sample Control and MacQuan version 1.5 software (MDS Sciex, Concord, ON, Canada). Quantitation was based on a linear least-squares regression analysis of calibration curves weighted 1/x2 using the area ratio versus concentration. The dynamic range of the assay was 1.00 to 500 ng/ml for both zoniporide and M1.
Determination of the Pharmacokinetic Parameters.
Pharmacokinetic parameters were determined by noncompartmental methods from individual concentration time profiles after intravenous infusion using WinNonlin (version 3.2; Pharsight, Mountain View, CA). The maximum plasma concentration (Cmax) and the time at which this concentration was achieved (Tmax, at the end of the infusion) were taken directly from the concentration data. The AUC0–tlast was calculated from 0 to the last quantifiable time point (tlast), using log-linear trapezoidal approximation. The plasma terminal elimination rate constant (kel) was estimated by linear regression analysis of the terminal slope of log plasma concentration-time curve. The terminal elimination half-life (t1/2) was estimated as ln2/kel. The total clearance (CL) was determined by the ratio CL = dose/AUC0–∞. The area under the plasma concentration-time curve from zero to ∞ (AUC0–∞) was estimated as the sum of AUC0–tlast and AUCtlast–∞. The AUCtlast–∞ was estimated from Ctlast/kel, where Ctlast represented the estimated plasma concentration at Tlast, based on the aforementioned regression analysis. The volume of distribution (Vss) was determined by CL · MRT, with MRT being the mean residence time after intravenous infusion.
Metabolic Profiling.
Urine.
Urine was pooled from 0 to 24 h so that >90% of the drug-related material excreted in urine was accounted for. The pooled urine samples were lyophilized overnight, and the residue was dissolved in 1 ml of water. The pooling was proportional to the volume of urine collected at each time point. The aqueous layer was centrifuged, and an aliquot (100 μl) was injected onto a column.
Plasma.
Plasma samples obtained were pooled to account for >90% of the radioactivity using the method reported by Hamilton et al. (1981) for profiling of circulating metabolites. The pooled samples were treated with 5 parts of acetonitrile to 1 part of plasma. The mixture was then centrifuged, and the supernatant was transferred to another tube. The pellets were washed once more to ensure that >90% of the radioactivity was recovered. The supernatants were mixed and evaporated to dryness in a TurboVap at 35°C under nitrogen. The residues were reconstituted in 100 μl of 5 mM ammonium formate (pH 3), and an aliquot (100 μl) was injected onto the column. Aliquots (30 μl) of the reconstituted samples were also counted on the liquid scintillation counter to determine the radioactivity extraction recovery.
Feces.
Fecal homogenates were pooled on a weight basis to account for 90% or greater of the drug-related material excreted in feces. Each pooled fecal sample was diluted with 30 ml of acetonitrile and vortexed. The sample was then centrifuged, and the supernatant was separated. The process was repeated several times until >90% of the radioactivity was extracted. All supernatants were mixed and evaporated to approximately 1 ml in a TurboVap at 35°C under nitrogen. The concentrated residue was extracted with 10 ml of hexane to remove all lipophilic material, and the aqueous layer was evaporated to dryness in a TurboVap at 35°C under nitrogen. The residue obtained was reconstituted in ∼300 μl of 3 mM ammonium formate solution, and a small sample was analyzed by liquid scintillation counting for radioactivity extraction recovery. A 100-μl aliquot of the reconstituted sample was injected onto the column.
Separation, Quantification, and Identification of Metabolites.
Metabolic profiling was performed using the HPLC system that consisted of an HP-1100 membrane degasser, HP-1100 autoinjector, and HP-1100 binary gradient pump (Agilent Technologies, Palo Alto, CA). Chromatography was performed on a Zorbax C8 column (5 μm, 4.5 × 150 mm) by injecting 100 μl of the reconstituted sample. The mobile phase was initially composed of acetonitrile (solvent B) and 10 mM ammonium formate (pH 3.0) (solvent A). The flow rate was 1.0 ml/min, and separation was achieved at ambient temperature. A 50-min gradient was used as follows: 0 to 10 min, 5% B; 10 to 20 min, 10% B; 20 to 35 min, 20% B; 35 to 40 min, 20% B; 40 to 44 min, 80% B; and 44 to 45 min, 5% B. The column was reequilibrated at 5% B for the next 5 min before next injection. The postcolumn eluate was split such that 95% of the flow was monitored continuously with a RAM fitted with a liquid scintillation cell (IN/US Systems, Riviera Beach, FL). The remaining 5% of the flow was diverted to the PE Sciex API 3000 triple quadrupole mass spectrometer. The RAM was operated in the homogeneous liquid scintillation counting mode with the addition of 3 ml/min of Tru-Count scintillation cocktail (IN/US Systems) to the effluent. The RAM response was recorded as a real-time analog signal by the MS data collection system.
The metabolites in the urine and feces were quantified by measuring radioactivity in the individually separated peaks in the radiochromatogram using Winflow software (IN/US Systems). The RAM provided an integrated printout in counts per minute and percentage of the radiolabeled material. The circulating metabolites were detected by an LC-ARC (AIM Research, Hockessen, DE). The LC-ARC was operated in the homogeneous liquid scintillation counting mode with the addition of 2.5 ml/min of Tru-Count scintillation cocktail (IN/US Systems) to the effluent. The circulating metabolites were quantified using the LC-ARC software (AIM Research) by measuring the radioactivity in the chromatographically separated peaks.
The metabolites were identified using a PE Sciex API 3000 mass spectrometer equipped with an electrospray ion source operated in the positive ion mode. The instrument settings and potentials (e.g., collision energy) were adjusted to provide optimal data for zoniporide. The PE Sciex API 3000 mass spectrometer was operated at an ionspray voltage of 4000 V and orifice voltage of 36 V. The collision-induced dissociation (CID) studies (precursor ion scan or product ion scan) were performed using nitrogen gas at a collision energy of 30 V and the collision gas thickness of 4 × 1014 molecules/cm2. The MS data were analyzed by MultiView 1.4 software (PerkinElmerSciex Instruments). The metabolites were identified using Q1 (full scan), neutral loss, and precursor ion-scanning techniques. The structures of metabolites were identified using a product ion scan of the molecular ions that were identified in the above scanning modes and multiple reaction monitoring scanning.
In Vitro Metabolism of Zoniporide.
