


Abstract
The diazepam (DZ)-omeprazole (OMP) interaction has been selected as a prototype for an important drug-drug interaction involving cytochrome P450 inhibition. The availability of an in vivo Ki value (unbound Ki, 21 μM) obtained from a series of steady-state inhibitor infusion studies allowed assessment of several in vitroderived predictions of this inhibition interaction. Studies monitoring substrate depletion with time were used to obtain in vitro Ki values that were evaluated against the more traditional metabolite formation approach using microsomes and hepatocytes. OMP inhibited the metabolism of DZ to its primary metabolites 4′-hydroxydiazepam, 3-hydroxydiazepam, and nordiazepam to different extents over a range of concentrations (0.3–150 μM), and a competitive inhibition model best fitted the data. The Ki values observed using the substrate depletion approach (16 ± 3 μM and 7 ± 2 μM in microsomes and hepatocytes, respectively) were in good agreement with the overall weighted Ki values obtained using the standard metabolite formation approach (12 ± 2 μM and 16 ± 5 μM in microsomes and hepatocytes, respectively). In vitro binding and cell uptake studies as well as human serum albumin studies in hepatocytes confirmed the importance of both intracellular and extracellular unbound concentrations of inhibitor when considering inhibition predictions. Both kinetic approaches and both in vitro systems predicted the in vivo interaction well and provide a good example of the ability of in vitro inhibition studies to quantitatively predict an in vivo drug-drug interaction successfully.
The inhibition of diazepam (DZ1) metabolism by omeprazole (OMP) has been well documented in vivo; therapeutic concentrations of OMP have been reported to decrease the plasma clearance of DZ both in humans (Gugler and Jensen, 1985; Andersson et al., 1990a,b) and in rats (Zomorodi and Houston, 1995). Zomorodi and Houston (1995) measured the in vivo clearance of DZ (administered by the hepatic portal vein) in rats receiving intravenous infusions together with matching bolus doses of OMP to achieve a wide range of steady-state plasma concentrations (10–50 μg/ml). The inhibition was modeled using a simple inhibition model (eq. 1) and the in vivo Ki was calculated to be 57 μM. 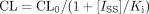 where CL and CL0 represent DZ clearance in the presence and absence of OMP, respectively, Iss represents the steady-state total plasma concentration of OMP, and Ki represents the inhibition constant. Figure 1 shows the relationship between DZ clearance and steady-state unbound concentration of OMP achieved in this study. Such a detailed in vivo inhibition study is only possible in animals. In humans, inhibition studies are normally performed using one inhibitor oral dosing regimen, and the ratio of the AUC in the presence and absence of the inhibitor is used to assess the degree of interaction (Gugler and Jensen, 1985; Andersson et al., 1990a,b).
where CL and CL0 represent DZ clearance in the presence and absence of OMP, respectively, Iss represents the steady-state total plasma concentration of OMP, and Ki represents the inhibition constant. Figure 1 shows the relationship between DZ clearance and steady-state unbound concentration of OMP achieved in this study. Such a detailed in vivo inhibition study is only possible in animals. In humans, inhibition studies are normally performed using one inhibitor oral dosing regimen, and the ratio of the AUC in the presence and absence of the inhibitor is used to assess the degree of interaction (Gugler and Jensen, 1985; Andersson et al., 1990a,b).

Observed and predicted effect of omeprazole on diazepam clearance in vivo in the rat.
Lines are defined as follows: solid black line represents model fit (eq. 1); dashed gray line represents microsomal metabolite formation prediction; dash-dot black line represents hepatocyte metabolite formation prediction; dash-dot-dot gray line represents microsomal depletion prediction; and dotted black line represents hepatocyte depletion prediction. N.B., hepatocyte metabolite formation prediction and microsomal depletion prediction overlap.
In vitro, the inhibition potential of a drug is traditionally assessed under initial rate conditions and the inhibition constant (Ki) for a particular metabolic pathway is obtained (Zomorodi and Houston, 1995; Komatsu et al., 2000; Ishigam et al., 2001). Methods involving substrate depletion with time are rarely used; however, it may be argued that this approach is analogous to the in vivo situation and may serve to predict the impact of drug interactions in cases where details of the metabolic pathways are not known or when several metabolic pathways are involved. The predicted degree of interaction caused by a reversibly acting inhibitor can then be determined based on the ratio of the inhibitor concentration at the active site in vivo to the Ki determined in vitro (Ito et al., 1998a,b). The majority of inhibition predictions in the literature have been performed using microsomal data (Yamano et al., 1999a,b, 2000, 2001; Komatsu et al., 2000; Ishigam et al., 2001; Tran et al., 2002; Yao et al., 2003). Although hepatocytes have been shown to give more realistic predictions of in vivo clearance compared with microsomes (Houston, 1994), there is, at present, a lack of evidence to demonstrate that hepatocytes are better predictors of inhibition potential.
There has been much controversy in the literature as to whether the total plasma, unbound plasma, or liver inhibitor concentration should be used for in vitro-in vivo extrapolations of inhibition (Bertz and Granneman, 1997; Ito et al., 1998a,b; Lin, 2000; Weaver, 2001). To quantitatively predict inhibition of metabolism in vivo, the concentration of inhibitor in the in vitro incubation should relate to the concentration of inhibitor in the plasma. Differences may result from the extensive binding of the inhibitor to microsomes, as demonstrated by Tran et al. (2002) and Margolis and Obach (2003). In addition, differences may occur as a consequence of accumulation of the inhibitor in the hepatocyte as a result of intracellular binding or active cell uptake. Attempts have been made to account for this cell accumulation by concentrative uptake (Yamano et al., 1999a,b, 2000, 2001) and liver partitioning techniques (von Moltke et al., 1994, 1998).
