


Abstract
The substrate depletion method is a popular approach used for the measurement of in vitro intrinsic clearance (CLint). However, the incubation conditions used in these studies can vary, the consequences of which have not been systematically explored. Initial substrate depletion incubations using rat microsomes and hepatocytes were performed for eight benzodiazepines: alprazolam, clobazam, clonazepam, chlordiazepoxide, diazepam, flunitrazepam, midazolam, and triazolam. Subsequent predictions of in vivo CLint (ranging from 3 to 200 ml/min) and hepatic clearance (CLH) (ranging from 0.3 to 15 ml/min) demonstrated that the general predictive ability of this approach was similar to that of the traditional metabolite formation method. A more detailed study of the substrate depletion profiles and CLint estimates indicated that the concentration of enzyme used is of particular importance. The metabolism of triazolam, clonazepam, and diazepam was monoexponential at all cell densities using hepatocytes; however, with microsomes, biphasic depletion was apparent, particularly at higher microsomal protein concentrations (2-5 mg/ml). Enzyme activity studies indicated that enzyme loss was more pronounced in the microsomal system (ranged from 8 to 65% activity after a 1-h incubation) compared with the hepatocyte system (approximately 100% activity after a 1-h incubation). For clonazepam (a low clearance substrate), these biphasic profiles could be explained by loss of enzyme activity. To ensure accurate predictions of in vivo CLint and CLH when using the substrate depletion approach, based on the results obtained for this class of drugs, it is recommended that low enzyme concentrations and short incubation times are used whenever possible.
The in vitro measurement of intrinsic clearance (CLint) using hepatic microsomes and/or hepatocytes is frequently used in both academia and the pharmaceutical industry to estimate the in vivo metabolic stability of new drug entities in both rat and human (Houston, 1994; Iwatsubo et al., 1997; Obach, 1999; McGinnity and Riley, 2001). The results of these assays are scaled and modeled, as described in eq. 1 for the well stirred liver model, to estimate in vivo hepatic clearance (CLH). 
where QH is the hepatic blood flow, SF represents the milligrams of microsomal protein or million cells per gram of liver multiplied by the grams of liver weight; fu b is the unbound fraction of drug in the blood, and fuinc is the unbound fraction in the incubation matrix.
Traditionally, the metabolite formation method has been used for measurement of in vitro CLint, where the initial rate of metabolite production using hepatic microsomes or hepatocytes is measured over a range of substrate concentrations under linear conditions with respect to protein concentration/cell density and time (Madan et al., 2002; Houston et al., 2003). In general, short incubation times and low enzyme (protein) concentrations are used in these studies, since compliance with the Michaelis-Menten equation assumes less than 10% substrate consumption. Therefore, issues such as the stability of the enzyme preparation (resulting from long incubation times), nonspecific binding (resulting from high enzyme concentrations), and end-product inhibition (resulting from the accumulation of the phase I hydroxylated metabolite in the microsomal incubation) are not generally limitations. However, the main disadvantage to this approach is that it requires prior knowledge of the particular metabolic pathway under study and its importance to the overall metabolic fate of the drug to ensure a true and accurate prediction of clearance; this may be particularly problematic if multiple metabolic pathways are involved. For many drugs, especially new drug candidates, this information is not known; therefore, the use of this approach is limited.
More recently, the substrate depletion approach has been adopted, where the consumption of parent drug is monitored over time. This method is particularly popular in the pharmaceutical industry because formal kinetic characterization and metabolite quantification are not required, allowing the rapid screening of compounds (Lavé et al., 1997; Obach, 2001; Obach and Reed-Hagen, 2002; Austin et al., 2002). However, compared with the metabolite formation approach, this method has been poorly defined. Although CLint is expressed per unit time and per unit enzyme (protein), initial linearity studies are usually not performed and arbitrary values for enzyme concentration and incubation time are used. Linearity is a necessary requirement when scaling CLint from in vitro incubations to the whole liver (Houston, 1994). What remains unresolved is the impact of this lack of formality on the predictive utility of this method. In contrast to the metabolite formation method, for analytical/sensitivity reasons, normally at least 20% of the substrate must be metabolized within the incubation period, so that any substrate depletion can be distinguished from baseline variability. Consequently, in general, longer incubation times and higher protein concentrations are used than for metabolite formation studies (e.g., substrate depletion incubation conditions of up to 90 min and 10 mg/ml have been reported by Austin et al., 2002, and Obach, 1999, respectively). For these reasons, the stability of the enzyme preparation, nonspecific binding, and end-product inhibition (resulting from the more extensive metabolism) must be considered.
The aim of this work was to evaluate the substrate depletion approach for the prediction of in vivo CLint and CLH and to examine in detail some of the limitations associated with this method, including the effects of enzyme concentration and incubation time. The rat was used as a model system to study eight benzodiazepines: alprazolam, clobazam, clonazepam, chlordiazepoxide, diazepam, flunitrazepam, midazolam, and triazolam. First, substrate depletion incubations were performed using microsomes and hepatocytes for all eight compounds, and subsequently, predictions of in vivo CLint and CLH were made. The general predictive ability of this approach was compared with the metabolite formation method (Ito and Houston, 2004). Triazolam, clonazepam, and diazepam were selected for further study to explore the effect of incubation time and enzyme concentration on enzyme activity over time, in vivo CLint and CLH predictions, and, in particular, the shape of the depletion time profile. The ultimate goal of this work was to establish a set of recommended incubation conditions for use with the substrate depletion approach in an attempt to optimize its predictive utility.
Materials and Methods
Materials. 3-Hydroxydiazepam, prazepam, and 4-hydroxytriazolam were obtained from Wyeth Pharmaceuticals (Maidenhead, UK), Warner Lambert Ltd. (Eastleigh, UK), and BIOMOL Research Laboratories (Plymouth Meeting, PA), respectively. 4′-Hydroxydiazepam, nordiazepam, and 3-hydroxyclonazepam were kindly supplied by F. Hoffman-La Roche (Basel, Switzerland). All other chemicals and reagents used were of the highest grade available and were purchased from Sigma Chemical (Poole, Dorset, UK) or BDH (Poole, Dorset, UK).
Animal Source, Housing, and Diet. Male Sprague-Dawley rats (220-270 g) were obtained from the Biological Sciences Unit, Medical School, University of Manchester. They were housed in groups of two to four, in opaque boxes on a bedding of sawdust. The accommodation was maintained at a temperature of 20 ± 3°C, with a relative humidity of 40 to 70% and a 12-h light/dark cycle. The animals were allowed free access to Chow Rat Mouse diet and fresh drinking water.
Preparation of Rat Liver Microsomes and Rat Hepatocytes. Rat liver microsomes and hepatocytes were prepared, as described previously by Hayes et al. (1995), from Sprague-Dawley rats. The protein content of the microsomes was determined using the method of Lowry et al. (1951). Hepatocyte viability was measured using the trypan blue exclusion test, and only preparations in excess of 85% viability were used.
Substrate Depletion in Rat Hepatocytes. Triazolam, clonazepam, diazepam, alprazolam, clobazam, chlordiazepoxide, flunitrazepam, or midazolam [2.5 μl; final concentration 2.5 μM; in dimethylformamide (DMF) or methanol (diazepam); final solvent concentration 0.5%] was incubated with Williams' medium E (pH 7.4) at 37°C. The reaction was initiated after 5 min by the addition of preincubated, freshly isolated rat cells [final concentration 0.5, 1, 2, or 4 × 106 cells/ml (triazolam), 0.5 and 4 × 106 cells/ml (clonazepam and diazepam), or 0.5 × 106 cells/ml (alprazolam, clobazam, chlordiazepoxide, flunitrazepam, and midazolam], giving a final incubation volume of 500 μl. Reactions were terminated at regular time intervals (0-120 min) by snap freezing in liquid nitrogen.