Zoniporide (10 μM) was incubated with human liver S9 fractions (protein concentration 2.5 mg/ml), MgCl2 (3.3 mM), in the presence of NADPH (3.0 mM) in a total volume of 1.0 ml of potassium phosphate (0.1 M, pH 7.4). Incubations were started by addition of NADPH and shaken in a water bath set at 37°C. Control experiments were carried out in a similar manner except that the NADPH solution was substituted with phosphate buffer. After 1 h, the incubations were quenched with acetonitrile (5 ml) and centrifuged, and the supernatant was evaporated to dryness in a TurboVap under nitrogen. The residue was reconstituted in 200 μl of acetonitrile and water mixture (1:3), and a 25-μl aliquot was injected onto the column for the identification of the oxidation products. Enzyme phenotying studies with zoniporide were carried out in a similar manner except that the incubation mixture was first treated with raloxifene (10 μM), an inhibitor of aldehyde oxidase, before addition of the substrate.
The metabolites in the in vitro samples were identified by a Finnigan LCQ Deca XP ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) equipped with an electrospray ion source. The mass spectrometer was operated in a positive ion mode, and the operating parameters for the ion trap were as follows: capillary temperature, 270°C; spray voltage, 4.0 kV; capillary voltage, 4.0 V; sheath gas flow rate, 90; and auxiliary gas flow rate, 30. The mass spectrometer was operated in a positive ion mode with data-dependent scanning. The ions were monitored over a full mass range of m/z 125 to 1000. For data-dependent scanning, the normalized collision energy was 40%. The MS data were analyzed using Xcalibur software (version 1.4; Thermo Fisher Scientific).
Enzyme Kinetics.
Zoniporide (1–500 μM) was incubated with pooled human liver cytosol (0.2 mg/ml; Sigma-Aldrich, St. Louis, MO) and EDTA (0.1 mM) in 0.2 ml of 25 mM phosphate buffer at 37°C for 60 min. (Preliminary experiments demonstrated that the protein concentration and incubation time used provided linear reaction velocity.) The incubations were commenced with addition of cytosol and terminated with the addition of 0.05 ml of 1 M formic acid. The terminated incubation mixtures were filtered through a Millipore Multiscreen-HA filter plate (0.45 μm), and filtrates were analyzed by HPLC-MS. Filtered incubation mixtures were injected (0.04 ml) onto a Luna C18 column (2.5 × 50 mm, 5 μ; Phenomenex, Torrance, CA) preequilibrated in 0.1% HCOOH containing 5% CH3CN at a flow rate of 0.4 ml/min. The HPLC system consisted of two Shimadzu LC-10AD pumps (Shimadzu, Columbia, MD), a CTC PAL autoinjector (CTC Analytics, Carrboro, NC), and a Micromass Ultima tandem quadrupole mass spectrometer (Micromass, Beverly, MA) operated in the multiple reaction monitoring mode. The mobile phase was maintained at initial conditions for 0.5 min followed by a linear gradient to 70% CH3CN at 3 min. The effluent was introduced into an ionspray source in positive mode, and tune file parameters were as follows: capillary, 3.5; cone, 25; source temperature, 135°C; desolvation temperature, 350°C; cone gas, ≈190; desolvation gas, ≈750; entrance, −5; collision, 20; and exit, 1, with other potentials optimized to maximize the signal. Metabolite M1 (Rt = 1.98 min) was monitored with the transition m/z 337 to 278. Quantitation was accomplished by extrapolation from a standard curve ranging from 0.1 to 10 μM. Assay standards were within ±9% of nominal values.
Because the initial velocity versus the substrate concentration plot showed a decrease in the rates at higher substrate concentrations, the enzyme kinetic data were fit to the uncompetitive substrate inhibition model using the enzyme kinetics module of SigmaPlot (version 9.01; Systat Software, Inc., San Jose, CA) (eq. 1):
 in which Vmax and Km are the terms for the standard Michaelis-Menten parameters maximum reaction velocity and substrate concentration at half-maximum velocity and Ksi is the constant describing the substrate inhibition interaction (Houston and Kenworthy, 2000). The intrinsic clearance value was then determined from the Vmax and Km using eq. 2:
in which Vmax and Km are the terms for the standard Michaelis-Menten parameters maximum reaction velocity and substrate concentration at half-maximum velocity and Ksi is the constant describing the substrate inhibition interaction (Houston and Kenworthy, 2000). The intrinsic clearance value was then determined from the Vmax and Km using eq. 2:
 The IC50 value was determined from eq. 3:
The IC50 value was determined from eq. 3:
 in which the terms A and B represent maximum and minimum control activities at [I] = 0 and [I] = ∞, respectively.
in which the terms A and B represent maximum and minimum control activities at [I] = 0 and [I] = ∞, respectively.
Results
Excretion Studies.
Healthy human volunteers.
Urine and feces were collected over 144 h (6 days) from four healthy male volunteers after intravenous infusion of 80 mg of [14C]zoniporide for 1 h. A mean 91% of the dose was recovered in the urine and feces over 144 h (Table 1). The majority of dose (>95%) was excreted within the first 96 h after administration of the radioactive dose in all subjects (Fig. 2A). Bile was the major route of excretion because 57% of the dose was excreted in the feces after an intravenous infusion of [14C]zoniporide (Table 1). Approximately 34% of the total dose was excreted in the urine (Table 1).
Percent recovery of the dose in healthy male volunteers, Sprague-Dawley rats, and Beagle dogs after intravenous administration of [14C]zoniporide
[14C]Zoniporide was administered intravenously to humans at a dose of 80 mg and to Sprague-Dawley rats and Beagle dogs at a dose of 10 and 3 mg/kg, respectively.

Mean urine and fecal cumulative recovery of total radioactivity versus time profile in healthy male volunteers (n = 4) (A), Sprague-Dawley rats (n = 6) (B), and Beagle dogs (n = 4) (C) after intravenous administration of [14C]zoniporide. The errors bars indicate S.D. Healthy male volunteers were administered [14C]zoniporide by intravenous infusion for 1 h at a dose of 80 mg (100 μCi), and the excreta were collected over 144 h. The rats were administered a bolus of [14C]zoniporide at a dose of 10 mg/kg (50 μCi), and the excreta were collected over 168 h. The dogs were administered a bolus of [14C]zoniporide at a dose of 3 mg/kg (150 μCi), and the excreta were collected over 168 h.