The aim of the present investigation was to address the issues discussed above relating to the prediction of drug-drug interactions. This was achieved using the rat as a test species and DZ and OMP as examples of substrate and inhibitor, respectively. Comparisons have been made between microsomes and hepatocytes and between the standard metabolite formation method and the substrate depletion approach. These in vitro prediction methods were evaluated by comparison with the Ki obtained from the in vivo study described above (Zomorodi and Houston, 1995). This study also investigated some aspects of OMP inhibition from in vitro systems, and we have extended these investigations and assessed the inhibition of the major pathway for DZ metabolism (4′-hydroxylation), incorporated inhibitor binding and cellular uptake in vitro, and related unbound OMP concentration to the inhibition of DZ clearance in hepatic microsomes, isolated hepatocytes, and in vivo.
Materials and Methods
Materials. All chemicals and reagents used were of the highest grade available and were purchased from Sigma Chemical (Poole, Dorset, UK) or BDH Chemicals Ltd. (Poole, Dorset, UK). 3-HDZ and prazepam were obtained from Wyeth Pharmaceuticals (Maidenhead, Berkshire, UK) and Warner Lambert Ltd. (Eastleigh, UK), respectively. 4′-HDZ and NDZ were kindly supplied by F. Hoffmann-La Roche (Basel, Switzerland). Astra (Hässle, Mölndal, Sweden) kindly supplied OMP and [14C]OMP.
Animal Source, Housing, and Diet. Male Sprague-Dawley rats (220–270 g) were obtained from the Biological Sciences Unit, Medical School, University of Manchester. They were housed in groups of two to four, in opaque boxes on a bedding of sawdust. The accommodation was maintained at a temperature of 20° ± 3°C, with a relative humidity of 40 to 70% and a 12-h light/dark cycle. The animals were allowed free access to Chow Rat Mouse diet and fresh drinking water.
Preparation of Rat Liver Microsomes and Rat Hepatocytes. Rat liver microsomes and hepatocytes were prepared as described previously by Hayes et al. (1995) from Sprague-Dawley rats. The protein content of the microsomes was determined using the method of Lowry et al. (1951). Hepatocyte viability was measured using the trypan blue exclusion test, and only preparations in excess of 85% viability were used.
Microsomal Studies. All incubations were performed in duplicate using microsomes prepared from four different livers. All kinetic and standard inhibition experiments were carried out under linear conditions with respect to incubation time and microsomal protein concentration. Substrate concentrations for standard depletion and inhibition depletion incubations were chosen based on published Km values (Zomorodi et al., 1995). Unless stated otherwise, all incubations contained 0.5% methanol.
Diazepam kinetic studies. DZ (2.5 μl; final concentration, 1–250 μM) was incubated with rat liver microsomes (final concentration, 0.1 mg/ml) in a shaking water bath at 37°C. The reaction was initiated after 5 min by the addition of 250 μl of a cofactor regenerating system (containing 0.37 mg of NADP+, 0.97 mg of isocitric acid, 0.6 units of isocitric dehydrogenase, and 8.1 μmol magnesium chloride in 0.1 M phosphate buffer, pH 7.4), giving a final incubation volume of 500 μl. Reactions were terminated after 5 min with 250 μl of ice-cold methanol containing an internal standard.
Diazepam depletion studies and the effect of organic solvents. DZ (2.5 μl; final concentration, 2.5 μM) in either methanol or DMF (final solvent concentration 0.5%) was incubated at a final microsomal concentration of 0.5 mg/ml as described above. Reactions were terminated at regular time intervals (0–30 min) with 250 μl of ice-cold methanol containing an internal standard.
Standard inhibition studies. DZ (2.5 μl; final concentration, 0.5–50 μM) and OMP (2.5 μl; final concentration 0.3–150 μM) or vehicle alone (methanol) were incubated at a final microsomal concentration of 0.1 mg/ml as described above. Reactions were terminated after 5 min with 250 μl of ice-cold methanol or 500 μl of ice-cold acetonitrile containing an internal standard.
Substrate depletion inhibition studies. DZ (2.5 μl; final concentration, 2.5 μM) and OMP (2.5 μl; final concentration 6–50 μM) or vehicle alone (methanol) were incubated at a final microsomal concentration of 0.5 mg/ml as described above. Reactions were terminated at regular time intervals (0–45 min) with 250 μl of ice-cold methanol containing an internal standard.
Hepatocyte studies. All incubations were performed in duplicate using hepatocytes isolated from four different livers. All kinetic and standard inhibition experiments were carried out under linear conditions with respect to incubation time and cell concentration. Substrate concentrations for standard depletion and inhibition depletion incubations were chosen based on published Km values (Zomorodi et al., 1995). Unless stated otherwise, all incubations contained 0.5% methanol.
Diazepam kinetic studies. DZ (2.5 μl; final concentration, 1–250 μM) was incubated with Williams medium E (pH 7.4) in a heater/shaker (Eppendorf Thermomixer Comfort; Eppendorf-5 Prime, Inc., Boulder, CO) at 900 rpm at 37°C. The reaction was initiated after 5 min by the addition of preincubated, freshly isolated rat cells (final concentration, 0.2 × 106 cells/ml), giving a final incubation volume of 500 μl. Reactions were terminated after 5 min by snap-freezing in liquid nitrogen.