Substrate Depletion in Rat Liver Microsomes. Triazolam, clonazepam, diazepam, alprazolam, clobazam, chlordiazepoxide, flunitrazepam, or midazolam [2.5 μl; final concentration 2.5 μM; in DMF or methanol (diazepam); final solvent concentration 0.5%] was incubated with 0.1 M phosphate buffer (pH 7.4) and rat liver microsomes [final concentrations 0.1, 0.5, 1, 2, and 5 mg/ml (triazolam), 0.5, 1, 2, and 5 mg/ml (clonazepam), 0.5 and 5 mg/ml (diazepam), or 1 mg/ml (alprazolam, clobazam, chlordiazepoxide, flunitrazepam, and midazolam)] in a shaking water bath at 37°C. The reaction was initiated after 5 min by the addition of a cofactor regenerating system [250 μl, containing 0.37 mg of NADP (nicotinamide adenine dinucleotide phosphate), 0.97 mg of isocitric acid, 0.6 unit of isocitric dehydrogenase, and 8.1 μmol of magnesium chloride in 0.1 M phosphate buffer (pH 7.4)], giving a final incubation volume of 500 μl. Reactions were terminated at regular time intervals (0-90 min) with 250 μl of ice-cold acetonitrile (triazolam and clonazepam), 250 μl of ice-cold methanol (diazepam), or 10 μl of 10 M HCl (alprazolam, clobazam, chlordiazepoxide, flunitrazepam, and midazolam).
Rat Microsomal Binding. Equilibrium dialysis was used to determine the extent of binding of triazolam, clonazepam, and diazepam (2.5 μM) to rat liver microsomes at microsomal protein concentrations between 0.1 and 5 mg/ml. All other compounds were measured at 1 mg/ml only. Incubation mixture (1 ml, without NADP) containing the compound was added to one side (microsomal protein side) of a dialysis cell (Diachema AG, Zurich, Switzerland) containing a dialysis membrane (Dianorm GMBH, Munich, Germany) and 1 ml of 0.1 M phosphate buffer (pH 7.4) to the other side (buffer side). The membrane had been presoaked in 0.1 M phosphate buffer (pH 7.4) overnight. The cells were sealed and the apparatus was fully immersed in water and rotated in a water bath at 37°C for 4 to 5 h. Initial time studies were performed to determine the time taken to establish equilibrium. Equipment binding was less than 10% and thus was considered to be minimal.
Diazepam Cell Uptake Studies. The uptake of 14C-radiolabeled diazepam (1-500 μM) into isolated hepatocytes (2 × 106 cells/ml) was determined at 0 and 37°C. After a 60-s incubation, the cells were separated by centrifugation through a layer of silicone oil and trapped in a lower layer of NaOH as described previously (Jones et al., 2004). The impact of binding for the other drugs within hepatocyte incubations was presumed to be negligible at the lower cell concentrations (0.5-1 × 106 cells/ml); however, at higher cell concentrations (4 × 106 cells/ml), binding may be more significant (see Results).
Time Linearity and Mechanism-Based Inactivation Studies. To determine time linearity for midazolam (2.5, 5, 10, and 50 μM), triazolam, clonazepam, and diazepam (2.5 μM) metabolism, incubations were performed at a microsomal protein concentration of 0.2 mg/ml (midazolam) or 0.5 mg/ml (triazolam, clonazepam, diazepam) for 0 to 15 min (midazolam), 0 to 20 min (triazolam), 0 to 60 min (clonazepam), and 0 to 40 min (diazepam) as described above to monitor metabolite formation.
Further inactivation studies were performed using midazolam with dextromethorphan as a probe for enzyme activity. Midazolam (2.5 μl; final concentration 0, 10, and 250 μM, in methanol; final solvent concentration 0.5%) was incubated as described previously, with 0.1 M phosphate buffer (pH 7.4) and a cofactor regenerating system at a microsomal concentration of 3.5 mg/ml. At selected time intervals (0, 5, 10, 20, and 30 min) 25-μl aliquots were transferred to a secondary reaction mixture [containing dextromethorphan (2.5 μl; final concentration 50 μM, in phosphate buffer), 0.1 M phosphate buffer (pH 7.4), and a cofactor regenerating system] giving a final microsomal protein concentration of 0.175 mg/ml. The secondary reaction was terminated after 7 min by the addition of 250 μl of ice-cold acetonitrile. These incubation conditions were within the linear range of metabolite production (Witherow and Houston, 1999). P450 activity with respect to methoxymorphinan (CYP3A) and dextrophan (CYP2D) production was measured.
Hepatocyte Enzyme Activity with Time. Rat hepatocytes (final concentration 0.5 and 1 × 106 cells/ml) and Williams' medium E (pH 7.4) were preincubated for 5 min at 37°C. The reaction was initiated at regular time intervals between 0 and 240 min by the addition of diazepam or 7-ethoxycoumarin [2.5 μl; final concentration 2.5 μM, in methanol (diazepam), 50 μM, in DMF (7-ethoxycoumarin); final solvent concentration 0.5%], giving a final incubation volume of 500 μl, and terminated after 2.5 min (diazepam) or 3 min (7-ethoxycoumarin). Viability was assessed at each of the time points by the trypan blue exclusion test.
Microsomal Enzyme Activity with Time. Rat liver microsomes (final concentration 0.5, 1, 2, and 5 mg/ml for triazolam; 0.5 and 5 mg/ml for clonazepam) were preincubated with 0.1 M phosphate buffer (pH 7.4) and a cofactor regenerating system in a shaking water bath at 37°C for 5 min. The reaction was initiated after 0 to 60 min by the addition of triazolam or clonazepam (2.5 μl; final concentration 2.5 μM, in DMF; final solvent concentration 0.5%), giving a final incubation volume of 500 μl. Reactions were terminated after 2.5 min with 250 μl of ice-cold acetonitrile.
Sample Analysis. Hepatocyte samples were thawed and 5 μl of internal standard (final concentration 5 μM) was added. Samples were mixed and then centrifuged at 11,600g for 15 min (Eppendorf centrifuge 5413; Eppendorf International, Hamburg, Germany). Triazolam, clonazepam, alprazolam, clobazam, chlordiazepoxide, and flunitrazepam samples were extracted from the supernatant by solid phase extraction (using Varian Bond Elut C18 Octadecyl 100 mg/1 ml cartridges; Phenomenex, Macclesfield, Cheshire, UK) and eluted with 400 μl of methanol. The methanol was evaporated to dryness (SpeedVac AES1010; Thermo Savant, Holbrook, NY), and the residue was reconstituted in mobile phase. Diazepam and midazolam samples were extracted with 5 ml of ethyl acetate after the addition of 1 ml of 0.1 M carbonate buffer (pH 10) by rotary mixing for 30 min and centrifuging at 900g for 10 min. The organic layer was removed and evaporated to dryness under nitrogen at 40°C, and the residue was reconstituted in mobile phase. In addition, hepatocyte enzyme activity samples were incubated with 0.5 ml of β-glucuronidase with sulfatase activity [200 units/ml (diazepam), 500 units/ml (7-ethoxycoumarin) in sodium acetate; 60 mM, pH 4.5] for 1.5 h in a shaking water bath at 37°C. All samples were analyzed by HPLC.
Internal standard (5 μl; final concentration 5 μM) was added to the microsomal samples, which were then centrifuged at 11,600g for 15 min. For triazolam, clonazepam, diazepam, and dextromethorphan samples, 200 μl of the supernatant was analyzed by HPLC, whereas alprazolam, clobazam, chlordiazepoxide, flunitrazepam, and midazolam microsomal samples were extracted as described above and then analyzed by HPLC.