Rats.
A mean 89% of the dose was recovered after a single bolus 10 mg/kg dose of [14C]zoniporide to Sprague-Dawley rats (Table 1). Most of the radioactivity was recovered in the initial 48 h in all rats (Fig. 2B). As in humans, the majority of the dose (61%) was excreted in the feces of rats and only 28% of the dose was excreted in the urine after 168 h. This result suggested that bile was the primary route of excretion of zoniporide material in rats as well.
Dogs.
Approximately 89% of the dose was recovered in the urine and feces of dogs 168 h after intravenous administration of [14C]zoniporide at a dose of 3 mg/kg. Bile was the primary route of excretion in dogs as well because 62% of the administered dose was excreted in the feces of this species, whereas 27% of the dose was recovered in the urine of dogs (Table 1). As in rats most of the dose was recovered in the first 48 h after dosing of the radiolabeled compound (Fig. 2C).
Pharmacokinetics of Total Radioactivity and Zoniporide.
Pharmacokinetics in healthy human volunteers.
The mean plasma concentration versus time profiles of total radioactivity and zoniporide after intravenous infusion of [14C]zoniporide in healthy male volunteers are presented in Fig. 3A. The Cmax of zoniporide (590 ng/ml) was approximately 66% of the Cmax of total radioactivity (899 ng-Eq/ml) and occurred at the end of infusion (1 h) of [14C]zoniporide (Table 2). The AUC0–tlast of zoniporide (833 ng-h/ml) accounted for ∼28% that of the AUC(0-tlast) of total circulating radioactivity (2925 ng-Eq-h/ml). The half-life of total radioactive material was 6.7 h. Unchanged zoniporide was rapidly cleared in humans after intravenous infusion of [14C]zoniporide with a clearance of 21 ml/min/kg and a half-life of 2.0 h (Table 2).

Mean plasma concentration-time profiles for total radioactivity and zoniporide after intravenous administration of [14C]zoniporide to healthy male volunteers (n = 4) (A), Sprague-Dawley rats (n = 6) (B), and Beagle dogs (n = 4) (C). The errors bars indicate S.D. Healthy male volunteers were administered [14C]zoniporide by intravenous infusion for 1 h at a dose of 80 mg (100 μCi). The rats were administered a bolus of [14C]zoniporide at a dose of 10 mg/kg (50 μCi). The dogs were administered a bolus of [14C]zoniporide at a dose of 3 mg/kg (150 μCi).
Mean pharmacokinetic parameters of total circulating radioactivity and zoniporide in healthy male volunteers, Sprague-Dawley rats, and Beagle dogs after intravenous administration of [14C]zoniporide
Healthy male volunteers were administered [14C]zoniporide by intravenous infusion for 1 h at a dose of 80 mg (100 μCi). The Sprague-Dawley rats and Beagle dogs were administered a bolus of [14C]zoniporide at a dose of 10 mg/kg (50 μCi) and 3 mg/kg (150 μCi), respectively.
Rat.
Figure 3B depicts the mean plasma concentration versus time profile for total radioactivity, unchanged zoniporide after a single bolus intravenous administration of [14C]zoniporide to rat. As in humans, zoniporide was rapidly cleared from rats (Cl = 113 ml/min/kg) with a half-life of 0.4 h (Table 2). The AUC0–tlast of unchanged zoniporide (1390 ng-h/ml) accounted for 39% of AUC0–tlast of total radioactivity (3570 ng-Eq-h/ml) in rats (Table 2). The mean terminal elimination half-life of total circulating radioactivity was 2.0 h and was 5-fold longer than that of zoniporide.
Dog.
After a bolus intravenous administration of 3 mg/kg [14C]zoniporide, the mean AUC0–tlast of total radioactivity and unchanged zoniporide were 3160 ng-Eq-h/ml and 2009 ng-h/ml, respectively (Table 2). The plasma concentration versus time profile for the total radioactivity and unchanged zoniporide is shown in Fig. 3C. Thus, in contrast to rats and humans, the exposure of unchanged zoniporide accounted for 64% of circulating total radioactivity in dogs. Furthermore, in contrast to rats and humans, the clearance of zoniporide was moderate (21 ml/min/kg), whereas the Vdss was higher (3.7 l/kg) in dogs. The mean terminal elimination half-lives of total circulating radioactivity and unchanged zoniporide were similar (2.8 and 2.5 h, respectively).
Metabolite Profiles of Zoniporide in the Urine and Feces.
Healthy male volunteers.
Only two metabolites (M1 and M2) were detected in the urine and feces of humans (Fig. 4). Metabolite M1 was the primary metabolite detected in the excreta (52% of the total dose), whereas zoniporide and metabolite M2 represented 18 and 17% of the total dose (Table 3).

Representative HPLC radiochromatogram of pooled urine (A) and feces (B) after intravenous infusion of [14C]zoniporide for 1 h at a dose of 80 mg (100 μCi) to healthy male volunteers.
Percentage of urinary and fecal metabolites of zoniporide after intravenous administration of [14C]zoniporide to healthy male volunteers, Sprague-Dawley rats, and Beagle dogs
Healthy male volunteers were administered [14C]zoniporide by intravenous infusion for 1 h at a dose of 80 mg (100 μCi). The rats were administered a bolus of [14C]zoniporide at a dose of 10 mg/kg (50 μCi). The dogs were administered a bolus of [14C]zoniporide at a dose of 3 mg/kg (150 μCi).
Rat.
Representative HPLC radiochromatograms of the extracted urinary and fecal (pooled from 0 to 24 h) samples from rat are shown in Fig. 5. As in humans, M1 was the primary excretory metabolite in rat and represented 35% of the total dose (Table 3). Unchanged zoniporide also constituted the majority of the dose in the urine and feces and accounted for 40% of the dose. The other metabolites M3 to M7 were minor metabolites and represented 0.8 to 5.9% of the dose (Table 3).
Representative HPLC radiochromatogram of pooled urine (A) and feces (B) after intravenous administration of [14C]zoniporide at a dose of 10 mg/kg (50 μCi) to Sprague-Dawley rats.
Dog.