Diazepam depletion studies and the effect of organic solvents. DZ (2.5 μl; final concentration, 2.5 μM) in either methanol or DMF (final solvent concentration, 0.5%) was incubated at a final cell concentration of 0.5 × 106 cells/ml as described above. Reactions were terminated at regular time intervals (0–45 min) by snap-freezing in liquid nitrogen.
Standard inhibition studies. DZ (2.5 μl; final concentration, 0.5–100 μM) and OMP (2.5 μl; final concentration, 0.3–150 μM) or vehicle alone (methanol) were incubated as described above at a final cell concentration of 0.2 × 106 cells/ml. Reactions were terminated after 5 min by snap-freezing in liquid nitrogen.
Substrate depletion inhibition studies. DZ (2.5 μl; final concentration, 2.5 μM) and OMP (2.5 μl; final concentration, 0.5–70 μM) or vehicle alone (methanol) were incubated at a final cell concentration of 0.5 × 106 cells/ml as described above. Reactions were terminated at regular time intervals (0–30 min) by snap-freezing in liquid nitrogen.
Inhibition studies in the presence of 4% HSA. DZ (2.5 μl; final concentration, 2.5 μM) and OMP (2.5 μl; final concentration, 0.3–150 μM) or vehicle alone (methanol) were incubated at a cell concentration of 0.5 × 106 cells/ml as described above, with/without the addition of HSA (final concentration, 4%). Reactions were terminated after 10 min by snap-freezing in liquid nitrogen.
Sample Analysis. HPLC was used for the analysis of all DZ kinetic and depletion incubations as well as microsomal depletion inhibition experiments. LC-MS/MS was used for the analysis of hepatocyte standard and depletion inhibition experiments, HSA incubations, and the microsomal standard inhibition experiments due to analytical and sensitivity problems on the HPLC.
Microsomal sample preparation. After termination of the reaction, microsomal samples were centrifuged at 11,600g AV for 15 min (Eppendorf centrifuge 5413). The supernatant was analyzed by HPLC or by LC-MS/MS.
Hepatocyte sample preparation. After termination of the reaction, hepatocyte samples were thawed and hydrolyzed with 0.5 ml of β-glucuronidase with sulfatase activity (500 units/ml in sodium acetate, 60 mM; pH 4.5) for 1.5 h in a shaking water bath at 37°C.
HPLC samples were extracted with 5 ml of ethyl acetate (containing an internal standard) after the addition of 1 ml of 0.1 M carbonate buffer (pH 10) by rotary mixing for 30 min and centrifuging at 900g for 10 min. The organic layer was removed and evaporated to dryness under nitrogen at 40°C. The residue was reconstituted in mobile phase and analyzed by HPLC. Ice-cold acetonitrile (500 μl, containing an internal standard) was added to the LC-MS/MS samples, which were centrifuged at 11,600g for 15 min (Eppendorf centrifuge 5413), and the supernatant was analyzed by LC-MS/MS.
HPLC method. The HPLC method used was based on the method described by Reilly et al. (1990). The HPLC system consisted of a Hypersil BDS C18 (150 × 4.6 mm) column (Phenomenex, Macclesfield, Cheshire, UK); mobile phase of 55.5% methanol and 44.5% water containing 0.02% (v/v) triethylamine adjusted to pH 7.0 with orthophosphoric acid, delivered by a Gilson 305 pump (Gilson Medical Electronics, Middleton, WI) at a flow rate of 1 ml/min; at a UV wavelength of 254 nm measured by a Milton Roy Spectromonitor 3100 detector (Milton Roy Company, Rochester, NY). DZ, 4′-HDZ, 3-HDZ, and NDZ concentrations were quantified by their peak area ratio relative to an internal standard (prazepam) with reference to a standard calibration curve.
LC-MS/MS method. The LC-MS/MS system comprised a Waters Alliance 2790 HPLC system coupled with a Micromass Quattro Ultima triple quadrupole mass spectrometer set to operate in atmospheric pressure electrospray ionization mode (Waters, Milford, MA). DZ and its metabolites were monitored using the following mass transitions: DZ, m/z 285.0→257.0 (cone voltage 70 V; collision energy 20 eV); NDZ, m/z 270.95→208.0 (cone voltage 74 V; collision energy 25 eV); 3-HDZ, m/z 301.1→254.9 (cone voltage 39 V; collision energy 12 eV); 4′-HDZ, m/z 301.1→153.9 (cone voltage 70 V; collision energy 20 eV). DZ, 4′-HDZ, 3-HDZ, and NDZ were quantified by their ratio relative to an internal standard (triazolam, m/z 343.0→308.0; cone voltage 80 V; collision energy 25 eV) by nonlinear regression of a calibration curve using MassLynx 3.5 quantification software (Micromass Ltd, Manchester, UK).
Determination of Omeprazole Binding. The extent of binding of OMP to microsomal protein, HSA, and rat plasma was determined by ultrafiltration. Samples containing microsomal protein (500 μl; final concentrations, 0.5, 1, and 2 mg/ml), HSA (final concentration, 4%), or rat plasma, together with 2.5 μl of OMP (final concentrations, 1, 10, 100 μM) and 5 μl of [14C]OMP (1.5 μCi) were preincubated in a shaking water bath at 37°C for 10 min. Aliquots (400 μl) were placed on 0.02-μm Anopore filters (VectaSpin Micro; Whatman, Maidstone, UK) and centrifuged at 7000g for 5 min. An equal aliquot of the filtrate and original sample was transferred to scintillation vials and scintillation fluid (10 ml, Optiphase HiSafe; PerkinElmer Life and Analytical Sciences, Boston, MA) was added. The samples were then analyzed by liquid scintillation spectrometry with spectral quench parametric analysis for 5 min.
