


Abstract
Over the last 5 years the quantification of transporter-protein absolute abundances has dramatically increased in parallel to the expanded use of in vitro–in vivo extrapolation (IVIVE) and physiologically based pharmacokinetics (PBPK)-linked models, for decision-making in pharmaceutical company drug development pipelines and regulatory submissions. Although several research groups have developed laboratory-specific proteomic workflows, it is unclear if the large range of reported variability is founded on true interindividual variability or experimental variability resulting from sample preparation or the proteomic methodology used. To assess the potential for methodological bias on end-point abundance quantification, two independent laboratories, the University of Manchester (UoM) and Bertin Pharma (BPh), employing different proteomic workflows, quantified the absolute abundances of Na/K-ATPase, P-gp, and breast cancer resistance protein (BCRP) in the same set of biologic samples from human intestinal and Caco-2 cell membranes. Across all samples, P-gp abundances were significantly correlated (P = 0.04, Rs = 0.72) with a 2.4-fold higher abundance (P = 0.001) generated at UoM compared with BPh. There was a systematically higher BCRP abundance in Caco-2 cell samples quantified by BPh compared with UoM, but not in human intestinal samples. Consequently, a similar intestinal relative expression factor (REF), derived from distal jejunum and Caco-2 monolayer samples, between laboratories was found for P-gp. However, a 2-fold higher intestinal REF was generated by UoM (2.22) versus BPh (1.11). We demonstrate that differences in absolute protein abundance are evident between laboratories and they probably result from laboratory-specific methodologies relating to peptide choice.
Introduction
Numerous laboratories utilizing a diverse range of techniques are quantifying the absolute abundance of proteins by quantitative targeted absolute proteomic (QTAP) strategies in mammalian tissues and in vitro cell systems. Up to 10-fold differences in absolute abundances have been observed for specific drug-transporter isoforms in independent samples between laboratories, for example, hepatic organic anion–transporting polypeptide (OATP) OATP1B1 (Prasad et al., 2014; Vildhede et al., 2014). Differences in the proteomic workflow may underlie this variability, including techniques required to: 1) select and generate the peptide standard(s); 2) generate an enriched membrane fraction; 3) denature and digest the proteins; 4) separate the peptides by chromatography; and 5) conduct mass analysis of the selected peptides and peptide fragments [liquid chromatography–tandem mass spectrometry (LC-MS/MS)].
Studies have investigated the impact of methodological differences on quantification of transporter-protein abundances. For example, the type of solubilizing agent used prior to protein digestion has been shown to influence the quantification of OATP proteins in human embryonic kidney 293-transfected cells (Balogh et al., 2012) and can be a significant source of variability between studies. The selection of the peptide standard is also likely to be important and its influence on protein abundance quantification requires verification. In a recent study, the choice of peptide standard was considered to bias end-point abundance quantification of hepatic OATP1B1 (Terasaki et al., 2014). However, in a comparable study by Prasad et al. (2014), there was also an impact of peptide choice on OATP1B1 abundance using two of the same peptides as Terasaki et al. (2014), yet little influence of peptide choice for P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), OATP1B3, and OATP2B1 was observed. The digestion strategy employed might also influence the capacity to quantify protein abundances and to generate peptides that suffer missed cleavage by trypsin (Chiva et al., 2014). Studies comparing different proteomic approaches within a laboratory found that when comparing immunoblotting to stable isotope labeling of amino acids in culture/mammals (SILAC/SILAM) techniques, there was a reasonable agreement within the same samples to the absolute quantification (AQUA) method routinely employed (Kamiie et al., 2008; Qiu et al., 2013; Prasad and Unadkat, 2014a).
After showing a direct relationship between activity and abundance, absolute transporter-protein abundance and the interindividual variability are required to generate representative populations within physiologically based pharmacokinetic (PBPK) models. Furthermore, a relative expression factor (REF), the ratio of the transporters’ in vivo expression to that in the in vitro system, enables the impact of transporter activity on oral drug absorption to be assessed using an in vitro–in vivo extrapolation (IVIVE)-PBPK strategy (Harwood et al., 2013). Establishing whether differences in abundances reported across laboratories are derived from interindividual variability of a sample set or population, variability associated with assay-specific techniques, and/or specific data-analysis methods within each laboratory are crucial for generating robust system parameters that can be employed in PBPK models that represent in vivo abundances. Considering the dramatic increase in the use of IVIVE-PBPK models for decision-making in pharmaceutical company drug development pipelines, as well regulatory submissions over the last 5 years (Huang et al., 2013; Shepard et al., 2015), better insight into the comparability of various quantifications of transporter-protein absolute abundance is becoming essential.
