


Abstract
The pregnane X receptor (PXR) has three known major transcript variants resulting from alternative splicing. The less well characterized variants T2 and T3 are identical to the well described variant T1 except for a 39-amino acid N-terminal extension in T2 and an internal 37-amino acid deletion in T3. We have developed reverse transcription-polymerase chain reaction (RT-PCR) methods to detect and quantify each human PXR (hPXR) in human liver and intestinal tissues and HepG2 and Caco-2 cell lines. All three isoforms were expressed in hepatic cells, whereas only T1 transcripts were found in Caco-2 cells. In general, most normal human liver and intestinal mucosa contained all three hPXR variants, but considerable interindividual variation in expression levels was found. The effect of each hPXR variant on expression of UDP-glucuronosyltransferase (UGT) UGT1A and UGT2B family isoforms was investigated in transiently transfected HepG2 and Caco-2 cells. As a family, UGT1A transcripts were up-regulated by T1 and T2 but not T3. Isoform-specific RT-PCR revealed that UGT1A1, 1A3, and 1A4 were the major isoforms induced in both cell lines. The levels of several UGT1A isoforms were also examined in human liver samples from a number of donors with characterized PXR expression. The data suggest that individual variation in PXR expression may account for differential expression of some UGT isoforms between subjects.
The pregnane X receptor (PXR)2, a ligand-regulated orphan nuclear receptor, has been identified as a species-specific xenobiotic receptor (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998). This receptor is activated by natural and synthetic pregnenolone derivatives and by a large number of structurally diverse compounds such as rifampicin (RIF), hyperforin (a constituent of St. John's wort), and bile acids (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer and Willson, 2002). Molecular studies have revealed that PXR is a key transcription factor responsible for CYP3A4 and CYP3A7 induction (Bertilsson et al., 1998; Blumberg et al., 1998; Pascussi et al., 1999; Xie et al., 2000; Staudinger et al., 2001; Kliewer and Willson, 2002) as well as some important efflux transporters, including multidrug resistant proteins 1 and 2 (Synold et al., 2001; Kast et al., 2002).
Several variants of human PXR (hPXR) have been previously identified. Blumberg et al. (1998), Bertilsson et al. (1998), and Lehmann et al. (1998) simultaneously isolated a cDNA variously termed SXR (steroid and xenobiotic receptor), hPAR-1, and hPXR (now known as T1). Northern blot analysis demonstrated that this mRNA is expressed at high levels in liver and moderate levels in intestine. Translation yields a protein of 434 amino acids, with a predicted molecular weight of 50,000. Concurrently, Bertilsson et al. (1998) isolated two cDNAs, T1 and hPAR-2 (T2). T2 cDNA differs from T1 at the 5′ end, resulting in an open reading frame 39 amino acids longer. A third hPXR variant mRNA (T3), containing an in-frame deletion of 111 nucleotides (823-933 relative to T1) was first described by Dotzlaw et al. (1999). T3, along with T1, was found to be expressed in normal and neoplastic breast tissue. T3 is similar to mouse PXR.2, which contains an in-frame 123-nucleotide deletion in a similar region of the ligand-binding domain (Dotzlaw et al., 1999). Mouse PXR.2, compared with mouse PXR.1 (analogous to T1), showed a reduced response to agents that could activate the wild-type receptor in transient transfection analyses. It is possible that human PXR variants may display a similar profile or be unable to bind ligand. Hustert et al. (2001) showed that T3 expressed in the human colon adenocarcinoma cell line, LS174T, did not direct transcription from the CYP3A4 promoter.
There is currently little information relating the function of nuclear receptors (NRs) to transcriptional activation of UGTs, and none regarding the roles of individual PXR variants. There are marked interindividual differences in the UGT content of the liver, intestine, and other organs, which are postulated to be the result of differential transcription. Depending on the substrate, variations in UGT activity of 6- to 15-fold in liver microsomes and 10- to 100-fold in intestinal microsomes have been found. Similar variations in UGT protein content have been demonstrated by Western blot in liver and intestinal microsomes (Burchell and Coughtrie, 1997; Little et al., 1999, 2002; Strassburg et al., 1999; Court et al., 2001; Antonio et al., 2003). A similar degree of variability in hepatic UGT mRNA levels has also been reported (Congiu et al., 2002). In addition, UGTs are distributed in a tissue-specific manner throughout the body. UGT1A1, 1A3, 1A4, 1A6, 1A9, and all the UGT2B isoforms have been shown to be expressed in the liver (Strassburg et al., 2000; Turgeon et al., 2001). In comparison, human intestine has been shown to express UGT1A1, 1A6, 1A8, 1A10 (Strassburg et al., 2000; Tukey and Strassburg, 2001), and UGT2B7 (Radominska-Pandya et al., 1998; Czernik et al., 2000). The intestine harbors many phase I and phase II enzymes and thus plays an essential role in the metabolism and detoxification of xenobiotics (see Tukey and Strassburg, 2001, for a review). Therefore, one factor influencing the tissue-specific UGT expression pattern may be the expression levels of NR proteins, in particular PXR, and the availability of their ligands.
In the present work, we have carried out the first systematic studies of hPXR variant mRNA levels in human liver and intestine as well as human hepatic and intestinal cell lines. We also describe for the first time the effect of the individual hPXR variants on UGT expression in HepG2 and Caco-2 cells. Two UGT isoforms, 1A3 and 1A4, were identified as new hPXR target genes. In contrast, the UGT2B isoforms were not responsive to PXR under the experimental conditions used. Finally, we investigated the association among the expression levels of T1, T2, and T3 hPXR mRNAs and UGT1A in human liver from several donors. We postulate that the varying levels of the natural PXR protein variants, combined with their differential transcription potentials, may have an important impact on both tissue-specific and interindividual target gene expression profiles. Furthermore, with the identification of two new target genes and elucidation of the PXR variants that are responsible for UGT regulation, our results give new insight into the role that PXR may play in UGT-related drug metabolism and clinical drug-drug interactions.
Materials and Methods
Cell Culture and Transient Transfections. The human hepatocellular carcinoma cell line, HepG2 (ATCC HB-8065) and human adenocarcinoma cells line, Caco-2 (ATCC HTB-37), were obtained from the American Type Culture Collection (Manassas, VA). Both cell lines were maintained at 37°C, 5% CO2 in high glucose Dulbecco's modified Eagle's medium (DMEM) with Earle's salts and l-glutamine (Invitrogen, Carlsbad, CA), supplemented with 1% nonessential amino acids, 1 mM sodium pyruvate, and 10% fetal bovine serum (Invitrogen or Trace Biosciences, Sydney, Australia). The culture medium was changed twice weekly during maintenance. Untransfected cells used for RNA isolation were harvested when they neared confluence. Caco-2 cells were received at passage number 18; the HepG2 cells were received at passage number 77. The Caco-2 cells were stable over the period of the experiments.