Determination of Omeprazole Cell Uptake. Rat hepatocytes (2 × 106 cells/ml, no concentration-dependent effect) were incubated (total incubation volume, 500 μl) with [14C]OMP (0.01–100 μM) for 0, 10, 20, 30, and 60 s. Then the cells were separated by centrifugation through a layer of silicone oil (100 μl, inserted before the incubate) and trapped in a lower layer of NaOH (50 μl, 1 M, for digestion). The tubes were frozen and the trapped cells were removed by cutting through the oil layer. The cell pellet was then collected in a scintillation vial and incubated at room temperature after diluting with water (1 ml). After digestion (for 16 h), scintillation fluid (5 ml of Optiphase HiSafe) was added and samples were allowed to rest for 4 h to exhaust chemiluminescence. These samples, together with medium samples, were analyzed by liquid scintillation spectrometry (Wallac 1409 Liquid Scintillation Spectrometer; PerkinElmer Wallac, Gaithersburg, MD) with spectral quench parametric analysis for 5 min. Cell uptake was corrected for the adherent fluid volume and, as measured by the concentration ratio, did not show any change with hepatocellularity.
Data Analysis.Kinetic data. Kinetic parameters were calculated from untransformed data by nonlinear least-squares regression. Analysis was performed assuming single-site Michaelis-Menten kinetics with no weighting factor. The log-average substrate concentrations were used to correct for the loss of substrate over time due to metabolism. In vitro CLint was calculated by dividing Vmax by Km. fm values (fraction of DZ metabolized to each metabolite) were determined using eq. 2: 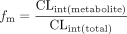 where CLint(metabolite) is the CLint of the metabolite, and CLint(total) is the total CLint for DZ.
where CLint(metabolite) is the CLint of the metabolite, and CLint(total) is the total CLint for DZ.
Substrate depletion data. Substrate depletion data were fitted to an i.v. bolus monoexponential decay model with 1/y weighting. In vitro CLint was calculated by dividing the dose by the AUC (from time 0 to infinity).
Standard inhibition data. Initially, IC50 plots were used to differentiate between competitive (inhibition decreases with increasing substrate concentration) and noncompetitive (inhibition is independent of substrate concentration) inhibition mechanisms. Subsequently, goodness of fit was assessed by visual examination of the data and residual plots and by the precision of the parameter estimates.
Using nonlinear regression analysis, a model for competitive inhibition (eq. 3) was used to fit the data.  where S is the substrate concentration, and I is the inhibitor concentration.
where S is the substrate concentration, and I is the inhibitor concentration.
A weighted Ki from the standard inhibition method was calculated by weighting the individual Ki values based on the relative importance of each of the metabolic pathways using the fm values derived from the kinetic experiments, using eq. 4. 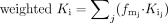
Substrate depletion inhibition data. The substrate depletion data in the absence and presence of OMP were modeled as described above using an i.v. bolus model. CLint was plotted against OMP concentration, and the IC50 determined. A weighted Km value (Km(w)) for DZ was determined using eq. 5, and the depletion Ki was calculated assuming competitive inhibition using eq. 6. 
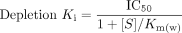
In addition, the metabolite data measured during the substrate depletion inhibition studies were fitted to either eq. 7 or 8. 
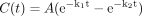
Binding data. The fraction unbound (fu) for OMP in rat liver microsomes, 4% HSA, and rat plasma was determined from the ratio of the concentration of OMP in the filtrate to the initial total concentration of OMP, as shown in eq. 9. Corrections were made for any binding to the apparatus.  where Cfiltrate is the concentration of drug in the filtrate, and Ctotal sample represents the concentration of drug in the total sample.
where Cfiltrate is the concentration of drug in the filtrate, and Ctotal sample represents the concentration of drug in the total sample.
Unbound in vitro and in vivo Ki values were calculated using eq. 10. 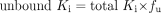
Cellular uptake data. The hepatocyte-medium partition coefficient (Kp) was calculated based on the ratio of the concentration of OMP in the cell to the concentration of OMP in the medium as shown in eq. 11. 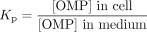
The unbound fraction of OMP in the cells was calculated using this hepatocyte-medium Kp as shown in eq. 12, assuming that the unbound concentration of OMP in the cell is equal to the unbound concentration in the medium. 
Based on the ratio of the cell volume to the incubation volume and the measured hepatocyte-medium Kp, the unbound fraction in the whole incubation was calculated using eq. 13,  where Vc represents the cell volume, and Vinc is the incubation volume. Vc (4 μl = 1 × 106 cells) was calculated from an average of five experiments using 14C-sucrose and 3H-water to measure the total volume (adherent water volume + cellular volume). The cellular volume was calculated by subtracting the adherent water volume from the total volume.
where Vc represents the cell volume, and Vinc is the incubation volume. Vc (4 μl = 1 × 106 cells) was calculated from an average of five experiments using 14C-sucrose and 3H-water to measure the total volume (adherent water volume + cellular volume). The cellular volume was calculated by subtracting the adherent water volume from the total volume.
Statistical analysis. A one-way analysis of variance was used to test any differences in the Ki prediction among the four in vitro methods/systems. In addition the Tukey multiple comparison method was used to assess pairwise comparisons.