For alprazolam, clobazam, chlordiazepoxide clonazepam, flunitrazepam, and triazolam, the HPLC system consisted of a Hypersil H5ODS-250A column (250 × 4.6 mm; Phenomenex), mobile phase of 68% 10 mM K2HPO4·3H2O (pH 3.7, with orthophosphoric acid), 29.8% acetonitrile, 2.2% methanol delivered by a Gilson 305 pump (Gilson Medical Electronics, Middleton, WI) at a flow rate of 1.5 ml/min and measured at a UV wavelength of 254 nm (triazolam and clonazepam) or 240 nm (alprazolam, clobazam, chlordiazepoxide, and flunitrazepam) by a Milton Roy Spectromonitor 3100 detector (Milton Roy Company, Rochester, NY). For diazepam, a Hypersil BDS C18 column (150 × 4.6 mm; Phenomenex) was used with a mobile phase of 55.5% methanol and 44.5% water containing 0.02% (v/v) triethylamine adjusted to pH 7.0 with orthophosphoric acid delivered at a flow rate of 1 ml/min and measured at a UV wavelength of 254 nm. For midazolam, an Ultra Techsphere C8 column (5 μm, 150 mm; Phenomenex) was used with a mobile phase of 60% 0.025 M ammonium acetate (pH 5) and 40% acetonitrile, delivered at a flow rate of 1.5 ml/min and measured at a UV wavelength of 240 nm. Finally, for dextromethorphan, an Ultra Techsphere C8 (5μm, 150 mm, Phenomenex) column was used with mobile phase of 66% water, 34% acetonitrile, 0.1% triethylamine, adjusted to pH 6.0 with orthophosphoric acid delivered at a flow rate of 0.6 ml/min, and measured at an excitation wavelength of 280 nm and an emission wavelength of 310 nm by a Jasco FP-920 fluorescence detector (Jasco UK Ltd., Essex, UK). Triazolam, clonazepam, diazepam (and their metabolites for time linearity and enzyme activity samples), methoxymorphinan, dextrophan, alprazolam, clobazam, chlordiazepoxide, flunitrazepam, and midazolam were quantified by their peak area ratio relative to the internal standard with reference to a standard calibration curve. For 7-ethoxycoumarin samples, 0.8 ml was added to 1.2 ml of 0.2 M glycine/NaOH buffer (pH 10.4). 7-Hydroxycoumarin production was measured using a technique modified by Carlile et al. (1998) from Greenlee and Poland (1978), using a PerkinElmer LS-2B/Luminescence Spectrometer LS50B fluorimeter (PerkinElmer Analytical Instruments, Beaconsfield, UK) at an excitation wavelength of 375 nm and an emission wavelength of 452 nm. Calibration was achieved by reference to a standard calibration curve.
All incubations were performed in duplicate using hepatocytes or microsomes prepared from four different rat livers. Substrate concentrations for depletion incubations were chosen based on published and in-house Km values (Zomorodi et al., 1995).
Data Analysis. All depletion data were fitted to the monoexponential decay model, with a 1/y weighting. Triazolam, diazepam, and clonazepam data were also fitted to the biexponential decay model. The monoexponential decay and biexponential decay models are shown in eqs. 2 and 3, respectively: 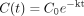
where C0 is the substrate concentration in the incubation media at time 0 and k is the terminal rate constant. 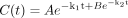
where A and B represent the back-extrapolated substrate concentration in the incubation media for phase 1 and 2, respectively, and k1 and k2 are the terminal rate constants for phase 1 and 2, respectively.
When two phases were apparent in the depletion profile, a biexponential fit was representative of a fit through both phases of the data, whereas when a monoexponential fit was used, the weighting resulted in emphasis on the first phase of the profile. Goodness of fit was assessed by the Akaike information criterion (AIC), visual examination of the data and residual plots, and the precision of the parameter estimates. In vitro CLint was calculated by dividing the dose by the area under the concentration-time curve. This value was then normalized for cell or microsomal protein concentration to obtain CLint in units of μl/min/106 cells or μl/min/mg protein.
The fraction unbound (fu) in microsomes was calculated using eq. 4, 
where Cbuffer is the concentration of drug on the buffer side and Cmicrosomal protein represents the concentration of drug on the microsomal protein side.
The fu values at different microsomal protein concentrations were fitted to eq. 5, 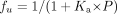
where Ka is the affinity constant and P is microsomal protein concentration.
In vitro CLint values were corrected for fu to obtain an unbound CLint using eq. 6, 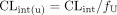
CLint(u) was scaled to an in vivo CLint by accounting for the microsomal recovery or hepatocellularity and liver weight using eq. 7 and to CLH using eq. 1: 
Assuming well stirred conditions, an in vivo CLint value for each compound was calculated from the observed CLH, blood/plasma ratio, and plasma fu using eq. 8: 
The value used for hepatic blood flow was 25 ml/min/250 g (Pang and Gillette, 1978; Pollack et al., 1999). The predicted and observed in vivo CLint and CLH values were compared.
Enzyme activity data (nmol/min) were fitted to eq. 9 (a nonlinear empirical function), 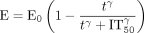
where E is enzyme activity, t is time, E0 is the enzyme activity at time 0, γ is a sigmoidicity parameter, and IT50 is the time at which 50% of the enzyme is inactivated.
The diazepam cell/media partition coefficient (Kp) was calculated based on the ratio of the concentration of diazepam within the hepatocyte to the concentration of diazepam in the media at each diazepam concentration as shown in eq. 10. 
The cell/media Kp values at different diazepam concentrations were fitted to a two-site binding model, which incorporates a high-affinity, low-capacity site and a nonsaturated linear component (eq. 11). 
where Kpmax is the maximum cell/media Kp value, Kpmin is the minimum cell/media Kp value, [S] is substrate concentration, and Kd is the specific binding constant.
Based on the cell and incubation volumes and Kp (at 2.5 μM, interpolated as 55), the unbound fraction in the whole incubation was calculated (Jones et al., 2004) using eq. 12, 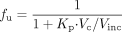
where Vc and Vinc represent the cell and incubation volumes, respectively.
Statistical Analysis. The average fold error was used to assess the accuracy of the prediction of in vivo CLint and CLH as shown in eq. 13 (Obach et al., 1997). 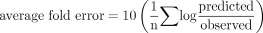
This parameter uses the prediction error (difference between predicted and observed in vivo values) for each drug and, when expressed as the geometric mean, provides a measure of bias with equal value to under- and over-predictions.
Dunnett's test was performed to determine any significant differences at the 5% level between 1) CLint values at the lowest and higher cell and microsomal protein concentrations (two-way ANOVA was used if only two concentrations were compared) and 2) P450 activity at time 0 and after preincubation for varying time periods.
Results
Initial Substrate Depletion Studies. Initial studies were performed to determine in vitro CLint for alprazolam, chlordiazepoxide, clobazam, clonazepam, diazepam, flunitrazepam, midazolam, and triazolam by measuring their depletion over time in both rat microsomes and hepatocytes from an initial substrate concentration of 2.5 μM. All microsomal CLint values were corrected for any nonspecific binding. The microsomal fu values for all compounds are shown in Table 1. Midazolam was the most highly bound compound followed by clonazepam, triazolam, and then diazepam; alprazolam, flunitrazepam, clobazam, and chlordiazepoxide exhibited negligible binding at the microsomal protein concentrations used. Therefore, for the latter four compounds, no corrections to CLint were made.
Microsomal and hepatocyte CLint and CLH predictions and observed values for eight benzodiazepines
Values are mean ± S.D. (n = 4-8).