Unlike the rat and humans, unchanged zoniporide was the primary constituent in the excreta of dogs (Fig. 6) and accounted for 37% of the total dose (Table 3). Major metabolites were M8 and M10, which accounted for 13 and 18% of the total dose, respectively. Other minor metabolites M3, M5, M7, and M9 detected in the excreta of the dog accounted for 8.6, 5.4, 4.1, and 2.1% of the total dose, respectively (Table 3). Most of these metabolites were excreted in the feces, suggesting biliary excretion of the radiolabeled material.

Representative HPLC radiochromatogram of urine (A) and feces (B) after intravenous administration of [14C]zoniporide at a dose of 3 mg/kg (150 μCi) to Beagle dogs.
Circulating Metabolites of Zoniporide.
Human circulating metabolites.
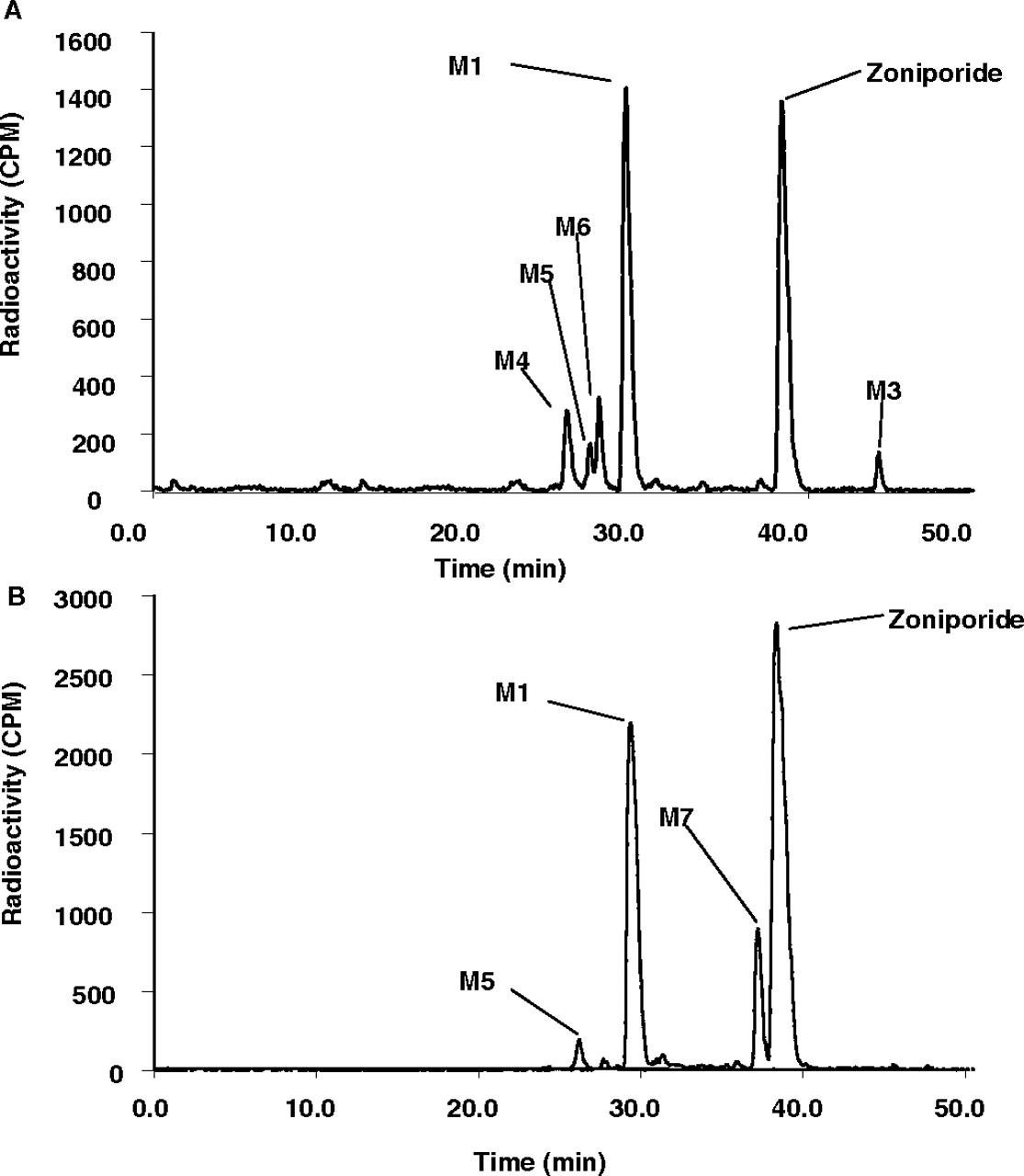
Circulating metabolites in humans were profiled by pooling the plasma samples obtained over 0 to 12 h using the method described by Hamilton et al. (1981) (Fig. 7A). This method gave a good assessment of the overall exposure of each metabolite over 0 to 12 h. Approximately 99% of the total circulating radioactivity could be identified. Three radioactive peaks were observed in plasma and corresponded to M1, zoniporide, and metabolite M3 (Fig. 7A). Metabolite M1 was the primary circulating metabolite and accounted for 60% of the circulating radioactivity, whereas zoniporide and M3 accounted for 30 and 6.4% of the total circulating radioactivity (Table 4).

HPLC radiochromatogram of pooled plasma of healthy male volunteers after intravenous infusion of 80 mg [14C]zoniporide for 1 h (A) and of Sprague-Dawley rats (B) and Beagle dogs (C) after intravenous administration of [14C]zoniporide at a dose of 10 and 3 mg/kg, respectively.
Percentage of circulating metabolites of zoniporide and their estimated exposure in healthy male volunteers, Sprague-Dawley rats, and Beagle dogs after intravenous administration of [14C]zoniporide
Healthy male volunteers were administered [14C]zoniporide by intravenous infusion for 1 h at a dose of 80 mg (100 μCi). The rats were administered a bolus of [14C]zoniporide at a dose of 10 mg/kg (50 μCi). The dogs were administered a bolus of [14C]zoniporide at a dose of 3 mg/kg (150 μCi). The estimated exposures were determined by multiplying percent circulating radioactivitymetabolite and AUC(0–tlast)radioactivity. The AUC(0–tlast)radioactivity is depicted in Table 2.
Rat.
Circulating metabolites in the rat were identified after the plasma samples obtained from 0 to 24 h were pooled according to the Hamilton pooling method. All circulating radioactivity in the rat plasma was accounted for. A representative HPLC radiochromatogram of rat plasma is shown in Fig. 7B. Of the four radioactive peaks identified in the plasma, zoniporide and M1 constituted 36 and 46% of the circulating radioactivity, whereas the other metabolites, M3 and M6 constituted 9.0 and 8.5% of the circulating radioactivity, respectively (Table 4).