To address whether there are laboratory-specific differences in generating system parameters for undertaking transporter-mediated IVIVE-PBPK, a multicenter study evaluating the consistency and comparability of the preparation steps and analytic outcome has been advocated (Harwood et al., 2014). However, to our knowledge, studies comparing the quantification of absolute transporter-protein abundances and the subsequent generation of REFs between laboratories for the same samples have not been reported in the literature yet. While not within the scope of this article, the impact of those laboratory-specific REF values on predicted pharmacokinetic outcomes in an IVIVE-PBPK model are provided in an accompanying study (Harwood et al., 2015). The aim of this study was to compare the absolute transporter abundances for three transporter proteins; Na/K-ATPase, P-gp, and BCRP, in matched Caco-2 cell and human intestinal total membrane fractions, quantified by two independent laboratories, the University of Manchester (UoM), Manchester, UK, and Bertin Pharma (BPh), Orleans, France, with each laboratory using different QTAP workflows. These data were used to generate REFs for intestinal Na/K-ATPase (REFiNa/K-ATPase), P-gp (REFiP-gp), and BCRP (REFiBCRP) on the basis of distal jejunal and Caco-2 absolute abundance specific to each laboratory.
Materials and Methods
Human Intestinal Tissue
Human intestinal tissue (n = 3 distal jejunum and n = 1 distal ileum) was obtained after informed consent from patients undergoing elective intestinal surgery at Salford Royal NHS Foundation Trust, Salford, UK. Prior ethics committee approval had been granted by the North West Research Ethics Committee, UK (12/NW/0306), and all procedures were carried out in accordance with the Declaration of Helsinki guidelines. Patients suffering from inflammatory bowel disease (i.e., Crohn’s disease or ulcerative colitis) and/or known to be affected by hepatitis B were excluded from participation. Donor demographics and drug history have are provided in the Supplemental Table 1.
Cell Culture
Caco-2 cells were purchased from the American Type Tissue Culture Collection (ATCC, Rockville, MD) at passage 18. Additionally, cryopreserved higher passage Caco-2 cells were used that were originally obtained from the ATCC and were subsequently cultured to a passage greater than 100 in the laboratories of the Biomedical Facility, Salford Royal Hospital Trust, Salford, UK. Cell culture reagents and transporter buffers, Dulbecco’s modified Eagle’s medium (DMEM), penicillin and streptomycin, l-glutamine, nonessential amino acids and Hanks’ balanced salt solution (HBSS) were purchased from Invitrogen Life Sciences (Paisley, Scotland). Newborn fetal calf serum gold (FCS) (heat inactivated) and trypsin-ethylenediaminetetraacetic acid (EDTA) were purchased from PAA Laboratories (Yeovil, UK). Low (LP, passage 25–35) and high passage (HP, passage 111) Caco-2 cells were maintained in complete DMEM growth media containing 10% FCS, 45 IU/ml penicillin, 45 μg/ml streptomycin, 1% nonessential amino acids, and 1.1% l-glutamine in an humidified atmosphere of 95% air and 5% CO2 at 37°C. The cells were fed every 2 days with fresh growth medium until 90% confluent, when they were subcultured routinely every 6 days with trypsin (0.05%)-EDTA and seeded into 75- or 175-cm2 adherent tissue culture flasks at 12,000 cells per cm2. Caco-2 cells were seeded onto 44-cm2 Transwell filters (CC3419; Corning, Lowell, MA) at 220,000 cells per cm2 (n = 3 pooled filters per experiment) and cultivated for 21 days (n = 4 experiments, including a single HP Caco-2 experiment) or 29 days (n = 3 LP experiments) with feeding every second day with complete DMEM. Caco-2 monolayer integrity was assessed by measurement of transepithelial electrical resistance (TEER) and permeability of the paracellular marker lucifer yellow (LY; 50 μM). LY permeability was measured in an apical-to-basolateral transport direction over 60 minutes at 37°C. Only monolayers showing significant TEER and LY apparent permeability (Papp) of ≤0.5 × 10−6 cm/s were used.
Human Enterocyte and Caco-2 Total Membrane Fractionation
Total membrane preparations were isolated by differential centrifugation after eluting human enterocytes by a calcium chelation protocol (Harwood et al., 2015). Caco-2 cell monolayers total membrane preparations were obtained after overnight lysing by differential centrifugation (Russell et al., 2013). Protein content was determined by the BCA assay.
Shipping/Transfer of Total Membrane Fractions
Total membrane samples were treated differently on the basis of the specific protocols employed within each laboratory. Total membrane fractions stored at –80°C were either retained at UoM for subsequent digestion and LC-MS/MS analysis, or shipped by freight overnight on dry ice to BPh. Upon receipt of the consignment of total membrane fractions by BPh, samples were immediately stored at –80°C. The total membrane fractions retained by UoM were also transferred on ice (approximately 45 minutes) from Salford Royal Hospital Trust to the Manchester Institute of Biotechnology where, following storage at –80°C, subsequent proteolytic digestion and LC-MS/MS analyses were performed. Analysis of total membrane protein samples by BPh was performed blind, whereas analysis at UoM was performed with knowledge of sample type. Final combined analysis of the quantified samples was performed at UoM.
Proteolytic Digestion
At BPh, total membrane proteins (50 μg) were digested by the MS2 Plex assay kit developed by BPh on the basis of techniques developed at Tohoku University, Sendai, Japan, including reduction, alkylation, and an overnight tryptic digestion step (Kamiie et al., 2008, Kunze et al., 2014). The digestion strategy at UoM was an adapted in-solution digest of 50 μg of total membrane protein derived from established methods (Balogh et al., 2012) and also accounts for peptide losses by a gravimetric method (Harwood et al., 2015). The digestion and analysis of the samples took place within a 1-month period between laboratories, a time period considered unlikely to impact on end-point abundance determinations (Sakamoto et al., 2011).