For transient transfection, HepG2 or Caco-2 cells were seeded in six-well plates at 1 × 106 and 2.5 × 105 cells per well, respectively. Transfections were performed in triplicate using 5 μg of expression plasmid or empty parent vector and 10 μl of LipofectAMINE 2000 (Invitrogen) according to the manufacturer's instructions. Six to seven hours after transfection, the cells were washed in DMEM and replacement medium containing 10 μM RIF (Sigma-Aldrich, St. Louis, MO) or vehicle (0.1% dimethyl sulfoxide) was added. At 24 h post-transfection, the cells were treated again with RIF or solvent in fresh DMEM. After a total of 40 h, all transfections were washed in phosphate-buffered saline and harvested for RNA extraction. Using the protocols of the manufacturer, the transfection efficiencies were on average about 70%.
Human tissue samples. The human intestinal tissues used in the present studies were obtained from organ donors (details are summarized in the legends to the appropriate figures) by transplant surgeons at University Hospital, Little Rock, AR, according to a protocol approved by the Human Research Advisory Committee of the University of Arkansas for Medical Sciences. The intestines were received from the surgeons in saline on ice. Working at 4°C, the small intestine was divided into four segments of 80 to 100 cm in length, and the colon was treated as a single segment. Each segment of intestine was opened, the contents were removed, and the tissue was rinsed in cold 0.9% NaCl. Mucosa was removed from each segment by scraping with a glass slide, and RNA was prepared as described below.
Two of the human livers used in these studies were also obtained from the transplantation program at the University of Arkansas for Medical Sciences. The remaining liver samples were a generous gift from Dr. Mary Relling at St. Jude Children's Research Hospital (Memphis, TN). Again, donor information is given in the legends to the appropriate figures. mRNA extraction is described below.
RNA Isolation. Total RNA was isolated from 50 mg of frozen tissue or untransfected cell culture using a phenol and guanidine isothiocyanate RNA extraction method following the instructions of the suppliers (Trizol; Invitrogen). Total RNA from transfection experiments was harvested using the RNeasy Mini Kit (QIAGEN, Valencia, CA), per the instruction manual. To avoid any contamination of the RNA by genomic DNA, RNA samples were treated with RNase-free DNase (RQ1; Promega, Madison, WI).
Plasmids and Cloning of hPXR Variants. Plasmids for expression of hPXR variants T1, T2, and T3 were constructed by insertion of PCR-amplified cDNAs (described below) into the XbaI and HindIII sites of pCMV5. Each cDNA was confirmed by sequencing and was subsequently transferred to the pCMX vector that was used to create the T1 expression vector. The pCMV5- and pCMX-based vectors showed no discernible difference in expression of the PXR variants. The sources of the pCMX and pCMV5 vectors have been described elsewhere (Andersson et al., 1989; Xie et al., 2000).
PXR T1 and T2 cDNAs were amplified from HepG2 cDNA using the common reverse primer PXRrc (agccattctagatcagctacctgtgatgccgaa) and specific primers for the 5′ ends, PXRT1 and 3 (agccataagcttatggaggtgagacccaaaga) and PXRT2 (agccataagcttatgacagtcaccaggactca). The CTG initiation codon of T1, and consequently T3, was replaced by the more conventional ATG through primer mismatch. Restriction sites are highlighted in bold. All PCRs for cloning were done with 1.25 units (U) of Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA) in 20 mM Tris-HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4, 1% Triton X-100, and 0.1 mg/ml bovine serum albumin with 0.5 mM dNTPs and 50 ng of each primer in a total volume of 20 μl. After an initial 4 min at 95°C, 40 cycles of 95°C, 45 s; 60°C, 45 s; and 72°C, 4 min were performed. Each PCR was finished with 5 min at 72°C.
T3 cDNA was synthesized from the newly cloned T1 cDNA by overlap PCR. T3 has a sequence identical to T1 except for an internal 111 base pairs. Primers spanning this junction, aagaatttccgggtctctctgcagctgcgg and ctgcagagagacccggaaattcttgaaatggga, were used in combination with PXRT1, -3, and PXRrc to amplify the remainder of T1 and fuse the two sections together.
Semiquantitative RT-PCR for hPXR Variants. cDNA was synthesized by mixing 1 μg of total RNA from each sample with 100 pmol random hexamers in 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, 100 U Moloney murine leukemia virus-reverse transcriptase, 20 U RNase inhibitor, and 1 mM each dNTP in a total volume of 20 μl (Promega). The samples were incubated at 37°C for 60 min and then diluted to 100 μl with sterile diethylpyrocarbonate-treated H2O. The reverse transcriptase was inactivated by heating at 95°C for 5 min.
The primers used to detect hPXR wild-type T1 (NM_003889), T2 (NM_022002), and T3 (NM_033013) variants are described in Table 1. In addition to a common primer set designed to amplify all three hPXR transcripts, specific primers to detect them individually were made, based on differences between the splice variants. To distinguish T2 from T1 and T3, the forward primer F2 was designed within the 5′ sequence unique to this transcript (Fig. 1). In contrast, T1 and T3 share all of the sequence except the deleted region in T3; thus, the amplification specificity between these variants was facilitated by the design of the reverse primers. To exclusively amplify T1, the F1 primer was positioned in the 5′ untranslated region (UTR) absent in T2, and the reverse primer was designed within the 111-base pair sequence deleted in T3. To detect T3 only, another primer in the 5′ UTR of F3 was used, in combination with an oligonucleotide which binds over the boundary of the sequence missing in T3 as shown in Fig. 1. GAPDH amplification was used as an internal control (see Table 1).
PCR primers and conditions for semiquantitative analysis of PXR variants and PXR-induced transcription Nucleotide sequences are reported for primer pairs used in RT-PCR analysis of PXR transfection experiments in HepG2 and Caco-2 cells. Two common primers are used for the UGT2B isoform PCR: cR1 has two mismatches to UGT2B4 and one mismatch each to UGT2B7, UGT2B11, and UGT2B15. cR2 has one mismatch to UGT2B10 and three mismatches to UGT2B28. The nucleotide sequences for UGT1A4F, UGT1A6F and R, UGT1A10R, and all UGT2B primers except UGT2B28F were obtained from Congiu et al. (2002). The sense primer for UGT1A10 was designed by Strassburg et al (1997). PXR variant primers were designed by J.-M. Heydel.

Structure of human PXR transcripts, design of transcript specific PCR primers, and natural protein variants of human PXR.
A, transcripts were amplified specifically by RT-PCR using transcript-specific primer pairs. T1 originates from exon 1A and corresponds to the cDNA published as PXR wild type. T2, which has a unique 5′ UTR and start codon, begins in exon 1B and is the cDNA published as PAR2. T3 is similar to T1 but has an in-frame deletion of 111 base pairs at the 5′ end of exon 5. B, T2 is similar to T1 but contains an additional 39 amino acids on its N terminus. T3 is similar to T1 but has an internal 37-amino acid deletion. F is the forward primer and R is the reverse. F1 is the T1 forward primer and R1 is the T1 reverse primer, etc. RC is the reverse control primer. Also, DBD is the DNA-binding domain, and LBD is the ligand-binding domain.