Dog.
The HPLC radiochromatogram of circulating metabolites in plasma pooled from 1 to 12 h is shown in Fig. 7C. At least 99% of the total circulating radioactivity in the pooled plasma was identified. Four radioactive peaks including zoniporide were observed in dog plasma. Zoniporide was the major component circulating and accounted for 62% of the circulating radioactivity (Table 4). The major circulating metabolite was M9 and represented 23% of the total circulating radioactivity, whereas other metabolites (M3, M4, M8, and M10) accounted for 2.3 to 5.1% of the circulating radioactivity (Table 4).
Assessment and Comparison of Exposures of Human Circulating Metabolites in Toxicology Species.
Both metabolites M1 and M3 detected in the plasma of humans were >10% of the parent as measured by the ratio of the percentage of radioactivity of the metabolite to the percentage of radioactivity of the parent drug as observed in the human circulating metabolic profile. Therefore, in accordance with the guidance, it was important to determine whether the exposure of these two metabolites was greater than or similar to the exposure in the toxicology species. To assess the coverage of M1 and M3, the exposures of these two metabolites in humans and preclinical species were estimated by eq. 4:
 The estimated exposures of all metabolites are depicted in Table 4. As shown in Table 4, the exposures (AUC0–tlast) of M1 and M3 were 1755 and 187 ng-Eq-h/ml, respectively, in humans and the estimated exposures of M1 and M3 in the rat were 1642 and 321 ng-Eq-h/ml, respectively. Metabolite M1 was not present in the dog; however, the estimated AUC(0–tlast) of M3 in this species was 73 ng-Eq-h/ml. The estimated exposures of other metabolites observed in the rat and dog are also shown in Table 4. Determination of circulating levels of zoniporide using this method resulted in AUC0–tlast values of 878, 1285, and 1959 ng-Eq-h/ml in humans, rat, and dog, respectively, which were within the range of experimental error compared with the levels that were determined by a validated bioanalytical method (Table 2).
The estimated exposures of all metabolites are depicted in Table 4. As shown in Table 4, the exposures (AUC0–tlast) of M1 and M3 were 1755 and 187 ng-Eq-h/ml, respectively, in humans and the estimated exposures of M1 and M3 in the rat were 1642 and 321 ng-Eq-h/ml, respectively. Metabolite M1 was not present in the dog; however, the estimated AUC(0–tlast) of M3 in this species was 73 ng-Eq-h/ml. The estimated exposures of other metabolites observed in the rat and dog are also shown in Table 4. Determination of circulating levels of zoniporide using this method resulted in AUC0–tlast values of 878, 1285, and 1959 ng-Eq-h/ml in humans, rat, and dog, respectively, which were within the range of experimental error compared with the levels that were determined by a validated bioanalytical method (Table 2).
To further confirm the coverage of M1 and M3 in the preclinical species, the exposures of these two metabolites were also estimated using eq. 5, and the results are depicted in Table 5:
 Because both human circulating metabolites, M1 and M3, were present in disproportionately lesser amounts in the dog, an assessment of the coverage of exposure of these two metabolites was made only in the rat. Using eq. 2, the exposure of M1 and M3 was 1666 and 178 ng-Eq-h/ml, respectively, in humans and 1776 and 348 ng-Eq-h/ml in the rat.
Because both human circulating metabolites, M1 and M3, were present in disproportionately lesser amounts in the dog, an assessment of the coverage of exposure of these two metabolites was made only in the rat. Using eq. 2, the exposure of M1 and M3 was 1666 and 178 ng-Eq-h/ml, respectively, in humans and 1776 and 348 ng-Eq-h/ml in the rat.
Estimation of exposure of metabolites M1 and M3 in rat plasma
Metabolite/parent ratio was determined from the abundance of the metabolite (% total circulating radioactivity) and parent (% total circulating radioactivity) in the plasma. Estimated exposure of metabolite = (metabolite/parent ratio)·AUCzoniporide. The AUC of zoniporide was determined after quantitation of the parent by a bioanalytical assay.
Assessment of coverage of M1 was also made by comparing the amount of metabolite excreted in the urine and feces of humans and rats, which provides a comparison of total body burden. The amount of M1 excreted in the urine and feces was determined using eq. 6:

As shown in Table 6, the amount of M1 in the excreta of humans after 1.14 mg/kg (assuming a 70-kg weight for humans) and rats after a 10 mg/kg dose was 594 and 3500 μg/kg, respectively. Thus, even though the circulating exposure to M1 in the rat is slightly lower than that in humans, the total body exposure in the rat is actually greater.
Estimation of total body burden of M1 from excretion data in humans and rats
Amount of M1 in the excreta was determined using eq. 3.
Identification of Metabolites.
The structures of metabolites were elucidated by LC-MS/MS using a combination of full, precursor ion, and neutral loss scanning techniques. All metabolites were further characterized using the product ion scans of the identified masses. Zoniporide gave a signal at m/z 321 [M + H]+ in a positive ion mode. The product ion mass spectrum of m/z 321 gave characteristic major fragment ions at m/z 262 (loss of guanidine) and 234 (further loss of carbon monoxide) and 220 (loss of the cyclopropyl ring) (Table 7). Wherever possible, the structures of the major metabolites were confirmed by comparing their retention time and mass spectra with those of the synthetic standards. The structures/proposed structures of metabolites of zoniporide are shown in Fig. 8.
Mass spectral fragmentation and structures of zoniporide and proposed metabolites

Proposed metabolic scheme of zoniporide in healthy male volunteers, Sprague-Dawley rats, and Beagle dogs after intravenous administration.
Metabolite M1.
Metabolite M1 showed a molecular ion at m/z 337, which was 16 amu higher than zoniporide suggesting oxidation of the parent compound. The mass spectrum of m/z 337 gave a fragment ion at m/z 278, 250, and 236 (Table 7). All of these ions were 16 amu greater than those observed in the mass spectrum of zoniporide. This finding suggested that the quinoline or the pyrazole moiety of the molecule was the site of metabolism. That the site of hydroxylation was the quinoline ring and M1 was 2-oxozoniporide was confirmed by comparing its retention time and mass spectrum with the authentic standard of 2-oxozoniporide.