Determination of Transporter Abundances
Different QTAP workflows were employed within each laboratory; BPh employed an AQUA approach whereas UoM used a quantification concatemer (QconCAT) approach. The key differences between the AQUA and QconCAT approaches are: 1) AQUA standard peptides are generated by chemical synthesis, whereas QconCAT peptides are metabolically generated and labeled in a biologic host vector (i.e., Escherichia coli); and 2) QconCAT constructs possess non-naturally-occurring peptide (NNOP) internal standards, enabling the concentration of the target labeled standards, released in equimolar concentrations upon proteolytic digestion, to be quantified, whereas AQUA peptide concentration(s) are known prior to their entrance into the QTAP workflow, and abundances are calculated against a calibration curve. Therefore, in this study the QconCAT construct is simultaneously digested with the total membrane fractions containing the analytes.
An overview of the key components of the QTAP workflows of each laboratory is shown in Table 1. At each stage, a different strategy, protocol, peptide choice, or analytical instrument was employed between laboratories as follows.
An outline of the methods constituting the QTAP workflow specific to each laboratory for the absolute quantification of Na/K-ATPase, P-gp, and BCRP
Bertin Pharma Methods.
Transporter-protein abundances for Na/K-ATPase, P-gp, and BCRP were determined by BPh utilizing an AQUA-based simultaneous multiple reaction monitoring (MRM) LC-MS/MS strategy. The stable isotope labeled (SIL) proteotypic peptides selected for analysis were AAVPDA[V13C,15N]GK (Na/K-ATPase), FYDPL[A13C,15N)GK (P-gp), and SSL[L13C,15N]DVLAAR (BCRP). The selected ions and transition schedules for these peptides have been published (Kamiie et al., 2008; Sakamoto et al., 2011). The assay mixture (60 μl) prior to analysis consisted of the digested peptides (50 μl) and labeled standards (10 μl). Isotope-labeled standards (10 μl) were added to the peptide mixture to obtain a mixture with a specific relationship after proteolytic digestion. Samples were analyzed by LC-MS/MS using a normal flow Series 200 autosampler/HPLC pump and a Flexar LC (PerkinElmer, Waltham, MA) coupled to a API5500 triple-quadrupole mass spectrometer (AB Sciex, Framingham, MA). The assay mixture (40 μl) for each sample was injected into the LC system analytical column (XBridge BEH C18, 130 Å, 100 mm × 1.0 mm, 3.5 μm; Waters, UK) with column oven maintained at room temperature. Peptides were eluted with a 50-μl-per-minute flow rate and a gradient of 2–60% acetonitrile over 60 minutes. The transition schedules were obtained by direct-flow injection of peptide solutions. The [M+2H]+2 ion was selected as parent ion (Q1) and the four most intense fragment ions obtained by collision-induced dissociation in Q2 were selected as the Q3 transitions.
University of Manchester Methods.
Transporter-protein abundances were determined by UoM using a QconCAT-based simultaneous MRM LC-MS/MS strategy (Russell et al., 2013; Harwood et al., 2015).
All protein abundances are given as femtomol of transporter per microgram (fmol/μg) of total membrane protein, where the denominator is the nominal protein mass (typically 50 μg) entering the digestion phase of the workflow for BPh analysis, and for UoM it is the peptide content entering the sample tube for injection onto the chromatography column after correction for losses by a gravimetric method (Harwood et al., 2015).
The choice of peptides within the entire protein sequence for Na/K-ATPase, P-gp, and BCRP is provided in Supplemental Figs. 1–3.
Statistical Analysis
The abundance data were tested for normality using the Kolmogorov-Smirnov normality test. Owing to the negative results of the normality test and the relatively small sample size (n = 10–11), only nonparametric statistical tests were used. Transporter absolute abundance data from each laboratory were compared by a Wilcoxon matched-pair analysis. Spearman rank order analysis was used to test for sample abundance correlations. The statistical significance of the correlations was assessed using t-distribution analysis. An α value of 0.05 was employed to indicate a significant difference in all analyses. The objectivity of the study was maintained by keeping the proteomic analysts at both BPh and UoM blinded as to the results of the abundance quantifications on the matched samples.
Relative Expression Factor Generation.
On the basis of abundance data in the distal jejunum samples (n = 3) and 21-day filter-grown Caco-2 monolayers (n = 3 experiments on different days, on the basis of n = 3 pooled filters per experiment), a REF for IVIVE was generated (using eq. 1) for all three proteins. The REFs were based on 21 days, rather than 29-day–grown Caco-2 monolayers, as a monolayer of the lesser age is more commonly used for drug transport assays in the industry.
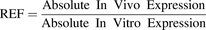 (1)
(1)Although absolute expression values were generated in this study, they were set relative to each other (eq. 1), thus one REF was obtained for each of the proteins (Na/K-ATPase, P-gp, and BCRP) for each laboratory.