The semiquantitative hPXR PCR reactions were performed as follows: a 10-μl cDNA aliquot was added to a reaction mixture containing 10 mM Tris-HCl buffer (pH 8), 20 mM KCl, 0.1% Triton X-100; 1.5 mM MgCl2, 0.2 mM each dNTP, 50 pmol of each primer, and 2 U of TaqDNA polymerase (Promega), in a total volume of 50 μl. The mixture was subjected to 34 cycles consisting of a 45-s denaturing step at 94°C, a 45-s annealing step at 59°C, and a 45-s elongation step at 72°C in a thermal cycler (MJ Research, Reno, NV). Amplification of the ubiquitously expressed GAPDH cDNA was performed under the same conditions in separate experiments. Amplification products were resolved by agarose gel (2%) electrophoresis and detected by ethidium bromide. The bands were visualized under UV light and photographed with a computer-assisted camera. Quantification of each band was performed by densitometric analysis using NIH Image software (National Institutes of Health, Bethesda, MD). The identities of all PCR products were confirmed by sequencing.
Semiquantitative RT-PCR for CYP3A7 and UGT Detection. cDNA from transfection experiments was made from 1 μg (HepG2) or 0.6 μg (Caco-2) RNA, using the Invitrogen Superscript system. First-strand synthesis was performed using the oligo(dT) primer method according to the supplier's instructions. All completed cDNA reactions were diluted in diethylpyrocarbonate-treated water to the equivalent of 10 μg/ml of the original RNA.
To confirm successful transfection of the PXR expression vectors and the ability of each individual construct to overexpress a PXR variant, PCR with a common primer set (Table 1) was used. All primers in Table 1 were obtained from Sigma-Genosys (Castle Hill, Australia) or Integrated DNA Technology (Coralville, IA). PCR for experiments with cultured cells were performed with 0.5 U of Taq polymerase (Amersham Biosciences Inc., Piscataway, NJ) on 4 to 10 μl of cDNA under the following conditions: 95°C, 5 min, followed by 25 to 42 cycles of 30 s at 95°C; 30 s at an appropriate annealing temperature; 1 min at 72°C; and a final 5 min at 72°C. Table 1 details the annealing temperature and cycle number required for each template. Primer pairs were designed to specifically amplify across exon boundaries in mRNA from β-actin, CYP3A7, UGT1A1, UGT1A9, UGT2B7, and the UGT1A family as a whole (Table 1). For CYP3A7, UGT1A, and UGT2B7, PCR for each reaction was paused at 72°C with 18 cycles remaining and 50 ng of both β-actin1 primers were added as an internal reference. For the remainder of the semiquantitative reactions, β-actin2 control PCR was performed as a separate reaction. The specificity of all primer pairs was confirmed through sequencing or restriction analysis of the PCR products. Semiquantitative analysis of each PCR product was as described above, using Molecular Analyst software (Bio-Rad, Hercules, CA).
Results
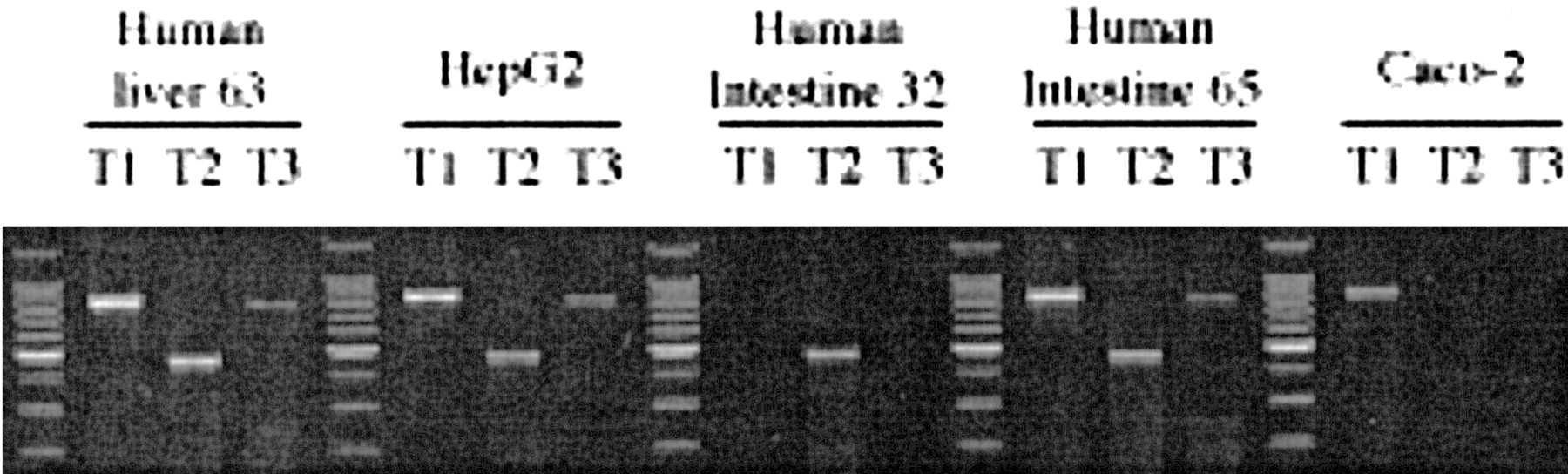
Identification of hPXR mRNA Variants in Human Tissues and Cell Lines. Estimation of mRNA levels of hPXR transcripts in human tissues and two human cell lines was carried out by semiquantitative RT-PCR. One human liver sample, intestinal mucosa from the jejunum of two donors, and HepG2 and Caco-2 cells were examined (Fig. 2). The expected PCR products for hPXR T1, T2, and T3 variants were detected in both the representative normal human liver and the human hepatoma cell line, HepG2 (Fig. 2). The expression pattern in these samples was almost identical. In contrast, the pattern of variant expression in the jejunal segments from the two selected donors was strikingly different: in one donor (human intestine 32), only the T2 variant was detected, whereas in the second donor (human intestine 65), the pattern of expression was similar to that of the liver (Fig. 2). Examination of Caco-2 cells showed that only T1 was present in this human intestinal cancer cell line, an obvious difference in comparison with human jejunal mucosa.