Metabolite M2.
Metabolite M2 gave a molecular ion at m/z 296. The mass spectrum of M2 at m/z 296 resulted in fragment ions at m/z 278 and 250 (Table 7). The ion at m/z 278 was a result of loss of water from M2, and the fragment ion at m/z 250 was a result of a loss of 46 amu from m/z 296. The loss of 46 amu, which is characteristic of a carboxylic acid group from the molecule, suggested that M2 was a carboxylic acid derivative. The presence of the carboxylic acid was further confirmed by esterification with methanolic HCl to the corresponding methyl ester (data not shown). The mass spectral fragmentation also indicated that the molecular ion of M2 was 16 amu higher than that of M3 (see below). Overall, the above data indicated that M2 was a carboxylic acid analog of the hydroxylated metabolite of zoniporide. The position of the hydroxyl group on the molecule and its structure was further confirmed by comparing its retention time and mass spectrum with the synthetic standard of M2.
Metabolite M3.
Metabolite M3 gave a signal at m/z 280 (41 amu less than the parent). A mass spectrum of M3 at m/z 280 showed major fragment ions at m/z 262 and 234, suggesting a loss of a water molecule followed by a loss of carbonyl functionality (Table 7). A difference of 46 amu between m/z 280 and m/z 234 suggested a loss of formic acid, which was characteristic of a carboxyl group. Esterification of the metabolite using methanolic HCl resulted in the formation of methyl ester, further confirming the presence of the carboxylic acid group in the molecule. Furthermore, these ions were similar to those observed in the mass spectrum of zoniporide, suggesting that the guanidine moiety of zoniporide was modified (Table 7). Together the above data suggested that M3 was a carboxylic acid derivative of zoniporide. Comparison of the retention time and mass spectrum of M3 with that of the authentic standard of carboxylic acid further confirmed its structure.
Metabolite M4.
Metabolite M4 showed a molecular ion at m/z 337, which was 16 amu greater than zoniporide. The mass spectrum of m/z 337 resulted in fragment ions at m/z 320, 278, 261, and 250 (Table 7). The fragment ions at m/z 278 and 250 were similar to those observed in the mass spectrum of M1 and resulted from the loss of guanidine (59 amu) followed by the loss of the carbonyl moiety (28 amu), suggesting that the addition had occurred on the pyrazolo-quinoline moiety. The fragment ions at m/z 320 and 261 resulted from the loss of 17 amu (a hydroxyl group) from m/z 337 and 278, respectively, and characteristic of an N-oxide. Treatment of the samples with 33% titanium chloride (TiCl3) (Kulanthaivel et al., 2004) resulted in the disappearance of the peak, which suggested that the metabolite was an N-oxide of zoniporide. The exact location of the oxygen atom (the quinoline nitrogen or pyrazole nitrogen) could not be discerned from the data. However, because previous studies have shown that quinoline-containing molecules can also undergo oxidation of the quinoline ring nitrogen in addition to the ring (Ehlhardt et al., 1998), the structure of M4 was proposed as the quinoline N-oxide of zoniporide. No further studies were attempted to isolate and fully characterize the metabolite.
Metabolite M5.
Metabolite M5 showed a protonated molecular ion at m/z 353, which was 32 amu greater than the molecular ion of zoniporide (m/z 321). Addition of 32 amu to the parent suggested further dihydroxylation of zoniporide. The mass spectrum of the metabolite showed major fragment ions at m/z 336, 294, 277, 266, and 249 (Table 7). The fragment ion at m/z 294 and 266 indicated a loss of 59 amu (loss of the guanidine moiety) followed by the loss of a carbonyl group, suggesting that the pyrazoloquinoline moiety of zoniporide was dihydroxylated. Other fragment ions m/z 277 and 249, observed in the CID spectrum, indicated a loss of 17 amu (hydroxyl group) from m/z 294 and 266, respectively. Because the loss of 17 amu is characteristic of an N-oxide as discussed above, M5 was proposed as an N-oxide of hydroxyzoniporide. No further characterization of this metabolite was done to ascertain the position of the hydroxyl group because the metabolite was present only in the preclinical species.
Metabolite M6.
Metabolite M6 showed a protonated molecular ion at m/z 513, which was 176 amu higher than m/z 337, the hydroxylated zoniporide. This suggested glucuronidation of hydroxyzoniporide. The mass spectrum of m/z 513 gave a major fragment ion at m/z 337, suggesting a loss of 176 amu from the molecular ion, and a fragment ion at m/z 278, suggesting further loss of guanidine. Based on these data, M6 was identified as the glucuronide conjugate of hydroxyzoniporide. The position of the hydroxy group could not be assessed from these data. No attempt was made to further characterize this metabolite because it was absent in the human matrices.
Metabolite M7.
Metabolite M7 showed a protonated molecular ion at m/z 337, suggesting hydroxylation of zoniporide. The fragment ions in the mass spectrum of the metabolite were similar to those observed in the mass spectrum of metabolite M1 (Table 7). The difference in the retention time of M1 suggested that M7 was a regio-isomer of M1; however, the exact position of the hydroxyl group could not be determined from the mass spectrum. Because the oxidative metabolite was lacking in humans, no further attempt was made to determine the position of hydroxylation in the metabolite.
Metabolite M8.
Metabolite M8 showed a molecular ion at m/z 355. The molecular ion of M8 indicated an addition of 34 amu to m/z 321, a molecular ion of zoniporide. The mass spectrum of m/z 355 gave a fragment ion at m/z 296 (a characteristic loss of the guanidine moiety) and at m/z 296, 278, 254, and 250 (Table 7). The loss of 59 amu that resulted in m/z 296 indicated that the guanidine moiety was intact. The fragment ion at m/z 278 and 250 possibly resulted from loss of water from m/z 296 followed by a loss of the carbonyl group (28 amu). The mass spectrum also showed ions at m/z 254 and 236, suggesting a loss of 42 amu (a cyclopropyl group) from m/z 296 and 278, respectively. The loss was similar to that observed in the CID spectrum of zoniporide and suggested that the pyrazoloquinoline group was the site of modification. Together, these fragment ions indicated that the M8 was probably a dihydrodiol metabolite of zoniporide (Fig. 8). Previous studies with quinoline-containing compounds have indicated that these motifs are capable of undergoing conversion to a dihydrodiol (Nicoll-Griffith et al., 1993). The definitive identification and characterization of the exact position of the dihydrodiol on the quinoline ring was not attempted.