Results
Interlaboratory Method Differences between Groups Quantifying Transporter-Protein Absolute Abundances
To give perspective to this cross-laboratory study, a literature analysis of the diversity of techniques used by different laboratories for transporter absolute abundance quantification was undertaken. Fifteen laboratories have reported quantification of transporter abundances in mammalian tissues or in vitro systems. Table 2 highlights the general differences in methods for sample preparation, peptide selection, digestion strategies, and analytical systems and shows that there is considerable variation in the methodologies used to quantify transporter abundances. The predominant approach has been to use synthetically made isotope-labeled standard peptides like AQUA. However, a combination of label-free and targeted (labeled) approaches have also been used simultaneously within the same study (Karlgren et al., 2012). BPh provides QTAP services and kits utilizing technologies developed at Tohoku University, and as a result the workflows from both laboratories share many similarities. In fact, several laboratories foster links with the forerunning developers of these methodologies, i.e., Tohoku University (Sendai, Japan) and Pfizer Ltd. (Groton, CT). Peptides that are used as surrogates for the quantification of the complete protein are selected on the basis of a variety of criteria (Ohtsuki et al., 2011; Oswald et al., 2013; Prasad and Unadkat, 2014b; Qiu et al., 2014). Consequently, for the frequently quantified transporter-protein P-gp, three peptides are routinely employed to quantify human P-gp (AGAVAEEVLAAIR, NTTGALTTR, FYDPLAGK; Table 2), highlighting a current lack of consensus among groups. Comparative studies testing all three peptides on matched samples between laboratories have yet to be reported. In-solution trypsin-based overnight digestion strategies dominate, but the utility of Lys-C to facilitate protein digestion prior to incubation with trypsin is favored by The Max Planck Institute (Martinsried, Germany) and UoM (Achour and Barber, 2013). Nanoflow and normal-flow LC approaches are used, but the advantages of one approach over another have not been discussed within these studies. The triple-quadrupole MS system is routinely employed for targeted approaches; however, the manufacturers and models differ between laboratories. For comparative purposes, the use of quantitative immunoblotting is also incorporated in this analysis. This approach does not employ peptide-selection strategies, protein digestion, and LC-MS/MS for analysis but relies on selective antibodies being available for target transporters, which is not always the case (Tucker et al., 2012).
A comparative analysis of methods used for quantification of transporter-protein absolute abundances across research groups
Cross-Laboratory Comparison of Transporter Absolute Protein Abundances Quality Control.
The analytical quality control for Na/K-ATPase, P-gp, and BCRP was established for linearity (R2 ≥ 0.999), precision (CV ≤ 15%), and accuracy (relative error, 71–112% using quality control samples), and the lower limit of quantification (LLOQ) was determined as 0.125 fmol/μg for all proteins by BPh (see Supplemental Tables 2–4). The linearity and LLOQ were derived from synthetic labeled standard peptides, with the precision determined by the coefficient of variation across the four selected transitions of each standard peptide quantified.