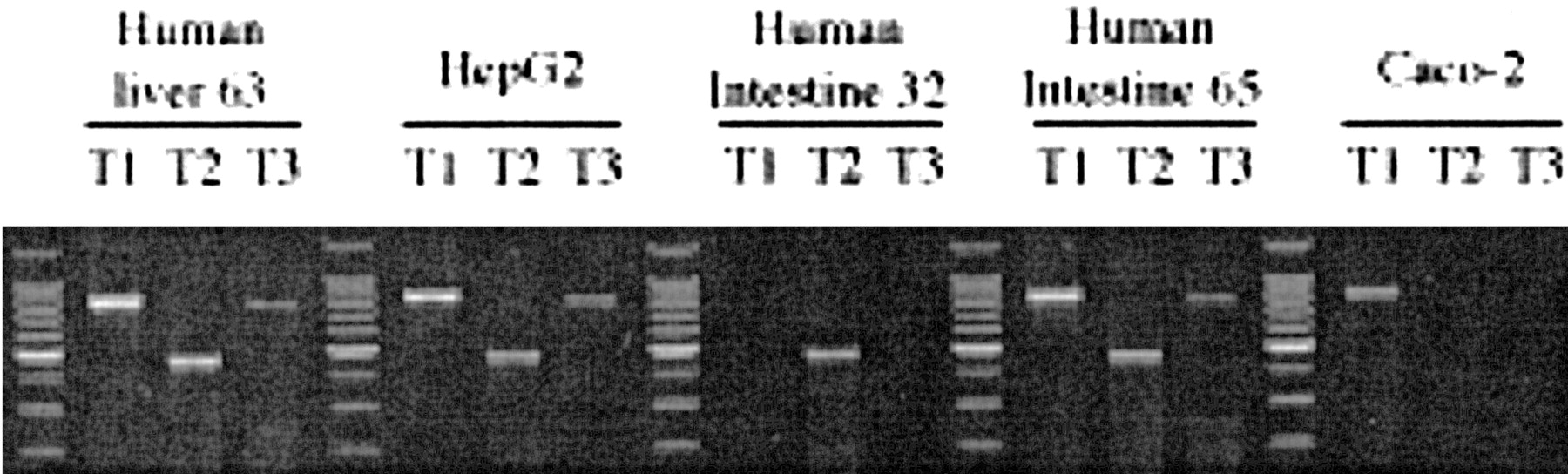
RT-PCR analysis of mRNA expression of hPXR variants in human tissues and cell cultures.
Total mRNA from human liver, human intestine, and cell cultures was analyzed by RT-PCR. Primers specific for the hPXR variants T1, T2, and T3 were used to generate the amplicons. A DNA standard ladder is positioned in the first lane and between each tissue sample group.
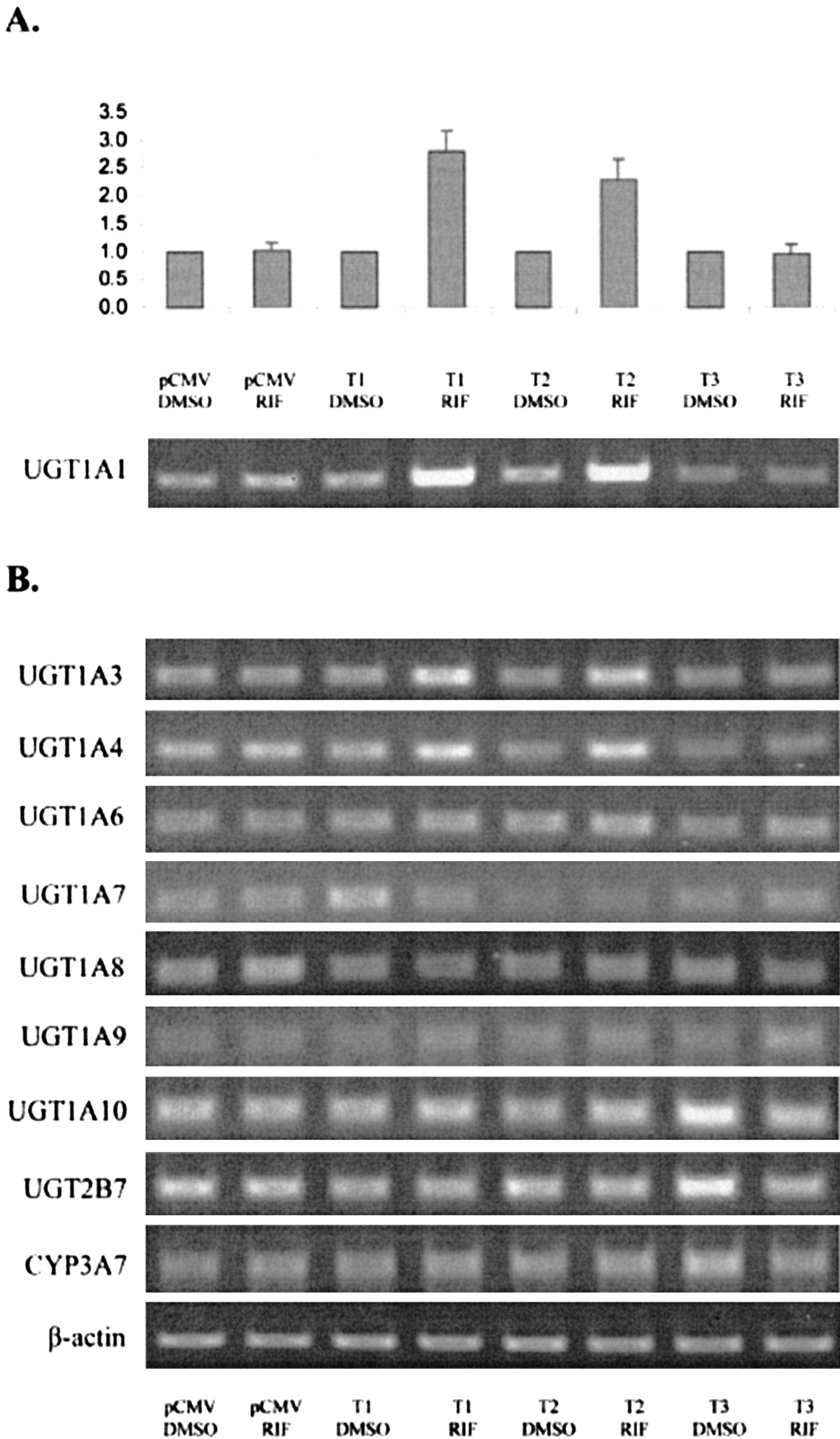
Induction of CYP3A7 and UGT Genes by PXR Variants in HepG2 Cells. Since it is well established that RIF, via PXR, induces P450s, we selected one isoform, CYP3A7, as a model for comparison with UGT induction studies. Cells transfected with control plasmid pCMV5 responded to the addition of RIF with a modest increase in CYP3A7 transcription (Fig. 3A). Cotransfection of PXR variant 1 in the presence of RIF enhanced this up-regulation approximately 4-fold relative to the original basal expression levels. Interestingly, hPXR T2 was as effective as T1 in mediating CYP3A7 induction, and T3 had no effect on transcription of any of the genes studied. This experiment was carried out to ensure that the HepG2 cells responded correctly to the inducer and no quantitation was attempted.