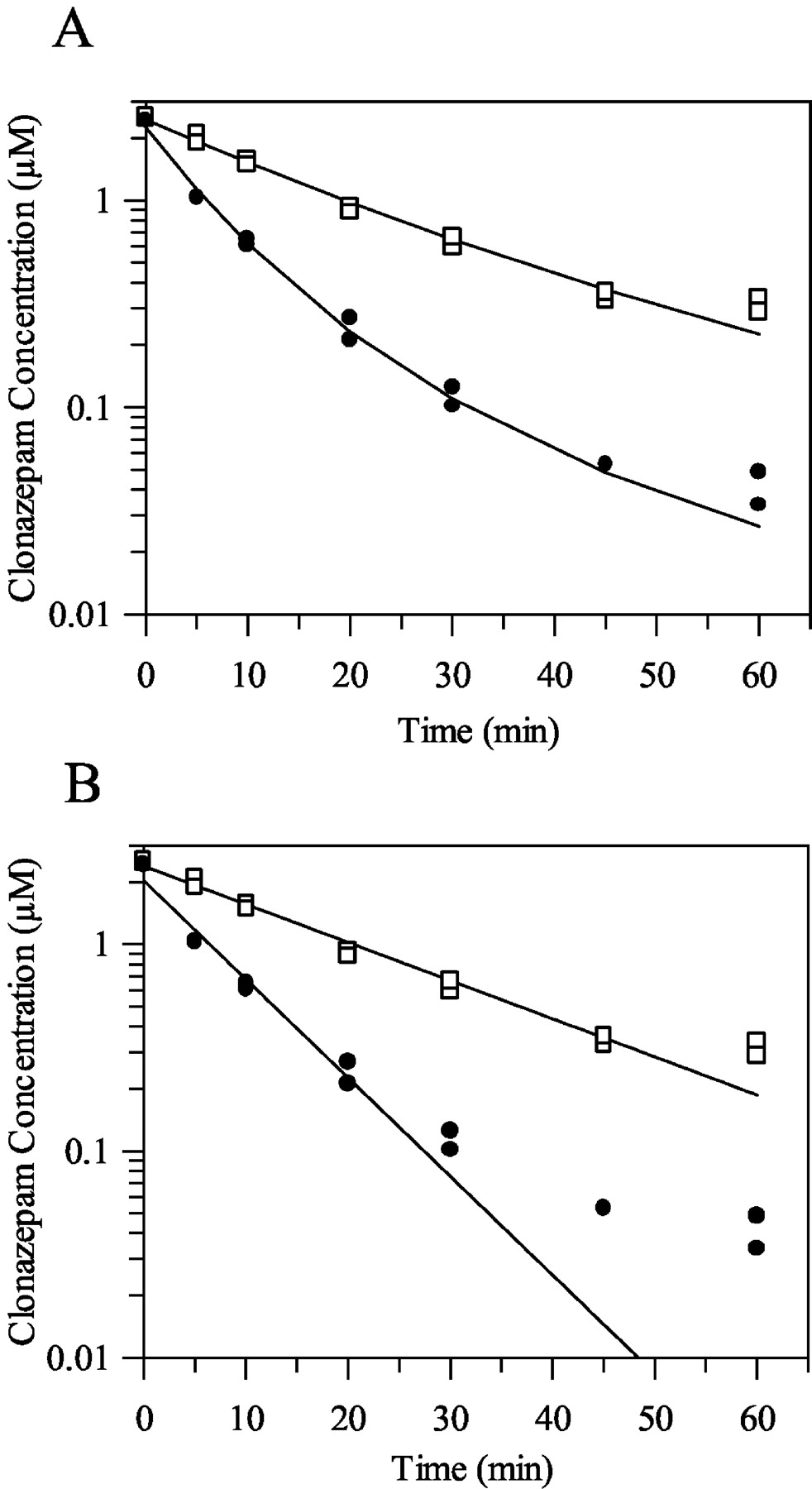
Diazepam was selected for investigation into the impact of cellular uptake on clearance determination since this drug has a log P similar to that of other benzodiazepines studied (see Table 1). Figure 1 shows substantial and concentration-dependent cellular uptake for diazepam (Kp 40-57). This was reduced by approximately 25% at 0°C compared to 37°C at low concentrations, indicating that the major component of the high Kp was intracellular binding rather than facilitated transport. The lack of any temperature effect (as well as no effect of the ATPase inhibitor rotenone or the membrane permeabilizer saponin; data not shown) at high concentrations (Kp approximately 40) is consistent with this interpretation. Given that the total volume of the incubation taken up by the cells was small (4 μl/106 cells), a Kp of 55 (corresponding to 2.5 μM diazepam incubation) results in a minimal loss of drug within the incubation (fu 0.9 for the standard 0.5 × 106 cells/ml incubation). Therefore, on the basis of these studies with diazepam, binding of the other benzodiazepines within hepatocyte incubations was presumed to be negligible at the lower cell concentrations (0.5 × 106 cells/ml), and no corrections to CLint were made. However, at higher cell concentrations (4 × 106 cells/ml), cell binding becomes more significant (fu 0.53 for diazepam) due to the increased cell volume (16 μl).

Concentration dependence of diazepam cell/media partition coefficient in rat hepatocytes at 37°C and 0°C. Mean ± S.D. (n = 3). Symbols are defined as follows: □, 37°C; ▪, 0°C.
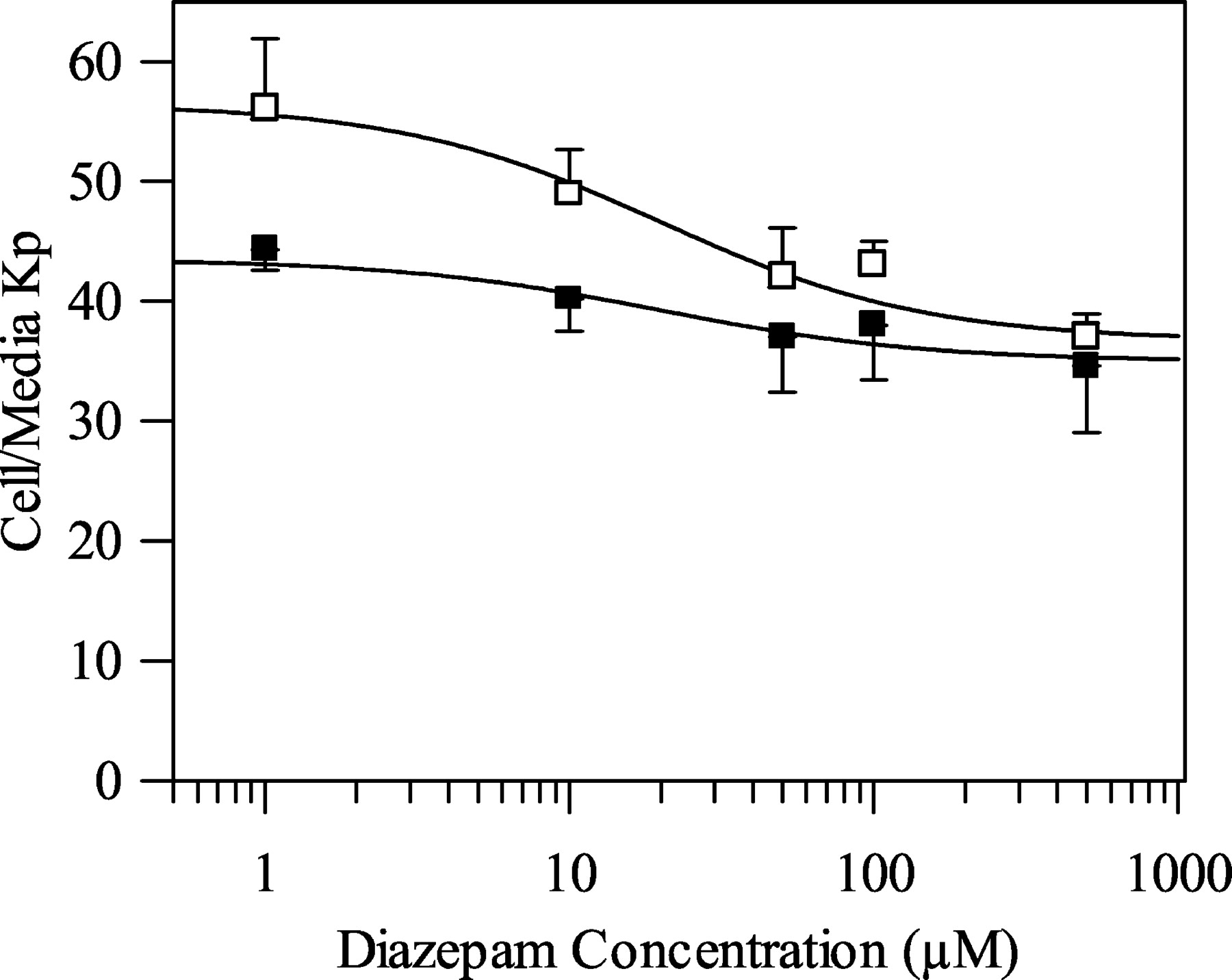
Table 1 shows the in vitro microsomal and hepatocyte CLint values, the scaled in vivo CLint values, and the predicted CLH obtained for the eight benzodiazepines studied, in addition to the observed CLH values, the plasma fu values, and the blood/plasma ratios. The microsomal in vitro CLint(u) values ranged from 4.5 μl/min/mg protein for chlordiazepoxide to 250 μl/min/mg protein for diazepam, whereas the hepatocyte in vitro CLint values ranged from 9.6 μl/min/106 cells for chlordiazepoxide to 170 μl/min/106 cells for triazolam. The rank order of in vitro and scaled in vivo CLint is similar for both in vitro systems, with the notable exceptions of clobazam and clonazepam, which were cleared faster in the microsomal system. Diazepam and triazolam were consistently the highest cleared compounds, with chlordiazepoxide being the lowest cleared. With regard to the prediction accuracy of in vivo CLint and CLH, the average fold error was 1.7 and 1.5 from hepatocytes and 2.3 and 1.9 from microsomes, respectively. Figure 2 illustrates the relationship between the predicted (from rat hepatocytes and microsomes) and observed CLint and CLH values using the substrate depletion approach described in this study, compared with previous predictions obtained using both the substrate depletion and metabolite formation approaches for a larger group of drugs (collated by Ito and Houston, 2004). The substrate depletion predictions were similar to those achieved using the traditional metabolite formation method.