The analytical quality control for the same proteins established at UoM has been reported in detail by Harwood et al. (2015). The linearity for abundance determination of each peptide in the presence of a biologic matrix (i.e., human intestinal total membrane protein) was established. In addition, linearity in the quantification of the QconCAT used by UoM was determined using the internal calibrator NNOP peptide Glu-Fibrinopeptide B (Harwood et al., 2015). The intraday and interday precision and relative error across biologic samples were also measured as part of the quality control procedure. The LLOQ for abundance determinations at UoM was 0.2 fmol/μg for all proteins.
Appraisal of Na/K-ATPase, P-gp, and BCRP Peptides Selected by BPh and UoM.
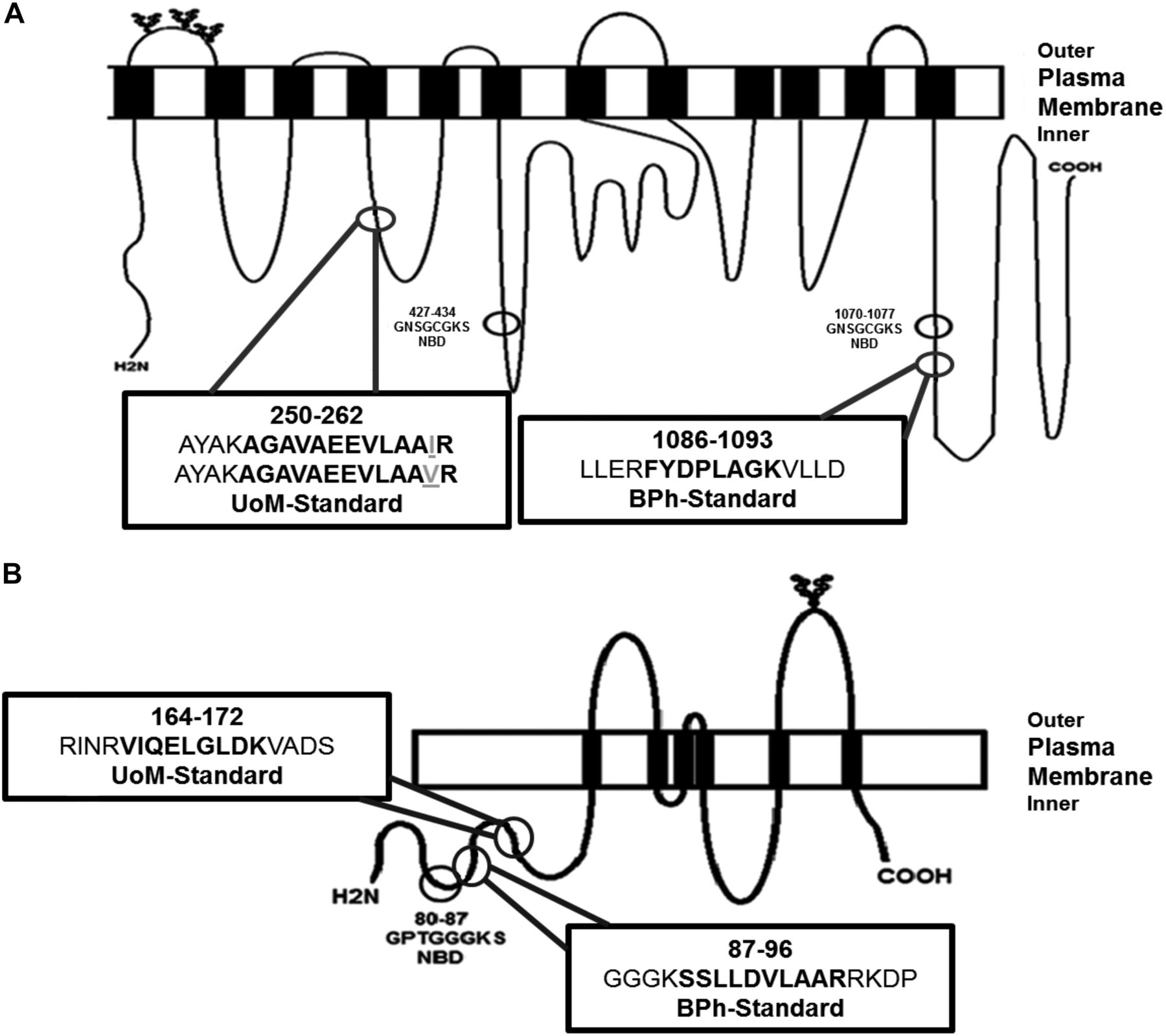
The proteotypic standard peptides analyzed by each laboratory for quantification of Na/K-ATPase, P-gp, and BCRP were selected using criteria published by Tohoku University and UoM (Kamiie et al., 2008; Russell et al., 2013). For Na/K ATPase (ATP1A1), the peptide selection by BPh (AAVPDAVGK) was not favorable for selection by UoM because of the potential for peptide miscleavage, as flagged the CONSeQuence program developed at the UoM (Lawless and Hubbard, 2012). Concerns were also raised regarding the peptides’ uniqueness for quantification purposes, as AAVPDAVGK also occurs in human Na/K-ATPase α2 subunit (Shull and Lingrel, 1987), a protein expressed at low levels in human liver tissue (http://www.proteinatlas.org/ENSG00000018625-ATP1A2/tissue). The position of the selected standard peptides within P-gp and BCRP protein structure is shown in Fig. 1. The criteria defined by Tohoku University would have led to rejection of the P-gp peptide selected by UoM, as a single-nucleotide polymorphism (SNP) is present at position 261 (p.I261V–c.A781G). This SNP has an allelic frequency in African Americans of 0.6% (Kroetz et al., 2003) and 6.9% in Ugandans (Mukonzo et al., 2010) but is not detected in other populations, including Caucasians (n = 100), Asian Americans (n = 30), Mexican Americans (n = 10), Pacific Islanders (n = 7), and Japanese (n = 145) (Kroetz et al., 2003; Ozawa et al., 2004). The standard peptide selected for P-gp quantification by BPh was flagged as having the potential for a trypsin missed cleavage event by ConSEQuence (Lawless and Hubbard, 2012). In the event of missed cleavage, the selected peptide would be elongated, changing its mass and resulting in the first mass filter (Q1) of a triple-quadrupole MS rejecting its selection for downstream collision fragmentation (Q2) and product ion monitoring (Q3). As a consequence, a native peptide signal lower in comparison with the standard peptide might be expected, in turn generating a lower biologic abundance.

The location and nomenclature of the selected peptides for quantifying P-gp (A) and BCRP (B) absolute protein abundances. Bertin Pharma (BPh)- and the University of Manchester (UoM)-selected peptide sequences (bold residues) are shown in the boxes. The amino acid residues immediately flanking the selected peptides are in plain text. UoM-selected peptide contains the potential to harbor a single-nucleotide polymorphism p.I261V (c.A781G) for P-gp, which is given as gray and underlined text. The variant “V” peptide is also provided. The nucleotide binding region (NBD) and N-glycosylated sites are shown in the first extracellular loop of P-gp and the third extracellular loop of BCRP.