Induction of CYP3A7 and the UGT1A gene family members by hPXR in a hepatocellular carcinoma cell line.
HepG2 cells were transfected with plasmids containing hPXR variants and treated with 10 μM RIF or solvent control. Total RNA harvested from cells exposed to each treatment was analyzed by RT-PCR for altered transcriptional regulation of CYP3A7 (A), UGT1A family (B), UGT1A1 (C), UGT1A3 (D), and UGT1A4, 1A6, and 1A9 (E). Results have been normalized by comparison with the expression of β-actin and are expressed as the mean (n = 3) ± S.D. The PCR shown in Fig. 3A is a control experiment carried out only once to ensure that the HepG2 cells responded correctly to the inducers as expected. The experiments in Fig. 3E were not quantitated since the bands were not clearly visible; therefore, only the raw data are presented.
There was a very marked increase in UGT1A transcripts in HepG2 cells transfected with either T1 or T2 and subsequently treated with RIF. When expressed in the presence of 10 μM RIF, these two PXR variants were each responsible for increased transcription of the UGT1A family by up to 7-fold (Fig. 3B). Interestingly, however, this augmented expression was found to be inconsistent among individual 1A family members, most likely due to the differential expression of UGT1A family members.
UGT1A1 appeared to contribute the most to the observed UGT1A family up-regulation in HepG2 cells by responding very considerably to the combined presence of either recombinant hPXR T1 or T2 and RIF. More than a 25-fold induction of UGT1A1 transcripts was observed under the conditions used (Fig. 3C). Similarly, UGT1A3 was strongly up-regulated by hPXR in response to RIF (Fig. 3D). Although baseline expression was low, UGT1A3 was observed to be induced to approximately 8-fold over controls. Again, T1 and T2 exhibited a similar potency for mediating up-regulation, whereas T3 showed no noteworthy activity (Fig. 3D).
Figure 3E illustrates that UGT1A6 and 1A4 were also responsive to PXR/RIF treatment, although to a much lesser extent than UGT1A1 and UGT1A3. In contrast, the level of UGT1A9 transcription remained unchanged by PXR transfection and RIF treatment. Production of UGT1A9 mRNA in HepG2 cells was not induced by cotransfection of constitutively active hPXR or human constitutive androstane receptor (data not shown). UGT1A7, 1A8, and 1A10 were also examined, but transcripts could not be detected in HepG2 cells. The data shown are representative of all replicates. However, the bands were too faint to quantify accurately, hence the raw data are shown.
In comparison with the UGT1A family, PXR-mediated induction of UGT2B transcripts was not detected. The ability of all three PXR variants to up-regulate UGT2B7 was examined, and no induction was observed. In addition, cotransfection of recombinant T1 had no effect on UGT2B4, 2B10, 2B11, 2B15, or 2B17 mRNA (data not shown). UGT2B28 transcripts could not be detected in HepG2 cells (data not shown), and the T2 and T3 PXR variants were not tested for regulatory interaction with any UGT2B gene other than UGT2B7. Therefore, it appears that hPXR, particularly variant T1, cannot direct transcription from the UGT2B promoter in HepG2 cells under the experimental conditions used. However, we cannot exclude the possibility that an assay of greater sensitivity may reveal subtle changes that currently remain undetected, but any such associations would be anticipated to be weak.
CYP3A7 and UGT Induction in Caco-2 Cells. Although the UGT1A expression profile in Caco-2 cells differed from that of HepG2 cells, the response of most individual genes to PXR/RIF treatment was similar, with some noteworthy exceptions (Fig. 4). UGT1A1 (Fig. 4A) was again strongly up-regulated by PXR variants T1 and T2, but to a lesser extent than in HepG2 cells. UGT1A3 and UGT1A4 also showed obvious responses to T1 and T2 but not T3 (Fig. 4B). These modest but clear up-regulations were consistent between replicates of the experiment shown in Fig. 4 and were confirmed in additional independent transfections. UGT1A9 and UGT2B7 expression again remained unchanged regardless of treatment. Unlike HepG2 cells, UGT1A7, 1A8, and 1A10 mRNAs were detectable in Caco-2 cells. Like the closely related UGT1A9 gene, UGT1A7, 1A8, and 1A10 were not detectably increased by PXR and RIF. This experiment was carried out several times, and the results were not consistent among replicates. On average, there was no effect.
Transcriptional regulation of CYP3A7 and UGT genes by hPXR in a colorectal adenocarcinoma cell line.
Caco-2 cells were transfected with expression constructs for hPXR variants T1, T2, or T3 and exposed to dimethyl sulfoxide or RIF as described in the text. After treatment, regulation of target genes was analyzed by RT-PCR using specific primers for the coding regions of UGT1A1 (A) and UGT1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7, and CYP3A7 (B). Up-regulation of UGT1A1 transcripts is presented as the mean (n = 3) ± S.D.
The only UGT1A isoform expressed in both HepG2 and Caco-2 cells, but found to differ in response to PXR/RIF treatment, was UGT1A6. UGT1A6 transcripts are present in Caco-2 cells at substantially higher levels than in HepG2 cells, yet the response seen in the latter to T1 and T2 (Fig. 3E) was not observed in Caco-2 culture (Fig. 4B). The other major difference observed between the two cell lines transfected with hPXR and subsequently exposed to RIF was the lack of CYP3A7 response in Caco-2 cells. CYP3A7 transcripts have been shown previously to be present at low levels in Caco-2 cells (Schmiedlin-Ren et al., 1997) and are known to be responsive to rifampicin in other cell lines (this study; Pascussi et al., 1999), yet CYP3A7 did not respond appreciably to the addition of RIF, with or without PXR cotransfection. Therefore, it is evident that hPXR variants display cell line- and, most likely, tissue-specific activity. It has also been demonstrated, in all our studies, that when PXR T1 was active, T2, but not T3, also showed activity. Thus, the behavior among the individual variants relative to one another was consistent among all genes and the two host cells tested.