Results
Diazepam Kinetic and Depletion Studies. As described previously by Zomorodi et al. (1995), the rates of 4′-HDZ, 3-HDZ, and NDZ formation were best described by the Michaelis-Menten equation. Table 1 shows the mean parameter estimates and the corresponding CLint and fm values obtained in these studies. The rank order for CLint in microsomes and hepatocytes was 4′-HDZ > 3-HDZ ≈ NDZ, in concordance with results obtained by Zomorodi et al. (1995) using DMF as the substrate solvent. However, in contrast, the magnitude of CLint was almost 2 to 3 times higher in the current study using methanol.
Kinetic parameters and corresponding CLint and fm values for 4′-hydroxydiazepam, 3-hydroxydiazepam, and nordiazepam in rat liver microsomes and rat hepatocytes Data represent a mean ± S.D. (n = 4).
The differential effect of 0.5% DMF compared with 0.5% methanol on DZ metabolism using rat liver microsomes and rat hepatocytes is shown in Fig. 2 for substrate depletion time profiles. DZ CLint was reduced by approximately 50% from 213 ± 60 μl/min/mg using methanol to 105 ± 14 μl/min/mg using DMF with microsomes and by around 66% from 159 ± 14 μl/min/106 cells with methanol to 54 ± 17 μl/min/106 cells with DMF using hepatocytes.

Effect of methanol and DMF on the depletion of diazepam in rat liver microsomes (A) and rat hepatocytes (B).
Symbols are defined as follows: DMF, •; methanol, ▪.
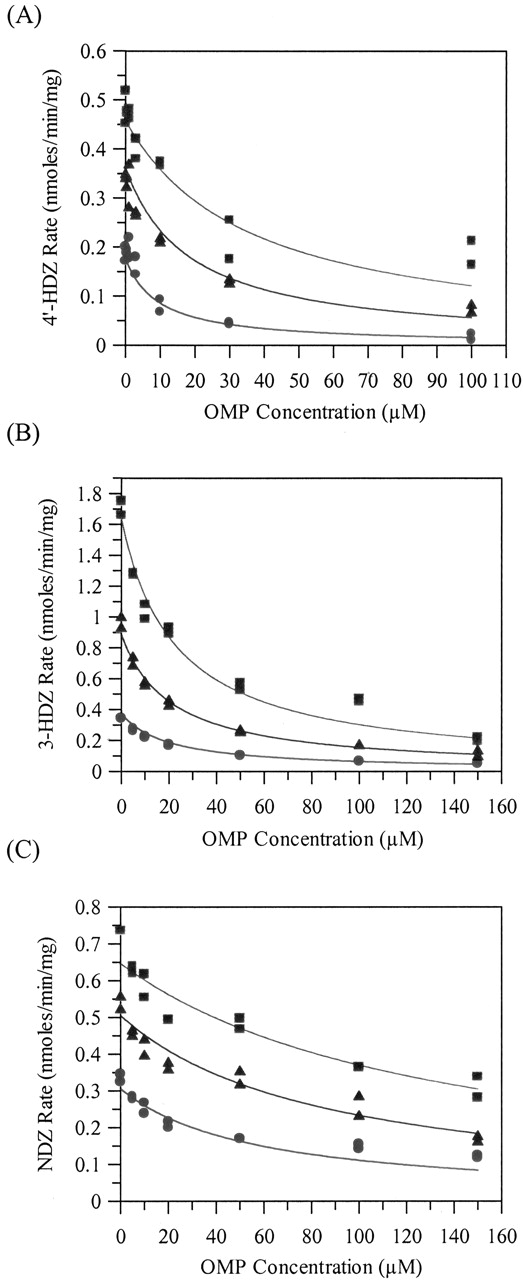
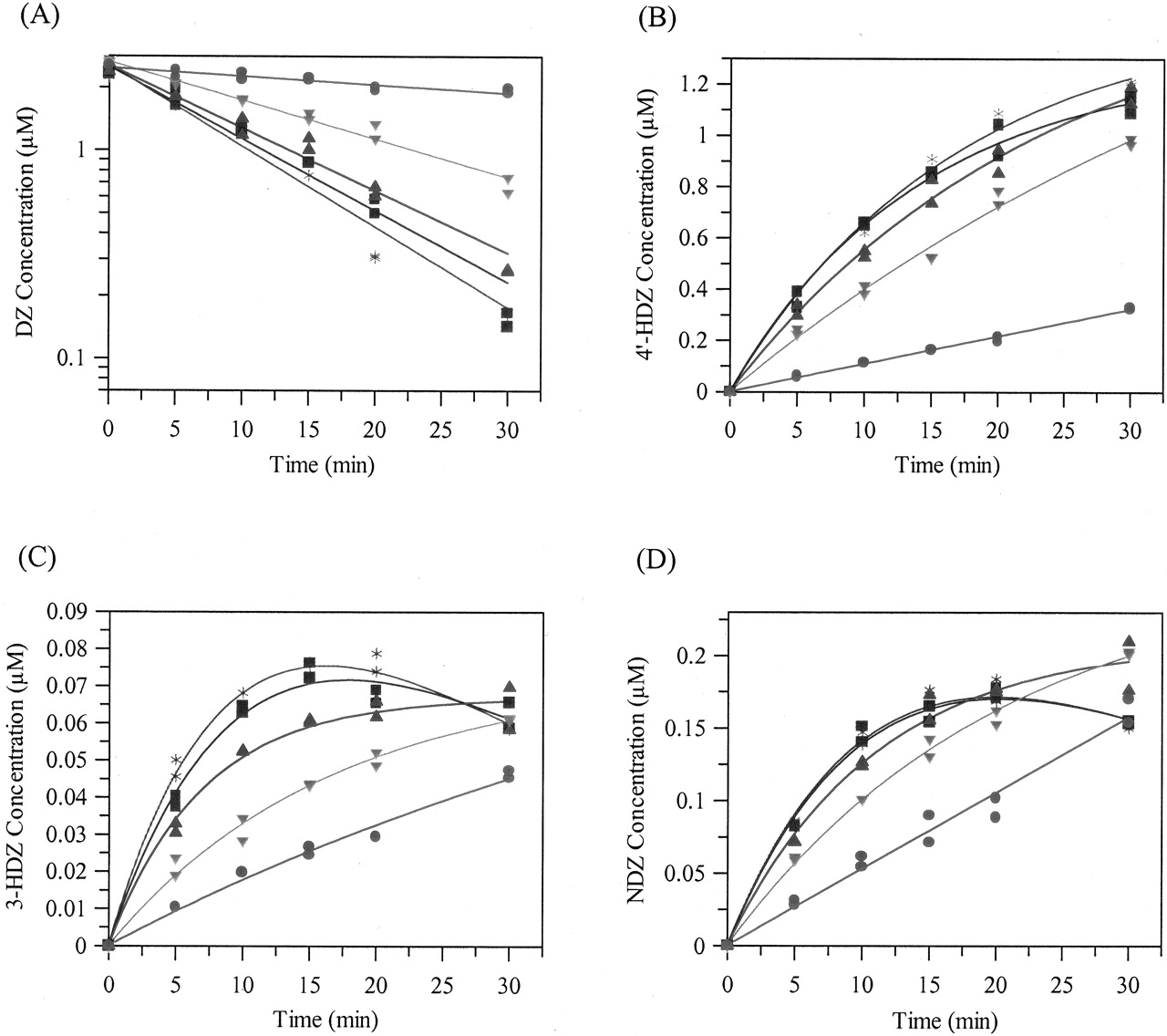
Inhibition Studies. For the standard microsomal and hepatocyte inhibition studies, using nonlinear regression analysis, the competitive inhibition model best fitted the 4′-HDZ, 3-HDZ, and NDZ data. The DZ concentration-dependent inhibition effect, as seen by the increasing IC50 values with increasing DZ concentration, is shown in Fig. 3A for 4′-HDZ. OMP inhibited all three metabolic pathways but to different extents. The 4′-HDZ pathway was most susceptible to inhibition, followed by the 3-HDZ pathway and the NDZ pathway. The extent of inhibition and the rank order was similar between microsomes and hepatocytes. Table 2 shows the Ki values for each pathway, together with an overall Ki obtained by weighting the relative importance of each pathway. Typical competitive inhibition fits for 4′-HDZ, 3-HDZ, and NDZ are shown in Fig. 4, A to C, for microsomal incubations.