The BCRP peptides selected by both groups showed no evidence of SNPs. The peptide-selection criteria applied by Tohoku University were unlikely to have given the UoM peptide a high score, owing to the presence of glutamine (Q), which has the potential to suffer a deamidation post-translational modification. This event was judged tolerable at UoM, given that the rate of glutamine deamidation is considerably lower than that for asparagine (N), with a reaction half-life of 660 days (Li et al., 2010b). The selection criteria employed by UoM would have rejected the BPh BCRP peptide standard owing to the anticipated difficulty of efficient trypsin cleavage at dibasic and tribasic tryptic sites, i.e., sequential lysine or arginine residues or lysine and arginine side-by-side at the N-terminal region of the BPh peptide (Lawless and Hubbard, 2012).
Transporter Abundances in Caco-2 and Human Intestinal Samples.
Total membrane fractions from seven Caco-2 cell monolayers, three human distal jejunums, and a single human distal ileum were analyzed in each laboratory (n = 11). The comparative absolute abundances and the correlations between BPh and UoM for Na/K-ATPase, P-gp, and BCRP are provided in Figs. 2 and 3, respectively. The individual abundance data for each sample are provided in Table 3. For Na/K-ATPase and BCRP, there was no difference in the mean abundances (P = 0.36 and 0.76, respectively, Wilcoxon matched-pair test) with a moderate correlation for Na/K-ATPase (Rs = 0.42) and no correlation for BCRP (Rs = 0.39) using a Spearman rank correlation analysis across all samples between laboratories. A systematically higher BCRP abundance was quantified by BPh compared with UoM in 21- and 29-day cultivated Caco-2 cells ranging from 20 to 129% in individual samples (Table 3). This was not reciprocated in the intestinal BCRP abundances, which were on average 17% higher (range –40 to 72%) in UoM analysis. A systematically and significantly higher P-gp abundance (63%, P = 0.001, Wilcoxon matched-pair test) was quantified by UoM compared with BPh across all samples; in contrast to Na/K-ATPase and BCRP, P-gp abundances were highly correlated (Rs = 0.72, P = 0.04, Fig. 3B). For the single distal ileum sample, P-gp abundance was below the LLOQ (<0.125 fmol/μg) in BPh determinations, but UoM determined a value of 0.2 fmol/μg total membrane protein. In both laboratories, lower mean abundances of P-gp were observed in the human intestinal samples than Caco-2 cell monolayers, irrespective of cultivation time. This is in contrast to BCRP, in which the opposite relationship was observed. The importance of correcting for losses of peptides during reduction, alkylation, and digestion steps when determining absolute transporter abundances has been established (Harwood et al., 2015). These corrections were applied only to abundance determinations at UoM, since they are not a routine component of abundance determinations at BPh. When peptide-loss corrections were not applied to samples quantified at UoM, P-gp abundances were still systematically and significantly higher (52%, P = 0.001, Wilcoxon matched-pair test), and Na/K-ATPase and BCRP abundances were also still not significantly different (P = 0.36 and P = 0.58, respectively, Wilcoxon matched-pair test).

Absolute protein abundances of Na/K-ATPase (A), P-gp (B), and BCRP (C) determined by Bertin Pharma (BPh, black) and the University of Manchester (UoM, white) in Caco-2 cell monolayers (n = 7) and human intestinal total membrane protein fractions (n = 4). The text above the bars indicates Caco-2 monolayer cultivation age. *p < 0.05. The mean ± standard deviation is given for the distal jejunum, 21-day, 29-day Caco-2 cells, and overall means are given across all samples for each protein. DI, distal ileum; DJ, distal jejunum.

Correlation analysis (Spearman rank order test) of the absolute protein abundances of Na/K-ATPase (A), P-gp (B), and BCRP (C) between Bertin Pharma and the University of Manchester. Diamonds denote human (white) and Caco-2 cell monolayers (black). Spearman rank correlation coefficients (Rs) are provided as text on the plot, as are the p values representing the t-distribution for assessing the significance of the correlation. The dashed line is the line of identity and the solid line is the line of best fit. A single sample (distal ileum) from BPh for P-gp was below the limit of quantification.
Individual abundances of human intestinal and Caco-2 cell monolayer, with REFs for Na/K-ATPase, P-gp, and BCRP
Standard deviations are given in parentheses. *Denotes mean relative error when accounting for individual samples.
Generation of Relative Expression Factors.
The potential impact of these results on IVIVE scaling was investigated by generating a REF (eq. 1) for human distal jejunum samples and 21-day cultivated Caco-2 cell monolayers (n = 3; Table 3). Owing to the high correlation of observations between laboratories for quantification of P-gp abundances, the systematically higher P-gp levels measured by UoM had no effect on the REF between laboratories (BPh-REFiP-gp = 0.37, UoM-REFiP-gp = 0.4). Na/K-ATPase, although exhibiting a moderate correlation (Rs = 0.42), also showed little difference in jejunal-REF between laboratories (BPh-REFiNa/K-ATPase = 0.68, UoM-REFiNa/K-ATPase = 0.58). However, BCRP showed distinct differences, with UoM-REFiBCRP being 2-fold higher than the BPh-REFiBCRP (REF were 2.22 versus 1.11, respectively), which probably resulted from the systematically lower Caco-2 cell abundance levels measured at UoM.