Metabolite M9.
Metabolite M9 showed a molecular ion at m/z 531, which was 176 amu higher than M8, indicating glucuronidation of M8. The mass spectrum showed fragment ions at m/z 472, 355, 296, 278, and 250 (Table 7). The fragment ion at m/z 472 was a characteristic loss of the guanidine moiety, suggesting that this group was not modified. The fragment ion at m/z 355 resulted from the loss of the glucuronide moiety. The remaining fragment ions were similar to those in the mass spectrum of M8. Previous studies have demonstrated that dihydrodiol metabolites of aromatic rings are capable of undergoing glucuronidation (Lantz et al., 2003). Thus, M9 was assumed to be a glucuronide conjugate of M8 (Fig. 8). No further characterization of this metabolite was performed because it was not detected in the human metabolic profiles.
Metabolite M10.
Metabolite M10 also showed a protonated molecular ion at m/z 337, suggesting hydroxylation of zoniporide. The fragment ions in the mass spectrum of the metabolite were similar to those observed in the mass spectrum of metabolite M1 and M7 (Table 7). The difference in the retention time of M10 from that of M1 and M7 suggested that M10 was an isomer of these two metabolites; however, the exact position of the hydroxyl group could not be determined from the mass spectrum. Because an oxidative metabolite was lacking in humans, no further attempt was made to determine the position of hydroxylation in the metabolite.
In Vitro Metabolism Studies.
To identify the enzymes responsible for the formation of M1, preliminary experiments were performed by incubating zoniporide with human liver S9 fraction at concentrations of 10 μM in the presence and absence of NADPH (Fig. 9, A and B). The presence of M1 in the incubations that lacked NADPH suggested that its formation was catalyzed by a non-P450 enzyme. Furthermore, inhibition of M1 in the presence of raloxifene, an aldehyde oxidase inhibitor, to the incubation mixture, indicated that aldehyde oxidase was responsible for its formation (Fig. 9C). Only trace amounts of M3 were detected in the mass spectral analysis of the above incubation mixture (data not shown) suggesting that hydrolysis constituted a minor pathway at least in incubations with liver S9 fractions.

Metabolic profile of zoniporide (10 μM) after incubation with human liver S9 fractions in the presence (W) of NADPH (A), absence (WO) of NADPH (B), and presence (W) of raloxifene, an inhibitor of aldehyde oxidase (C).
Based on the above information, enzyme kinetics studies for formation of M1 by aldehyde oxidase were performed using pooled human liver cytosol, and the kinetic parameters were determined. Analysis of the relationship between reaction velocity and substrate concentration revealed substrate inhibition kinetics with kinetic parameters of Km = 3.4 ± 0.2 μM and Vmax = 74 ± 2 pmol/min/mg protein and a Ks value of 152 μM (Fig. 10A). A detailed assessment of the molybdenum hydrolases (aldehyde oxidase or xanthine oxidase) was also conducted by determining the IC50 for the inhibition of M1 formation using specific inhibitors of aldehyde oxidase (raloxifene and menadione) and xanthine oxidase (allopurinol). Raloxifene potently inhibited the reaction (IC50 = 0.012 ± 0.002) and menadione also inhibited the reaction (9.8 ± 1.2 μM), further confirming the involvement of aldehyde oxidase in M1 formation (Fig. 10B). Allopurinol did not inhibit the reaction (IC50 > 1 mM), indicating that xanthine oxidase did not metabolize zoniporide.

Enzyme kinetics for the formation of 2-oxozoniporide (M1) from zoniporide in pooled human liver cytosol (A) and inhibition of this reaction by raloxifene, menadione, and allopurinol (B). The enzyme kinetics experiments were done in triplicate, and the inhibition studies were done in duplicate. The errors bars in A indicate S.D.
Discussion
In the present study, the routes of elimination and metabolism and the excretion mass balance of zoniporide in humans were investigated after an intravenous infusion of [14C]zoniporide. A dose of 80 mg was administered in this study, which was approximately equal to the anticipated clinical dose to be used in the phase III program. The comparison of metabolite profiles of a drug candidate in animals and humans is essential to ensure that animal species used in toxicological evaluations are appropriate models of humans and to confirm that all human circulating metabolites are covered in these species (Food and Drug Administration, 2008). The metabolic profiles in humans, rat, and dog were compared to assess whether all metabolites observed in human matrices were detected in these two toxicology species. The doses used in the preclinical species were equivalent to the dose in the toxicology studies in which minimum adverse effects were observed.
After intravenous administration, the majority of the radioactivity was excreted in feces, suggesting that biliary excretion was the principal route of elimination of zoniporide-related material in the humans. Metabolism was the primary route of clearance for zoniporide in humans because only 18% of the dose was excreted unchanged in the urine and feces. In addition, the majority of the total circulating radioactivity comprised metabolites because the exposure of unchanged zoniporide accounted for ∼28% of exposure of total circulating radioactivity in plasma. 2-Oxozoniporide (M1), which was formed via oxidation of the quinoline ring of zoniporide, was the primary circulating and excretory metabolite in humans. Its exposure was 2-fold greater than that of unchanged zoniporide in plasma, and approximately 52% of the dose was excreted as M1 in the urine and feces. Metabolite M1 was also pharmacologically active against the NHE-1 receptor. However, the activity was ∼3-fold less than that of the parent compound. Because the systemic exposure of M1 was greater than that of the parent drug and the protein binding was approximately the same as that of zoniporide, the possibility of the metabolite exerting pharmacological activity in addition to the parent in vivo could not be ruled out. Given that M1 was the major circulating metabolite in humans and was pharmacologically active, in vitro studies using human liver subcellular fractions were performed to identify the enzymes responsible for the formation of M1. Furthermore, this was important in light of the fact that inhibition of the enzyme responsible for the conversion of zoniporide to M1 would increase the AUC of the parent drug significantly and would also possibly affect the efficacy of the compound. Phenotying studies revealed that formation of M1 was primarily catalyzed by aldehyde oxidase (Clint = 22 μl/min/mg cytosolic protein). Aldehyde oxidase is a cytosolic molybdo-flavoenzyme that is expressed predominantly in the liver, lung, and kidney and plays a major role in the oxidation of aldehydes and nitrogen-containing heterocyclic compounds (Kitamura et al., 2006). Some heteroaromatic compounds of pharmacological and toxicological importance that are metabolized by aldehyde oxidase include carbazeran (Kaye et al., 1985), famciclovir (Rashidi et al., 1997), methotrexate (Jordan et al., 1999), zaleplon (Lake et al., 2002), brimonidine (Acheampong et al., 1996), and N-[(2′-diethylamino)ethyl]acridine-4-carboximide (Schofield et al., 2000). Thus, it is not surprising that zoniporide was a substrate of aldehyde oxidase.