A comparison of the prediction of CLint from rat hepatocytes (A) and microsomes (B) and of CLH from rat hepatocytes (C) and microsomes (D) using the metabolite formation and substrate depletion approaches. Symbols are defined as follows: ○, database (metabolite formation method); □, database (substrate depletion method); ▪, present study (substrate depletion method); the solid line represents the line of identity and dashed lines represent the propagation of a 2-fold error in CLint on the CLint and CLH prediction.
Effect of Enzyme Concentration on Substrate Depletion Profiles. Triazolam, diazepam, and clonazepam hepatocyte CLint values were determined by measuring their depletion over time at varying cell concentrations. Typical concentration-time profiles for the metabolism of triazolam, diazepam, and clonazepam in rat hepatocytes are shown in Fig. 3, A to C. At all cell concentrations, their depletion over time was linear on a log scale, and in terms of the AIC, the monoexponential model best described their metabolism. The corresponding CLint values obtained by fitting this model are shown in Table 2. Normalization of CLint to 1 × 106 cells/ml produced CLint values that decreased on increasing cell concentration (p < 0.05); triazolam, diazepam, and clonazepam CLint values decreased between 0.5 and 4 × 106 cells/ml by approximately 50, 40, and 35%, respectively.

Typical depletion concentration-time profiles in rat hepatocytes for triazolam (A), diazepam (B), and clonazepam (C) and in rat microsomes for triazolam (D), diazepam (E), and clonazepam (F). For hepatocytes a monoexponential fit is shown at all cell densities. For microsomes a monoexponential fit is shown at 0.1, 0.5, and 1 mg of protein/ml and a biexponential fit at 2 and 5 mg of protein/ml for triazolam and diazepam, and a biexponential fit at all microsomal protein concentrations for clonazepam. Symbols are defined as follows. A: □, 0.5 × 106cells/ml; ▪, 1 × 106cells/ml; ○, 2 × 106 cells/ml; •, 4 × 106cells/ml; B: ▴, 0.1 mg/ml; □, 0.5 mg/ml; ▪, 1 mg/ml; ○, 2 mg/ml; •, 5 mg/ml.
Effect of cell concentration on triazolam, diazepam, and clonazepam in vitro CLint in rat hepatocytes
Values are mean ± S.D. (n = 4). CLint is expressed as μl/min/106 cells.
Triazolam, diazepam, and clonazepam CLint values were determined by measuring their depletion over time at varying microsomal protein concentrations. Typical concentration-time profiles for the metabolism of triazolam, diazepam, and clonazepam are shown in Fig. 3, D to F. At lower microsomal protein concentrations (0.1, 0.5, and 1 mg/ml), the depletion of triazolam and diazepam was linear on a log scale, and in terms of the AIC, the monoexponential model best described their metabolism. However, in contrast to hepatocytes, at higher microsomal protein concentrations (2 and 5 mg/ml), their depletion was linear on a log scale for only 5 to 10 min. At these microsomal protein concentrations, the biexponential model best described their metabolism. For clonazepam, which was incubated for an hour, at all microsomal protein concentrations its depletion was nonlinear on a log scale, and a biexponential model best described its metabolism.
CLint values obtained by fitting the monoexponential model, expressed in terms of both total and unbound CLint in rat microsomes for all three substrates, are shown in Table 3 (biexponential estimates not shown). Triazolam, diazepam, and clonazepam microsomal binding was measured over a range of microsomal protein concentrations; their protein concentration-dependent binding is shown in Table 3. Normalization of CLint values to 1 mg/ml of microsomal protein produced CLint values that decreased on increasing protein concentration. Triazolam, diazepam, and clonazepam CLint decreased between 0.5 and 5 mg/ml by 60 to 70%. Correction for microsomal binding reduced this trend, although it did not fully eliminate it at 5 mg/ml in the case of triazolam and diazepam and at 2 and 5 mg/ml in the case of clonazepam (p < 0.05).
Effect of microsomal protein concentration and binding on triazolam, diazepam, and clonazepam in vitro CLint in rat microsomes
Values are mean ± S.D. (n = 4). CLint is expressed as μl/min/mg, and CLint values represent those obtained from monoexponential fits in all cases.
Mechanism-Based Inactivation Studies. To establish whether mechanism-based inactivation occurred, two concentrations of midazolam were preincubated with rat microsomes and a cofactor regenerating system. Aliquots from this primary reaction were diluted into secondary incubations with dextromethorphan, to assess P450 activity with respect to methoxymorphinan (CYP3A) and dextrophan (CYP2D) formation. At both midazolam concentrations, and in the absence of midazolam, there was a similar decrease in enzyme activity over time with respect to both methoxymorphinan and dextrophan production (data not shown). Therefore, there was no evidence for mechanism-based loss in enzyme activity. In addition, time linearity for midazolam (2.5, 5, 10, and 50 μM) for 5 min and for triazolam, clonazepam, and diazepam (2.5 μM) for 10 min was established in rat microsomes (data not shown).