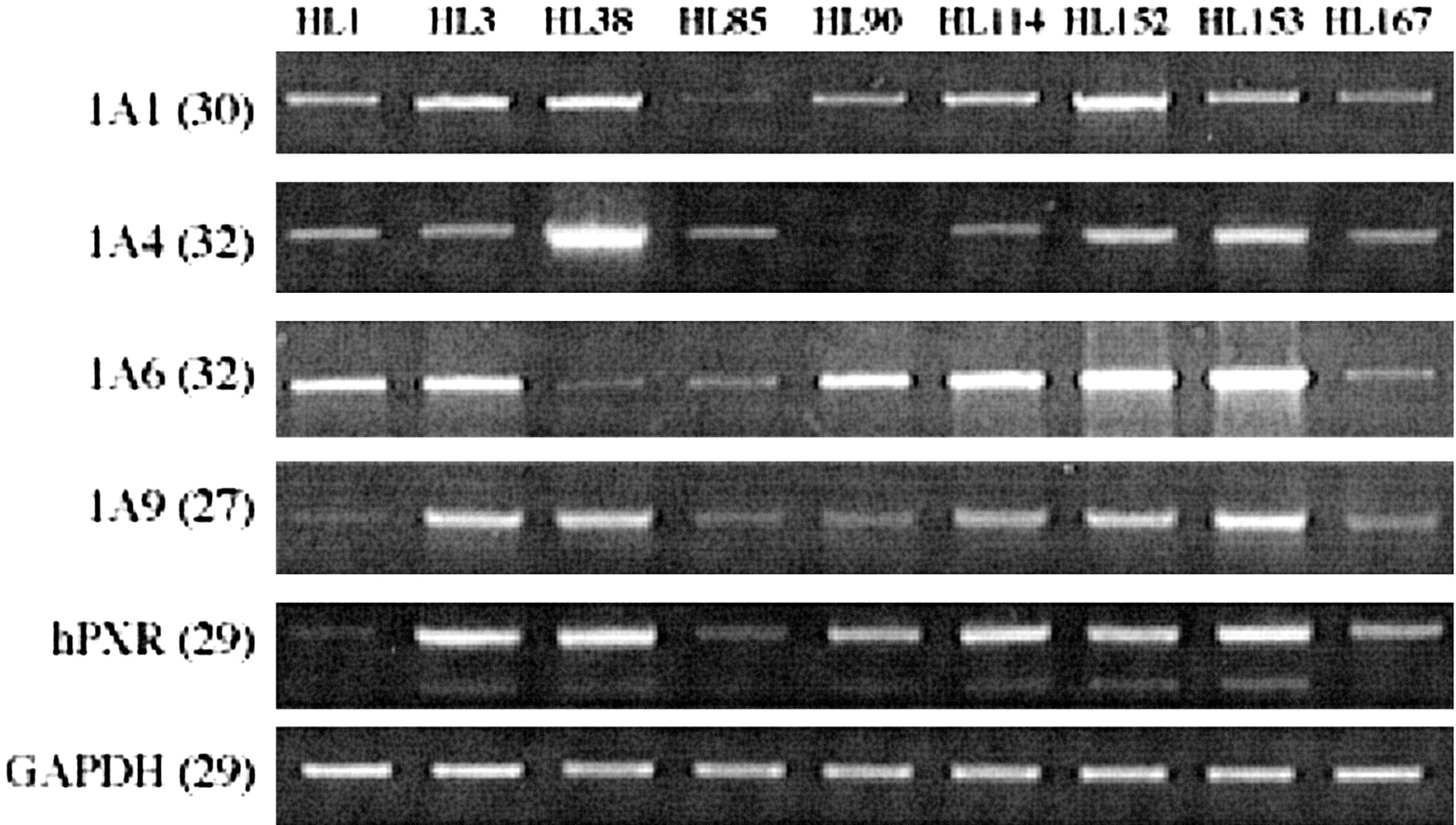
Variability of hPXR mRNA Expression Levels in Human Liver and Intestine. As variability in expression of hPXR T1, T2, and T3 variants could potentially influence the transcriptional regulation of target genes in human liver, we investigated mRNA expression in nine human livers derived from generally healthy donors. The mRNA levels of hPXR variants were measured by RT-PCR and normalized to the GAPDH mRNA level in each sample. The results obtained are shown in Fig. 5. RT-PCR with common primers, as well as those for the individual transcripts, showed great individual variations among donors. Overall, livers with high expression of one variant also had high expression of the other two variants. Interestingly, the UGT1A isoform whose expression correlates most with the expression of PXR was UGT1A9. However, we have not identified this enzyme as being up-regulated by any of the hPXR variants in HepG2 and Caco-2 cells. This phenomenon could be explained by the possibility that these cells do not have all the cofactors necessary for UGT1A9 transactivation by PXR.

Individual variation of hPXR transcript expression in human liver samples.
General and variant specific expression of hPXR in total RNA purified from nine human liver samples were analyzed by RT-PCR. To optimize transcript detection, each RT-PCR profile was obtained from reactions with differing numbers of cycles (in parentheses). H1 and H2 were, respectively, a 66-year-old female and a 49-year-old male, both of whom died of strokes. The remaining liver samples were from normal females ranging in age from 23 to 64. No further information was available for these donors. The PCR shown is representative of three to seven estimations.
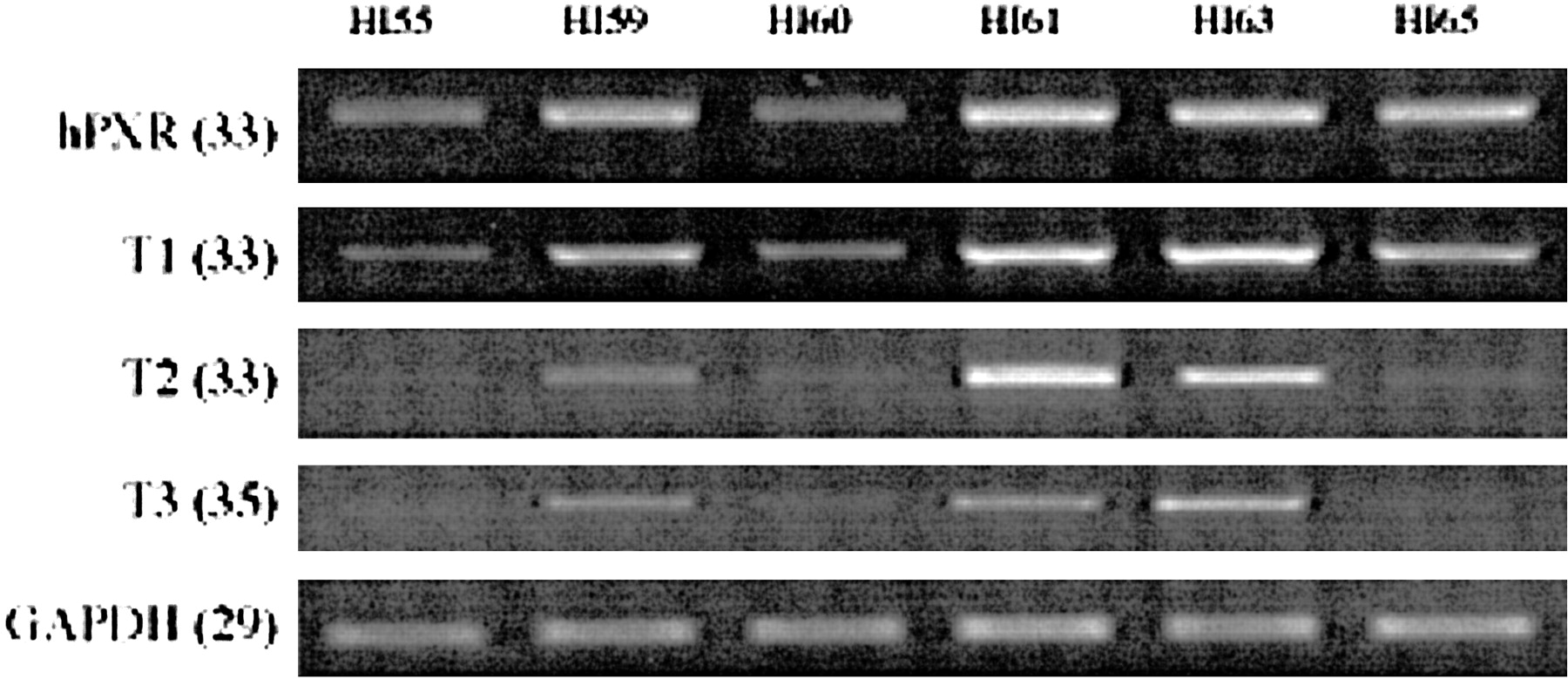
We also investigated mRNA expression in six human intestinal (jejunum) segments from healthy donors. As noted previously, the mRNA levels of hPXR variants were measured by RT-PCR and normalized to the GAPDH mRNA level in each sample. As with the liver samples, the intestinal mucosa showed great individual variations between donors. mRNA expression for the individual donors was considerably different in terms of amount and distribution pattern (Fig. 6).