Discussion
Several laboratories have developed and validated protocols for quantifying transporter-protein abundances in biologic systems using proteomic approaches (Table 2). With this raft of abundance data generated across multiple laboratories now being incorporated into IVIVE-PBPK strategies (Bosgra et al., 2014; Kunze et al., 2014; Prasad et al., 2014; Vildhede et al., 2014), it is critical to establish if laboratory-specific methodologies contribute to a bias in abundance determination. To address this, membrane protein samples from human intestine and Caco-2 monolayers were analyzed independently by two laboratories, Bertin Pharma and the University of Manchester. For P-gp, there is a systematic bias in abundances between laboratories. For Na/K-ATPase and BCRP, mean abundances reported by BPh and UoM were similar across all samples; however, the limited correlation in abundances of these transporters in individual samples suggests a potential influence of technical and/or analytical variability in postmembrane processing between laboratories.
Efficient protein digestion is critical for abundance determination, and integral membrane transporter proteins are notoriously challenging to digest because of their poor solubility (Mirza et al., 2007). Therefore, different solubilizing agents are often used to enhance protein solubility and facilitate target protein digestion. On the basis of findings by Balogh et al. (2013), UoM employed sodium deoxycholate as the solubilizing agent. However, BPh does not disclose the components of their MS2Plex digestion kit, and therefore a systematic evaluation of the effect of potential differences in the procedures employed in both laboratories to denature, reduce, alkylate, and digest the proteins cannot be undertaken.
Critical to the quantification of transporter-protein abundances is completion of enzymatic digestion (Ji et al., 2012). Digestion efficiency for selected peptides has been assessed in several studies (Kamiie et al., 2008; Li et al., 2008; Zhang et al., 2011; Balogh et al., 2013; Ji et al., 2012; Gröer et al., 2013; van de Steeg et al., 2013). However, assessing digestion completeness is a significant challenge owing to the limited availability of purified transporter proteins used to monitor peptide digestion (Prasad et al., 2014; Harwood et al., 2015). Peptide instability over the duration of digestion should be considered and could be a factor in mass spectrometry signal reduction (van den Broek et al., 2013). The digestion strategy at UoM incorporated a lysyl-endoproteinase as a priming step prior to trypsin incubation, cleaving the C-terminal side of lysine residues, generating lysine-terminated peptides to improve trypsin digestion efficiency (Achour and Barber, 2013). Within this study, it was assumed, on the basis of prior optimization, that digestion achieves a steady state over the time course of incubation with the proteolytic enzymes within the biologic matrices under investigation for both laboratories; therefore, any source of variability between samples should not result from inefficient digestion.
Selecting peptides that uniquely identify the targeted protein on the basis of trypsin digestion are required. The peptide (AAVPDAVGK) selected by BPh to quantify Na/K-ATPase (ATP1A1) is not unique, as the sequence is also found in human Na/K-ATPase α2 subunit (ATP1A2) (Shull and Lingrel., 1987). Peptides possessing a lower potential for missed cleavage events are also advantageous (Lawless and Hubbard, 2012). This is pertinent given that in relative quantification analysis, up to 46% of peptides generated miscleaved events after in-solution trypsin digestion in E. coli (Chiva et al., 2014). For the peptide (AAVPDAVGK) selected for Na/K-ATPase quantification by BPh, the C-terminal lysine (K) is closely followed by an arginine (R), separated by a cysteine (C) residue [K.CR. (in which the period denotes potential cleavage by trypsin)]. As a result, the P-gp peptide selected at Tohoku University did not score as favorably as other P-gp peptides selected and used by UoM (Russell et al., 2013). Peptide selection through the triple-quadrupole mass spectrometer is based on predefined masses, where any violation of these masses, i.e., by missed cleavage of target peptide, leads to the peptide’s exclusion in mass filter 1 (Q1), resulting in lower abundance measurements. These events may explain the differences in P-gp abundance quantification between laboratories shown in this study. Interestingly, Miliotis and coworkers (2011) used the same peptide selected by UoM (AGAVAEEVLAAIR) to assess the abundance of P-gp in 29-day–grown Caco-2 monolayers, reporting an abundance of 7.89 fmol/μg total membrane protein, similar to UoM (6.92 fmol/μg total membrane protein) and much higher than quantification by BPh (1.97 fmol/μg total membrane protein). This lends support to the conjecture that peptide selection may contribute to bias in abundance measurements.
These observations reinforce the critical importance of peptide choice in abundance determination as demonstrated in studies of hepatic OATP1B1 abundance (Terasaki et al., 2014). A previous study from that group used a peptide located in the ninth transmembrane domain of OATP1B1, a region associated with lower tryptic digestion efficiency (Kamiie et al., 2008), and reported OATP1B1 abundances below detection limits in 8 of 17 human liver samples (Ohtsuki et al., 2012). This was unexpected given that OATP1B1 exhibits the highest abundance of all liver OATP transporters as shown in a recent meta-analysis (Badée et al., 2015). Re-evaluation of the original peptide against four other peptides located in intracellular or extracellular loop domains of OATP1B1 showed that this peptide underestimates mean abundance by more than 20-fold in comparison with the best performing peptide (Terasaki et al., 2014). The systematic difference in P-gp abundances quantified by UoM and BPh in this study is smaller (63%, Table 3); however, this level of methodological bias in abundance determination is of concern when incorporating population abundances into PBPK models, given the potential effect on predicted transporter-mediated drug disposition (Harwood et al., 2013).