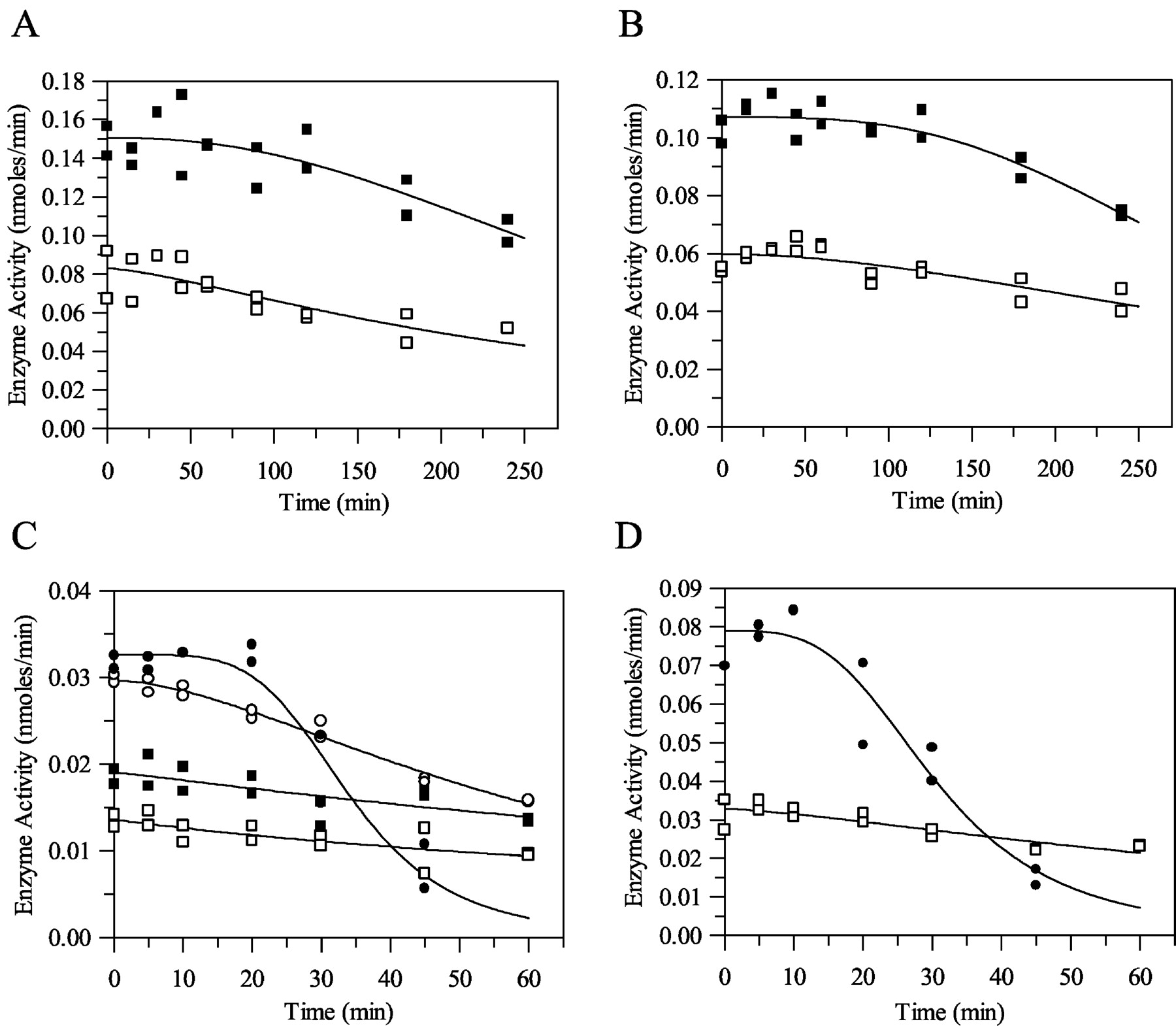
Hepatocyte Enzyme Activity with Time. The activity of freshly prepared rat hepatocytes was measured over a 4-h incubation period at 37°C with respect to 4′-hydroxydiazepam production from diazepam and 7-hydroxycoumarin production from 7-ethoxycoumarin. Typical results for diazepam and 7-ethoxycoumarin are shown in Fig. 4, A and B, respectively. Table 4 shows the mean results obtained in this study (at selected time intervals only). At both cell concentrations, the enzyme activity with respect to 4′-hydroxydiazepam from diazepam and 7-hydroxycoumarin from 7-ethoxycoumarin was reasonably stable for at least 1 h. However, after 1 to 2 h, there was a time-dependent decrease in P450 activity over the 4-h incubation period. This decline in P450 activity was fitted to eq. 9. This time-dependent decease in enzyme activity was statistically significant (p < 0.05) at the later time points beyond 180 and 240 min; the cell viability remained constant over time (data not shown).
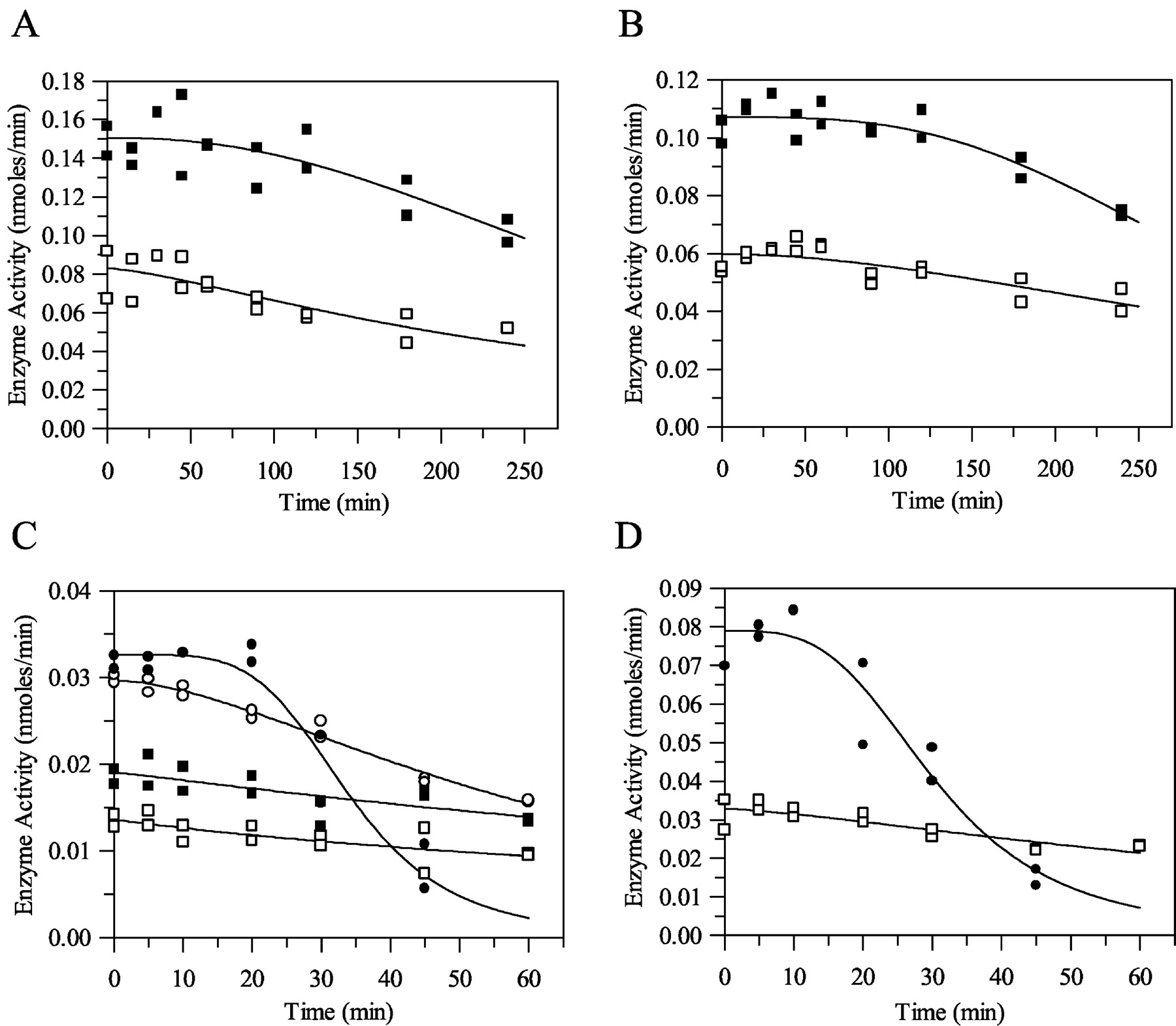
Cytochrome P450 activity in rat hepatocytes with respect to production of 4-hydroxydiazepam (A) and 7-ethoxycoumarin O-deethylase activity (B) and in rat microsomes with respect to production of 4-hydroxytriazolam (C) and 3-hydroxyclonazepam (D). Symbols are defined as follows. A: □, 0.5 × 106cells/ml; ▪, 1 × 106cells/ml; B: □, 0.5 mg/ml; ▪, 1 mg/ml; ○, 2 mg/ml; •, 5 mg/ml.