Individual variation of hPXR transcript expression in human intestine.
General and variant specific expression of hPXR in total RNA purified from six human intestinal (jejunum) samples were analyzed by RT-PCR. To optimize transcript detection, each RT-PCR profile was obtained from reactions with differing numbers of cycles (in parentheses). Donors were as follows: H55, 49-year-old male, died of a stroke; H59, 19-year-old male; H60, 21-year-old male; H61, 47-year-old male; H63, 39-year-old male; H65, 22-year-old male; all of whom died in motor vehicle accidents.
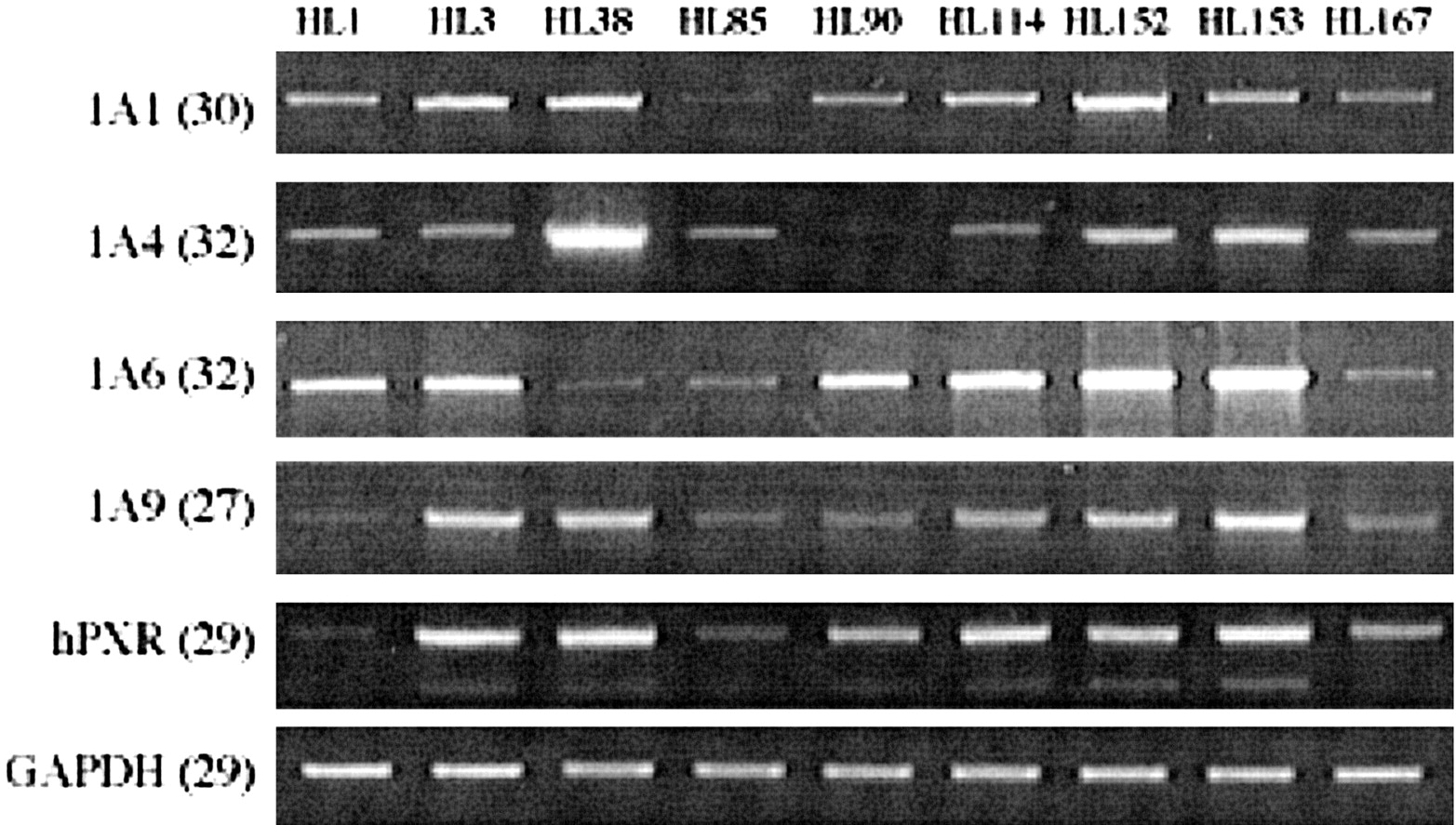
Finally, the expression of hPXR and UGT1A isoforms was compared, and the results are shown in Fig. 7. There was noticeable interindividual variation observed in the expression of UGT1A isoforms in different donors. In general, donors with a low level of one UGT isoform also had low levels of the other isoforms, and the reverse was also true. Although quantitative evaluation was not possible for this experiment, the level of expression of some UGT1A isoforms appeared to mimic hPXR transcript expression.
Expression of several UGT1A family members mimics hPXR transcript expression.
RNA isolated from human liver samples was analyzed by RT-PCR to examine levels of expression for several UGT1A family members. For comparison, hPXR expression was also analyzed from the same liver samples. To optimize transcript detection, each RT-PCR profile was obtained from reactions with differing numbers of cycles (in parentheses). The source of the liver samples is described in the legend to Fig. 5.