IC50 plot for the inhibition of 4′-hydroxydiazepam formation from diazepam by omeprazole in rat hepatocytes: effect of substrate concentration (A) and 4% HSA (B).
A, symbols are defined as follows: 0.5 μM DZ, •; 2.5 μM DZ, ▴; 7.5 μM DZ, ▪. B, symbols are defined as follows: control, •; + 4% HSA, ▪. Data points represent the average of duplicate incubations.
Inhibition of diazepam metabolism by omeprazole in rat liver microsomes and rat hepatocytes

Inhibition of the formation of 4′-hydroxydiazepam (A), 3-hydroxydiazepam (B), and nordiazepam (C) from diazepam by omeprazole in rat liver microsomes.
Symbols are defined as follows: low DZ, •; medium DZ, ▴; high DZ, ▪. Exact DZ concentration varies depending on the metabolite.
Using the depletion approach, there was a decrease in DZ CLint as OMP concentration was increased. Assuming competitive inhibition, the Ki calculated using this method was similar between in vitro systems and was in reasonable agreement with those measured using the standard metabolite formation method (Table 2). The effect of OMP on the individual metabolic concentration-time profiles was similar to, and had the same rank order as, the standard inhibition approach previously described. Typical depletion profiles for DZ in the absence and presence of OMP together with the individual metabolite time profiles using hepatocytes are shown in Fig. 5, A to D.

Inhibition of diazepam depletion (A) and the simultaneous formation of 4′-hydroxydiazepam (B), 3-hydoxydiazepam (C), and nordiazepam (D) by omeprazole in rat hepatocytes.
Symbols are defined as follows: control, *; 0.5 μM OMP, ▪; 2.5 μM OMP, ▴; 10 μM OMP, ▾; 70 μM OMP, •.
The analysis of variance showed that there was a statistically significant difference between the Ki values determined using the four in vitro methods/systems (p < 0.01). Following the Tukey multiple comparison procedure, the hepatocyte depletion Ki value was found to be significantly different (p < 0.05) from the microsomal depletion Ki value and the hepatocyte metabolite formation Ki value.
Hepatocyte inhibition studies in the presence of 4% HSA were performed to mimic the potential impact of OMP plasma binding on the observed inhibition in vivo. HSA shifted the IC50 curve (with respect to 4′-HDZ production) to the right (see Fig. 3B). This resulted in a 4-fold increase in IC50 from 15 ± 2 μM in the absence of HSA to 59 ± 14 μM in the presence of HSA.