Cytochrome P450 Activity (nmol/min) with respect to diazepam 4'-hydroxylation and 7-ethoxycoumarin O-deethylase activity in rat hepatocytes
Values are mean ± S.D. (n = 4).
Microsomal Enzyme Activity with Time. Microsomal P450 activity with respect to the formation of 1′-hydroxytriazolam and 4-hydroxytriazolam from triazolam and 3-hydroxyclonazepam from clonazepam was investigated over a 1-h incubation period at 37°C. Typical results for triazolam and clonazepam are shown in Fig. 4, C and D, respectively. Table 5 shows the mean results obtained in this study (at selected time intervals only). At all microsomal protein concentrations, the enzyme activity with respect to 1′-hydroxytriazolam and 4-hydroxytriazolam from triazolam and 3-hydroxyclonazepam from clonazepam was stable for approximately 10 min. However, after 10 min, there was a time-dependent decrease in P450 activity over the 1-h incubation period. This time-dependent decrease was more significant (p < 0.05) at the higher microsomal protein concentrations (2 and 5 mg/ml). This decline in P450 activity was fitted to eq. 9. Mean parameters were obtained for eq. 9 by modeling all four replicates together at each microsomal protein concentration for 4-hydroxytriazolam and 3-hydroxyclonazepam. These mean parameter values were incorporated into an enzyme decay depletion model (eq. 14, which represents the differential form of the monoexponential model accounting for loss in enzyme activity). 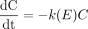
Cytochrome P450 activity (nmol/min) with respect to triazolam 4-hydroxylation and clonazepam 3-hydroxylation in rat microsomes
Values are mean ± S.D. (n = 4).
where E represents the enzyme decay for the particular substrate at a particular microsomal protein concentration (i.e., eq. 9).
The triazolam, diazepam, and clonazepam microsomal depletion data were then refitted using this model to correct for any enzyme loss. The triazolam depletion profiles were corrected using the 4-hydroxytriazolam enzyme loss data, the clonazepam profiles were corrected using the 3-hydroxyclonazepam enzyme loss data, and the diazepam profiles were corrected using the 4-hydroxytriazolam enzyme loss data, assuming the same enzyme loss would occur if diazepam had been used.
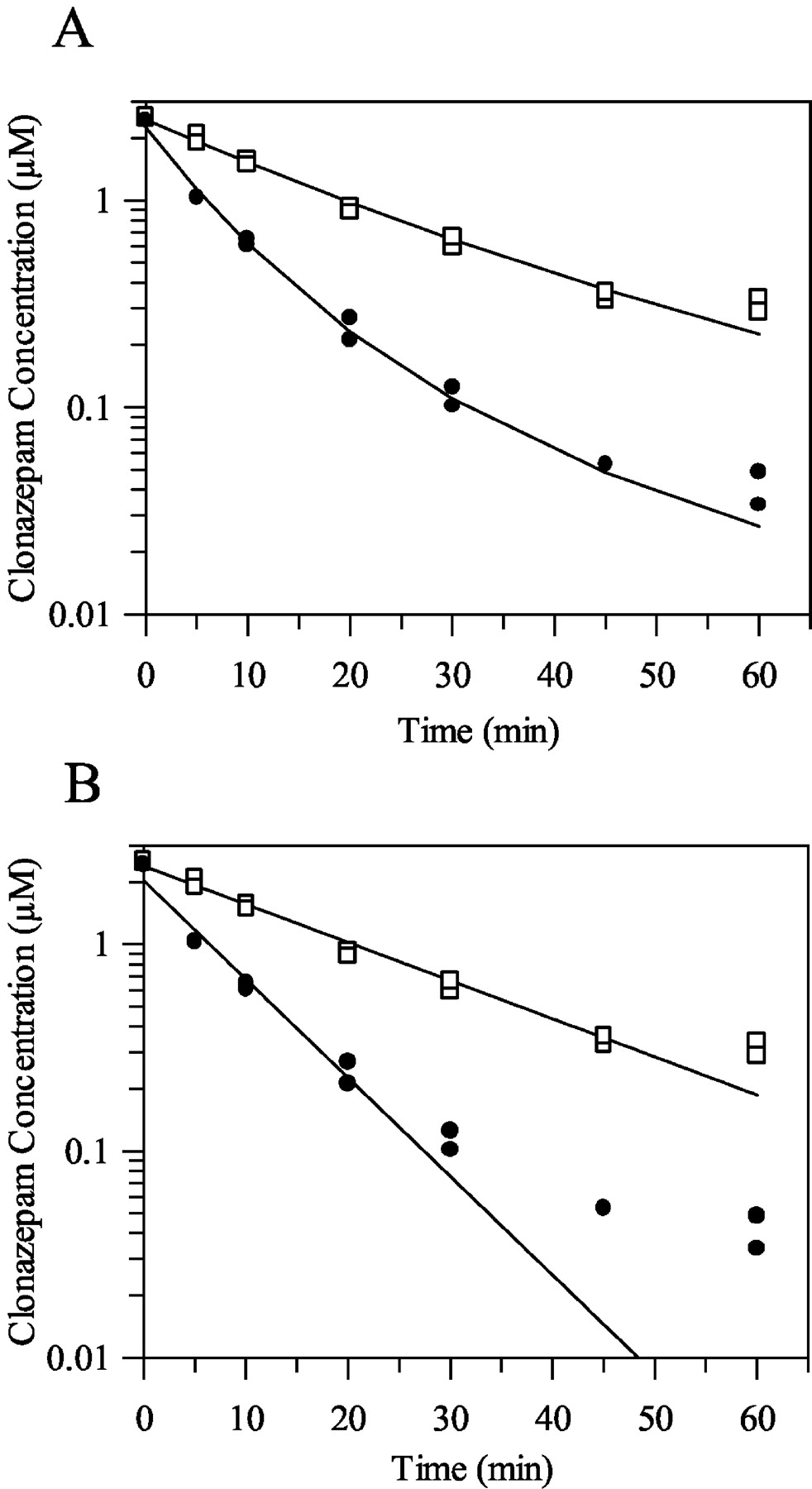
In the case of clonazepam, eq. 14, in terms of the AIC, described the data significantly better than the monoexponential model and as well as the more complex biexponential model. A typical profile of this fit compared with the traditional monoexponential fit is shown in Fig. 5, A and B, respectively. In the case of triazolam and diazepam, eq. 14 offered no advantage over the monoexponential fit at the lower microsomal protein concentrations (0.1, 0.5, and 1 mg/ml). At the higher microsomal protein concentrations (2 and 5 mg/ml), in terms of AIC, this model was equivalent to the monoexponential fit, and the biexponential model best described the data.
A typical clonazepam depletion concentration-time profile in rat microsomes fitted using eq. 14 (A) and fitted using a monoexponential model (B). Symbols are defined as follows: □, 0.5 mg/ml; •, 5 mg/ml.
Discussion
The substrate depletion approach is frequently used in the pharmaceutical industry for rapid screening of new drug entities, since formal kinetic characterization and quantification of the specific metabolites are not required (Obach, 2001). However, the incubation conditions used in these experiments may vary across studies; the consequences of these variations have not been thoroughly investigated. The aim of this work was to assess the predictive ability of the substrate depletion method and to produce a set of optimal incubation conditions.