Discussion
The results presented here support the recent report that PXR is capable of regulating the expression of UGT1A family isoforms and are in agreement with our previous results (Xie et al., 2003). Here, we show that two additional UGTs, UGT1A3 and UGT1A4, are also up-regulated by PXR variants. This finding suggests an important role for this xenobiotic receptor in the ligand-dependent activation of human UGTs. Our data also demonstrate that there are different mechanisms involved in the regulation of hepatic and intestinal UGTs. This would be consistent with the presence of distinct liver- and intestine-specific UGT isoforms.
Several laboratories have demonstrated the presence of natural variants of hPXR (Bertilsson et al., 1998; Kast et al., 2002), some of which possess altered transactivation activity toward P450 genes (Lehmann et al., 1998; Hustert et al., 2001). However, no systematic studies have been performed yet on the tissue distribution and function of these variants or their role in regulation of human UGTs. In these studies, we have analyzed the available PXR amino acid sequences and designed primers for the identification of three human PXR mRNAs, corresponding to the wild-type hPXR (T1) and variants with 39 extra N-terminal amino acids (T2) or an internal 37-amino acid deletion (T3). We have also cloned all three PXR variants and investigated their effect on regulation of UGTs from both the UGT1A and 2B families, as well as CYP3A7. Moreover, we have carried out the first systematic studies of the expression of these variants in human liver and intestine and in the human derived cell lines, HepG2 and Caco-2. It has been suggested that hPXRs are expressed in a restricted number of tissues, with the highest expression observed in the liver, followed by small intestine and colon (Bertilsson et al., 1998). Generally, hPXR variant expression has been analyzed in human tissue by Northern blot, and hPXR mRNAs of different sizes have been detected in both liver and intestine; however, in the latter tissue, hPXR expression was limited to the cells of the intestinal mucosal layer (Bertilsson et al., 1998). In addition, one earlier study has described the expression of PXR T1 and T3 by RT-PCR in normal and cancerous breast tissue (Dotzlaw et al., 1999).
Our data demonstrate that variants of human PXR are expressed in human liver, HepG2 cells, intestinal mucosa, and CaCo-2 cells; however, the pattern of distribution varies considerably between hepatic and intestinal tissue. All the variants were expressed in human liver and HepG2 cells. However, distribution in human intestine and Caco-2 cells was very different. In jejunum from H32, only T2 was expressed while, in the same tissue from H65, the pattern was identical to that of liver. Variants T2 and T3 were missing in Caco-2 cells. This indicates important tissue-specific distribution of hPXR variants and reflects the differential regulatory function of these variants.
We also investigated the effect of hPXR variant expression on the regulation of UGTs and CYP3A7 in HepG2 and Caco-2 cells. Interestingly, the same genes were up-regulated in both cell lines. However, the rate of up-regulation and response to RIF was somewhat different. In HepG2 cells, UGT1A1 responded quite strongly to the combined presence of T1/T2 and RIF. These data are consistent with a recent report by Xie et al. (2003), which showed that UGT1A1 is up-regulated in mice bearing constitutively activated hPXR. The same investigators also showed that hPXR and human constitutive androstane receptor can induce UGT1A1 promoter reporter gene expression in HepG2 cells. The UGT1A1 response to PXR T1 and T2 in the presence of RIF mirrors that of the UGT1A family as a whole. This is because, first, UGT1A1 is strongly induced by RIF and, second, UGT1A1 appears to be the major UGT isoform expressed in these cells. Thus, UGT1A1 up-regulation may be the major contributor to the up-regulation of the UGT1A family as a whole. However, UGT1A3 expression was also extensively increased, and UGT1A4 and 1A6 expression may contribute to the up-regulation as well.
Interesting data were also obtained on the regulatory effect of PXR variants in Caco-2 cells. When transfected with either T1 or T2 and treated with RIF, there was a marked increase in UGT1A1 transcription in these intestine-derived cells. Also, UGT1A3 transcription was increased by PXR/RIF. Like UGT1A3, UGT1A4 transcripts in CaCo-2 cells were clearly increased in the cells in response to T1 and RIF. Interestingly, UGT1A6, which, in our hands, is the most highly expressed UGT1A isoform in Caco-2 cells, was found not to be regulated by T1, with or without RIF. Thus, the high levels of endogenous UGT1A6, in conjunction with its unresponsiveness to PXR regulation, are likely to be responsible for the smaller response of the UGT1A family seen in Caco-2 cells relative to that of HepG2 cells. UGT1A8 and 1A10, whose expression is limited to the intestine, did not respond to PXR/RIF in Caco-2 cells. Moreover, none of the PXR variants in combination with RIF was able to influence UGT1A9 expression in Caco-2 cells.
In summary, the cell culture studies demonstrated that PXR T1 and T2 can up-regulate the UGT1A family of isoforms, in particular UGT1A1, UGT1A3, and UGT1A4. PXR T3 does not mediate upregulation of any of the genes studied. None of the UGT2B isoforms responded to any of the hPXR variants. Although HepG2 and Caco-2 cells have relatively low expression of endogenous UGT genes, they seem to be excellent models for studying hPXR-mediated regulation. This is especially true for UGT1A3 and UGT1A4, as both genes are expressed at low levels in these cell lines; however, both isoforms can be effectively up-regulated by PXR. This work has also shown, for the first time, that PXR regulates not only UGT1A1 but also UGT1A3 and UGT1A4. The different responses of UGT1A isoforms, varying from strong to undetectable, suggests differing regulation of these is under the control of its own unique promoter (Ritter et al., 1992). From these observations, it can be concluded that, like P450 enzymes and drug transporters, UGTs are also strongly regulated by xenobiotic NRs. In addition, the present studies have defined an important role of PXR in tissue specific regulation of UGTs.
Footnotes
-
↵2 Abbreviations used are: PXR, pregname X receptor; RIF, rifamppicin; hPXR, human PXR; NR, nuclear receptor; UGT, UDP-glucuronsytransferase; DMEM, Dulbecco's modified Eagle's medium; RT-PCR, reverse transcription-polymerase chain reaction; dNTP, deoxynucleoside-5′-triphosphate; UTR, untranslated region; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; P450, cytochrome P450.
-
This work was supported in part by National Institutes of Health Grants DK45123, DK60109 (A.R.-P.), 20 RR016460 (T.L.), and tobacco settlement funds (A.R.-P.). P.M. is a National Health and Medical Research Council Senior Principal Research Fellow and is supported by grants from the Cancer Council South Australia and the National Health and Medical Research Council of Australia. W.X. is supported by NIH Grant ES12479 and Department of Defense Grant BC23189. T.L. is also supported by funds from the Biomedical Research Infrastructure Network.
-
J.-M.H. and D.G.-S. contributed equally to this work.
-
↵1 Present address: Unité de Biochimie-Pharmacologie-Toxicologie, Université de Bourgogne, Dijon, France.
- Received August 27, 2003.
- Accepted November 11, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}