Omeprazole Binding and Cell Uptake. The extent of OMP binding to rat liver microsomes, 4% HSA, and rat plasma was measured using substrate concentrations of 1, 10, and 100 μM to cover the range used for the inhibition studies. Equipment binding was deemed to be less than 10% and was therefore considered to be minimal. There was minimal binding of OMP to rat liver microsomes (fu = 0.92 ± 0.03; no concentration dependence), so no correction was made to the microsomal Ki.
Cellular uptake of OMP was determined over the concentration range 0.01 to 100 μM. Results showed that OMP accumulates in the cell resulting in a hepatocyte-medium Kp of 16. Over the concentration range 0.01 to 10 μM, this accumulation was substrate concentration-independent. Assuming that this accumulation was a result of cellular binding rather than active uptake, the intracellular fu was calculated to be 0.06 (using eq. 12). At the concentration of cells used, using eq. 13 assuming rapid equilibrium, the OMP accumulation resulted in an unbound fraction in the whole incubation only marginally lower than the total OMP concentration (fu = 0.97–0.99). Therefore, no correction was made to the hepatocyte Ki.
The binding of OMP to 4% HSA and rat plasma was substantial (fu = 0.66 ± 0.10 and 0.36 ± 0.13, respectively), but showed no concentration dependence over the range of OMP concentrations used. The in vivo Ki measured by Zomorodi and Houston (1995) was therefore corrected for this unbound fraction in plasma (using eq. 10), resulting in an unbound, in vivo Ki of 21 μM.
Discussion
The purpose of this study was to compare different in vitro methods (metabolite formation versus substrate depletion) and different in vitro systems (microsomes versus hepatocytes) for the quantitative prediction of drug-drug interactions, using the DZ and OMP interaction as an example. Availability of an in vivo Ki obtained from a series of steady-state inhibitor infusion studies (Zomorodi and Houston, 1995) allowed the evaluation of these in vitro techniques. To directly correlate the in vitro and in vivo Ki values, microsomal binding, hepatocyte binding, hepatic uptake, and in vivo plasma binding were considered.
Formation of all three DZ metabolites followed Michaelis-Menten kinetics as described previously by Zomorodi et al. (1995). Although the rank order for the different metabolites was consistent with the Zomorodi et al. (1995) study, where 1% DMF was used as the substrate solvent, the CLint values obtained in the present study (using 0.5% methanol) were higher. Hickman et al. (1998), using human liver microsomes, reported that 1% DMF inhibits CYP2C19, CYP2D6, and CYP3A4 to approximately 10, 60, and 80% of their control activity. This, together with the observation reported here that DMF (when compared with methanol) inhibits DZ metabolism by 50 and 66% in rat microsomes and hepatocytes, respectively (Fig. 2), indicates that the lower CLint values in the Zomorodi et al. (1995) study may be a result of the substrate solvent used.
As shown in Table 2, the 4′-HDZ pathway was most susceptible to inhibition by OMP, followed by the 3-HDZ pathway, and the NDZ pathway, implicating the CYP2D, CYP3A, and CYP2C enzymes in the inhibition of DZ by OMP (Neville et al., 1993). This is consistent with earlier studies in rats and humans, in which CYP3A enzymes were implicated more than CYP2C enzymes (Zomorodi and Houston, 1995, 1996). However, in humans, Andersson (1991) attributes the inhibition of DZ by OMP to inhibition of CYP2C19 rather than CYP2D6 or CYP3A4, whereas Venkatakrishnan et al. (2001) describe OMP as an inhibitor of CYP2C19, CYP2C9, CYP3A4, and CYP2D6.
The present results represent an extension to the previous rat study performed by Zomorodi and Houston (1995), where only the inhibition of 3-HDZ and NDZ formation was monitored and not the major metabolite (4′-HDZ). In the study by Zomorodi and Houston (1995), the 3-HDZ pathway was most susceptible to inhibition (Ki values of 108 ± 30 and 28 ± 11 μM in microsomes and hepatocytes, respectively), followed by the NDZ pathway (Ki values of 226 ± 76 and 59 ± 27 μM in microsomes and hepatocytes, respectively). Although, the respective Ki values between the two studies vary in their magnitude, the rank order is the same. These differences in magnitude, particularly for the microsomal Ki values, may be due to the use of 1% DMF by Zomorodi and Houston (1995). Lower DMF concentrations (0.3%) were used for the hepatocyte incubations producing Ki values more comparable with the current findings.
Many compounds like DZ are metabolized by several cytochrome P450 isoforms. In these circumstances, it is important to consider all metabolic pathways involved in drug clearance to accurately predict the inhibition of in vivo metabolism. For such compounds the substrate depletion approach may act to predict the inhibitory effects more effectively than the traditional initial rate approach. In agreement with data obtained by Hallifax et al. (2000) for the inhibition of codeine metabolism by dextromethorphan predictions based on depletion data and the weighted total from metabolite formation studies were more representative of the in vivo unbound Ki. For example, if only the 4′-HDZ pathway had been considered, the interaction would have been overpredicted, whereas consideration of all three pathways gave the best prediction. In addition, the depletion results (Table 2) were consistent with those from Chenery et al. (1988), who demonstrated that the addition of 50 μM OMP to DZ rat hepatocyte incubations results in a 67 to 79% reduction of DZ clearance. However, one complication associated with the prediction of inhibition using this method is the inability to distinguish between inhibition caused by the inhibitor and associated inhibitory effects from substrate/inhibitor metabolites.