Initial substrate depletion studies were performed with eight benzodiazepines. A detailed comparison of the predictive ability of microsomal versus hepatocyte substrate depletion data was made using formal scaling procedures (see Materials and Methods). Although predictions have been made using the substrate depletion approach in the literature (Lavé et al., 1997; Obach, 1999; Suzuki et al., 1999; Austin et al., 2002; Obach and Reed-Hagen, 2002), to the best of our knowledge, no comparisons between in vitro systems have been reported. In agreement with data reported by Houston and Carlile (1997) and Ito and Houston, (2004), predictions obtained from hepatocytes were more accurate than those obtained using microsomes. This may be because the microsomal system lacks certain enzyme and transporter activities, which are present in the more sophisticated hepatocyte system and in vivo. The overall accuracy of the predictions obtained in this study compared well with previous predictions obtained using both the substrate depletion and metabolite formation approaches for a larger but mixed group of drugs (Ito and Houston, 2004). Also, with both clearance approaches there is bias toward under-prediction of in vivo CLint and CLH, and this is particularly evident with the microsomal data (Fig. 2).
This systematic study of the effects of enzyme concentration on substrate depletion profiles and CLint estimates for triazolam, diazepam, and clonazepam indicates key differences between microsomes and hepatocytes. In general, the monoexponential model best described the metabolism of all three substrates at all hepatocyte concentrations. A significant decrease in CLint is evident at the highest cell density (4 × 106 cells/ml), and this is probably a result of cellular binding (fu value for diazepam 0.53); however, this was not investigated further because cell concentrations above 1 × 106 cells/ml are rarely used and because no biphasic effects were observed.
In contrast, the monoexponential model best described the metabolism of triazolam and diazepam only at low microsomal protein concentrations. At higher microsomal protein concentrations for triazolam and diazepam and at all microsomal protein concentrations for clonazepam, the biexponential model better described their metabolism; a similar effect has been observed in our laboratory for basic compounds including propranolol. In all cases the increase in microsomal protein concentration (even after correction for the microsomal fu) resulted in a significant decrease in CLint. Similar decreases in CLint were observed by Venkatakrishnan et al. (2000) when heat-inactivated human liver microsomal protein was added to incubations with amitriptyline involving metabolite formation, and by Austin et al. (2002) for drug depletion incubations; however, no effect on the shape of the depletion time profile was reported.
The loss of enzyme activity over time also differs between microsomes and hepatocytes. In hepatocytes, enzyme activity (measured with respect to 7-ethoxycoumarin and diazepam metabolism) was stable for between 1 and 2 h and decreased slightly between 2 and 4 h; similar results are evident in the literature. Steinberg et al. (1999) showed that 7-ethoxycoumarin metabolism and 6β- and 2β-hydroxytestosterone formation rates decline in a time-dependent manner to about 55, 57, and 28% of the activity at time 0 by 2 h. In contrast, 7-ethoxyresorufin metabolism and 16α-, 2α-, 7α-, and 15β-hydroxytestosterone activities do not decrease over the 2-h incubation period, suggesting that individual P450 isoforms are not equally stable when held in suspension. However, in microsomes, enzyme activity (with respect to triazolam and clonazepam metabolism) decreased substantially within a 1-h period and was more extensive at higher microsomal protein concentrations. Explanations for this may include poor mixing at the higher protein concentrations, resulting in oxygen availability limitations and enhanced enzyme degradation.
Thus, in the case of the hepatocyte incubations, enzyme activity was retained over the time period used for the depletion assays, whereas for the microsomal depletion incubations, enzyme activity decreased substantially over the incubation period used. The incorporation of this enzyme activity information into the differential form of the monoexponential depletion model (eq. 14) described the clonazepam (the lowest cleared substrate) depletion data well, providing an explanation for the biphasic depletion in this case. However, for triazolam and diazepam (higher clearance substrates), the biphasic depletion occurred at too early a stage in the incubation time to be explained by enzyme loss.
One plausible explanation for the biphasic effects in the case of triazolam and diazepam is the end-product inhibition phenomenon. In the microsomal system, the absence of functioning phase II conjugation enzymes results in an accumulation of the phase I hydroxylated metabolite(s). This metabolite, in theory, may compete with the parent for binding to the same P450, resulting in inhibition of the parent metabolism. Examples of compounds in the literature believed to demonstrate this phenomenon both in vitro and in vivo include phenytoin (Gerber et al., 1971; Ashley and Levy, 1972; Ashforth et al., 1995) and diazepam (Savenije-Chapel et al., 1985; Zomorodi et al., 1995; Carlile et al., 1997). This phenomenon will be addressed in more detail in a subsequent paper.
There is evidence to indicate that midazolam is an inactivator of CYP3A4 in human liver microsomes (Podoll et al., 1995; Schrag and Wienkers, 2001) and in recombinant CYP3A4 purified from Escherichia coli (Khan et al., 2002). The possibility that midazolam (and, indeed, triazolam, clonazepam, and diazepam) may be an inactivator of CYP3A in rat tissue was investigated. Using dextromethorphan as the reference substrate, there was no evidence for any time- or concentration-dependent inactivation effects in rat microsomes by midazolam. It would appear that the different CYP3A isoforms involved in midazolam metabolism in human (CYP3A4) and in rat (CYP3A1/3A2) (Kronbach et al., 1989; Kobayashi et al., 2002) show a different propensity to mechanism-based inactivation. Thus, this is an unlikely explanation for the time-dependent effects observed in this study.
In summary the prediction accuracy of the substrate depletion approach was reasonable for eight benzodiazepines. However, detailed hepatocyte and microsomal studies (with triazolam, clonazepam, and diazepam) indicated the importance of the careful selection of substrate depletion incubation conditions. For these compounds, we have confirmed that the use of arbitrary incubation conditions can result in biphasic depletion profiles and/or underestimations of in vivo CLint and CLH. In general, for both microsomal and hepatocyte incubations, the use of lower enzyme concentrations gave statistically better predictions of the in vivo value. Therefore, to achieve optimal utility of the substrate depletion approach, low enzyme concentrations and short incubation times should be used. Based on the class of compounds studied, the ideal conditions would be an enzyme concentration below 0.5 mg of protein/ml (microsomes) or 0.5 × 106 cells/ml (hepatocytes) and an incubation time less than 30 min (microsomes) or 1 to 2 h (hepatocytes). However, for compounds that are slowly cleared, these conditions may result in little or no substrate turnover; in these cases, enzyme concentrations and incubation times should be as low as is realistically possible. Interestingly, in a recent study designed to investigate the relative stability of various CYP2D6 mutants, Foti and Fisher (2004) recommended similar incubation times for microsomal incubations involving bufuralol. Following the above conditions will reduce the likelihood of biphasic depletion profiles and under-prediction of in vivo CLint and CLH by decreasing the potential for microsomal binding and by maintaining enzyme stability.
Acknowledgments
We thank David Carlile, David Hallifax, Susan Murby, and Glynis Nicholls for assistance in these studies.
Footnotes
-
↵1 Current address: Department of Non-Clinical Drug Safety, Drug Metabolism and Pharmacokinetics, F. Hoffmann La Roche, Basel, Switzerland.
-
This work was funded by a consortium of pharmaceutical companies (Astra-Zeneca, Bristol Myers Squibb, GlaxoSmithKline, F. Hoffmann-La Roche, Novartis, Pfizer, and Servier) within the Centre for Applied Pharmacokinetic Research at the University of Manchester.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.000125.
-
ABBREVIATIONS: CLint, intrinsic clearance; CLH, hepatic clearance; DMF, dimethylformamide; HPLC, high-performance liquid chromatography; AIC, Akaike information criterion; NADP, nicotinamide adenine dinucleotide phosphate.
- Received April 7, 2004.
- Accepted June 8, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}