Potential drug-drug interactions due to inhibition of aldehyde oxidase have not been established. Although cytochrome P450 enzymes have been and continue to be a major focus of drug interactions, alterations in the activities of other drug-metabolizing enzymes can also be an underlying mechanism of drug-drug interactions. Only one clinically relevant drug-drug interaction between cimetidine and zaleplon has been ascribed to inhibition of aldehyde oxidase in the literature so far (Renwick et al., 2002). Because several drugs have been demonstrated to be human aldehyde oxidase inhibitors in vitro (Obach, 2004; Obach et al., 2004), it is possible that these drugs could potentially increase the levels of zoniporide in humans. No such drug-drug interactions studies have been conducted and it remains to be determined whether aldehyde oxidase inhibitors identified using in vitro methods could potentially cause a more profound interaction of clinical significance in vivo.
Metabolites M3 and M2, the carboxylic acid analogs of zoniporide and quinolone metabolite (M1), were also observed in humans. This finding suggested that the hydrolytic cleavage was another pathway by which zoniporide was metabolized. Although M3 accounted for ∼20% of parent drug in humans, M2 was found only in the excreta in amounts of ∼17% of the dose. The exact metabolic pathway for the formation of M2 was difficult to discern because the metabolite can be formed via two parallel pathways (Fig. 8). One pathway could involve hydrolysis of M1 to M2. Alternatively, zoniporide could undergo hydrolysis to M3, which could subsequently undergo an aldehyde oxidase-mediated or a P450-mediated oxidation of the quinoline ring to yield M2. Because M2 was not detected in the urine or the plasma of humans, it is possible that the formation of this metabolite in vivo was possibly mediated by microflora in the gastrointestinal tract.
The metabolism and excretion studies using [14C]zoniporide in the rat and dog suggested that the route of excretion of zoniporide in these two species was similar to that in humans and the majority of the dose was also eliminated into the feces via the bile. In addition, as in humans, zoniporide metabolism was the primary route of clearance in the rat and dog. Only 40 and 37% of the total dose constituted unchanged zoniporide in the rat and dog, respectively. However, comparison of plasma exposure of unchanged zoniporide and total radioactivity in rat and dog revealed that even though circulating radioactivity comprising metabolites in rats just as in humans, unchanged zoniporide was the primary constituent of total radioactivity in dogs.
Analysis of the metabolic profile of zoniporide in the rat revealed that both human circulating metabolites (M1 and M3) were present in the rat in addition to several other metabolites. Although dogs metabolized zoniporide extensively, in contrast to humans and rats, this species showed differences in its metabolic profile. Metabolite M1, which was a major metabolite in rats and humans, was not detected in the dogs, whereas M3 was a minor circulating metabolite (6% of the parent) but present in ∼9% of the dose in the excreta. The absence of M1 in the dog was not surprising given that dogs lack aldehyde oxidase activity (Kitamura et al., 2006). Even though oxidation was a primary pathway of metabolism in dogs, the major metabolites observed were the dihydrodiol (M8) and hydroxyzoniporide (M10). Because the other oxidative metabolites detected in the rat and dog were absent in humans, no further consideration was given to these metabolites with respect to potential safety implications.
An assessment of the coverage of major circulating metabolites in humans was made in this study by comparing estimated exposure levels of M1 and M3 in humans with those in preclinical species. Assessment was done only in the rat because the dog did not produce the M1 metabolite, and M3 was produced in <10% of the parent in vivo. Because levels of M3 in rat exceeded those estimated in humans by 1.7- to 2.0-fold (Table 4), the exposure to this human metabolite was considered to be covered in the rat. The estimated exposure level of M1 in humans, on the other hand, was comparable with that estimated in rats as depicted in Tables 4 and 5. Because zoniporide was administered intravenously to humans and the preclinical species, the coverage of metabolite M1 in the preclinical species was also ascertained by estimating the amount of metabolite excreted in the rat and human. This estimation indicated that the amount of M1 to which the rat was exposed was 5.9-fold greater relative to that in humans. Furthermore, M1 did not have any structural alerts that would pose a risk to humans. Thus, M1 was also considered to be adequately evaluated in the toxicology species, and no further assay validation and toxicological evaluation of this metabolite was undertaken in nonclinical safety species. Metabolite M2 was detected only in human feces and was not present in rat and dog, which indicated that this metabolite was unique to the humans. However, because M2 was not present in circulation, was pharmacologically inactive, and did not have a functionality that could potentially be a toxicophore, it was not considered for further safety evaluation.
In summary, this study demonstrates the primary routes of excretion and metabolism of zoniporide in humans, rats, and dogs. Furthermore, the study also demonstrates the strategy that was used to assess whether the further toxicological evaluation of the human circulating metabolites was necessary in the preclinical species. Zoniporide is among the few compounds that are almost exclusively metabolized by aldehyde oxidase in humans (with approximately three-fourths of its clearance mediated by this enzyme) and hence can be used as a model substrate for this enzyme. Studies are in progress to assess the species differences in the metabolism of aldehyde oxidase and understand the structure-metabolism relationship of the compound.
Acknowledgments.
We thank the radiochemistry group in Groton for synthesizing [14C]zoniporide.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.109.030783.
-
ABBREVIATIONS:
- NHE-1
- sodium/hydrogen exchanger
- CL
- total clearance
- AUC
- area under plasma concentration versus time curve
- SBE
- sulfobutylether
- HPLC
- high-performance liquid chromatography
- MS
- mass spectrometry
- RAM
- radioactivity monitoring detector
- LC-ARC
- liquid chromatography-accurate radioisotope counting system
- CID
- collision-induced dissociation
- amu
- atomic mass units
- P450
- cytochrome P450.
- Received October 13, 2009.
- Accepted December 29, 2009.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}