An important issue in the prediction of in vivo drug-drug interactions is the accurate measurement of the inhibitor concentration in vitro and in vivo at the enzyme site (Lin, 2000). In the current study, there was no binding of OMP to microsomes; therefore, it was assumed that the concentration of OMP added to the incubation was representative of the unbound concentration available to the enzyme. Hepatocyte uptake studies generated a hepatocyte-medium Kp of 16 (representative of total OMP concentration, i.e., bound + unbound). Based on the similarity of the Ki values between both in vitro systems, there appears to be no direct evidence for active uptake of OMP into the cell. This was further confirmed by results from Kanamitsu et al. (2000b), who measured an unbound hepatocyte-medium Kp for OMP of 1 (representative of unbound OMP concentration only). In addition, results from Sewell et al. (1994) indicate that OMP accumulation in the hepatocyte is due to intracellular binding of OMP to cytosolic proteins. Although the intracellular binding of OMP was high, consideration of the proportion of total cellular volume in the total incubate and the hepatocyte-medium Kp resulted in an unbound concentration of OMP only marginally lower than unity. Therefore, as with microsomes, the hepatocyte Ki was representative of the unbound OMP concentration.
There is no agreement in the literature as to whether total or unbound inhibitor concentrations in vivo should be used for the in vitro/in vivo extrapolations of inhibition (Tucker et al., 2001). Some drug-drug interactions are better predicted when the total inhibitor concentration is used, e.g., the triazolam and ketoconazole interaction (von Moltke et al., 1996), but others give better predictions when unbound concentrations are used, e.g., the tolbutamide and sulfaphenazole interaction (Komatsu et al., 2000). To address these conflicting views, in vitro hepatocyte inhibition studies were performed in the presence of 4% HSA to mimic the in vivo plasma binding effect. The 4-fold increase in IC50 with addition of 4% HSA indicates that a decrease in the unbound concentration of OMP due to albumin binding results in a reduction in the extent of inhibition. An analogous effect was reported by Tran et al. (2002) in microsomes; the inhibitory capacity of ketoconazole and OH-itraconazole on 3-HDZ formation from DZ was reduced using heat-inactivated protein. In the present study, correction of the IC50 value for the unbound fraction of OMP resulted in an IC50 that was 2.5-fold larger than the control value; therefore, this reduction in inhibition cannot be totally accounted for by the binding of OMP to HSA. Addition of albumin to the incubation may result in an increase in DZ reaction rate by increasing the binding affinity of DZ for the enzyme, thereby reducing its susceptibility to inhibition by OMP. This effect has been reported previously, where the addition of albumin resulted in a decrease in unbound Km and an increase in reaction rate for phenytoin and tolbutamide (Ludden et al., 1997; Carlile et al., 1999; Tang et al., 2002; Wang et al., 2002).
Because albumin binding reduces the extent of inhibition observed and because the concentration of OMP in vitro is representative of the unbound concentration, a correction for plasma binding was made for a more accurate in vivo comparison. Correction of the in vivo Ki for the unbound fraction of OMP in plasma resulted in an unbound Ki of 21 μM. The in vitro Ki values (Table 2) are more comparable with this value than with the total Ki of 57 μM, consistent with the “free drug hypothesis.” The in vivo predictions (from microsomes and hepatocytes using both kinetic approaches) are presented together with the observed in vivo data in Fig. 1. One might expect the metabolite formation method (using initial rates) to give the best predictions; however, there is little difference between the predictions based on either of the two microsomal approaches (metabolite formation and substrate depletion) or the metabolite formation approach using hepatocytes. It is unknown why the substrate depletion approach in hepatocytes provides a significantly lower Ki value (see Table 2) and hence an overprediction of the in vivo effect.
The ultimate aim of these in vitro inhibition studies was to assess their in vivo predictive capability. In this study we were able to predict the in vivo interaction of DZ and OMP with a reasonable degree of accuracy using both in vitro methods (metabolite formation and substrate depletion) and both in vitro systems (microsomes and hepatocytes). However, in this case the in vivo comparator was obtained under carefully controlled experimental conditions, where a range of inhibitor concentrations were maintained in vivo at steady state. This allowed the calculation of an in vivo Ki that could be directly correlated with the in vitro Ki values. In humans, such in vivo studies are not experimentally possible, so Ki values cannot be readily determined, and any inhibitory effect is assessed by an increase in the AUC of the substrate. The current study is valuable in demonstrating the principles of quantitative drug-drug interaction prediction and provides encouragement for the challenge presented by the more complex human situation.
Footnotes
-
↵1 Abbreviations used are: DZ, diazepam; OMP, omeprazole; AUC, area under the plasma concentration-time curve; 3-HDZ, 3-hydroxydiazepam; 4′-HDZ, 4′-hydroxydiazepam; NDZ, nordiazepam; DMF, dimethylformamide; HSA, human serum albumin; HPLC, high-performance liquid chromatography; LC-MS/MS, liquid chromatography/mass spectrometry; fm, fraction of parent compound metabolized to each metabolite; fu, fraction unbound in plasma; Kp, partition coefficient.
-
This work was funded by the Centre for Applied Pharmacokinetic Research (CAPKR), consisting of a consortium of pharmaceutical companies (AstraZeneca, Bristol Myers Squibb, GlaxoSmithKline, F. Hoffmann-La Roche, Novartis, Pfizer, and Servier).
- Received December 9, 2003.
- Accepted February 11, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}