The consistently lower BCRP abundance in Caco-2 cells reported by UoM in this study (Table 3, Figs. 2C and 3C) does not appear to be peptide-specific since BCRP abundances in three out of four human samples were higher at UoM than BPh. One possible explanation is that the Caco-2 cell membrane matrix may affect digestion of the selected BCRP peptide (Fig. 1), impacting on abundance quantification. Chromatographic effects may also contribute to these differences, as reported for NTTGALTTR, a peptide commonly used to quantify P-gp (Gröer et al., 2013). Chromatographic interference is probably not be the source of the observed lower BCRP abundances in Caco-2 cells, as coelution profiles (i.e., identical retention times) of native and standard peptides were generated. However, the methodological validation of these peptides was initially performed in human intestine (Harwood et al., 2015). Further studies assessing the optimal digestion conditions for the selected BCRP peptides in Caco-2 cells are warranted. Additionally, this study does not control for different membrane fractionation techniques, which was recently suggested as a source of variability between and within studies (Heikkinen et al., 2015). To test this, repeated membrane extraction, protein digestion, and subsequent abundance quantification are required on the same tissue over multiple days. This would remove variability associated with technical/analytical procedures from the intrinsic biologic variability, a requirement for building representative IVIVE-PBPK frameworks with population variability.
In addition to obtaining transporter abundances in tissues of individuals for incorporation into PBPK models, abundance data from mammalian tissues and in vitro systems are used to generate IVIVE scaling factors (Li et al., 2010a; Karlgren et al., 2012; Vildhede et al., 2014). This study generated laboratory-specific REF-IVIVE scaling factors for all three transporters, using abundance data for jejunum and Caco-2 monolayers. This enabled the assessment of laboratory-specific biases in abundance measurements on REF. REFiP-gp was the same between laboratories owing to the consistent bias in P-gp abundances for in vivo and in vitro systems. However, if bias in abundance quantification occurs selectively within a biologic system between laboratories (i.e., Caco-2 cell BCRP abundances being nearly 2-fold higher at UoM), the laboratory-specific REF is affected and can be significantly different.
The results of this study for the first time show significant differences in transporter-protein abundance reported on the same samples by different laboratories, implying methodological bias is likely to be a significant source of variability. Thus, caution should be exercised when utilizing absolute mammalian tissue abundances from literature sources to estimate whole organ abundances and REFs in IVIVE-PBPK strategies. Cross-laboratory protein abundance comparisons on the same samples can reveal biases that are integral to a particular laboratory, or a laboratory-specific step within a workflow. We recommend further comparison studies using matching samples across a wider range of laboratories, including the complete workflow specific to each laboratory, to establish the critical factors influencing protein abundance quantification. Subsequently, by selecting the technique/operating procedure with the most favorable precision, accuracy, and reproducibility, a further study using the same biologic samples as the first comparative analysis can commence. The overall aim is to reduce method-dependent variability to an acceptable minimum, so the intrinsic biologic variability can be determined with high accuracy in any laboratory using the same workflow.
Finally, to what extent do variations in REF caused by differences in transporter abundance data between laboratories influence the prediction of oral drug absorption by IVIVE-PBPK models? Assessing the impact of REFs from different sources on PK parameters by IVIVE-PBPK strategies is addressed in the accompanying study (Harwood et al., 2015).
Acknowledgments
The authors acknowledge Professor Douglas Kell, Professor Royston Goodacre, and Dr. David Ellis (University of Manchester) for kindly allowing laboratory access and The Michael Barber Centre for Mass Spectrometry, University of Manchester, for allowing access to the LC-MS/MS instrument.
We would also like to thank Eleanor Savill for her assistance in formatting the manuscript for submission.
Authorship Contributions
Participated in research design: Harwood, Achour, Neuhoff, Russell, Warhurst, Rostami-Hodjegan.
Conducted experiments: Harwood, Achour.
Contributed new reagents or analytic tools: Russell, Carlson.
Performed data analysis: Harwood, Achour.
Wrote or contributed to the writing of the manuscript: Harwood, Achour, Carlson, Neuhoff, Russell, Warhurst, Rostami-Hodjegan.
Footnotes
- Received September 25, 2015.
- Accepted December 1, 2015.
This study was supported by an Industrial Fellows Grant awarded to M.D. Harwood from The Royal Commission for Exhibition of 1851, which has contributed to OrBiTo IMI project (http://www.imi.europa.eu/content/orbito) as a side-ground. Manchester Pharmacy School, The University of Manchester, funded work undertaken by B. Achour and M.R. Russell.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AQUA
- Absolute quantification
- BCRP
- breast cancer resistance protein
- BPh
- Bertin Pharma
- DMEM
- Dulbecco's modified Eagle's medium
- EDTA
- trypsin-ethylenediaminetetraacetic acid
- FCS
- newborn fetal calf serum gold
- HBSS
- Hanks' balanced salt solution
- IVIVE
- in vitro–in vivo extrapolation
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- LLOQ
- lower limit of quantification
- MRM
- multiple reaction monitoring
- OATP
- organic anion–transporting polypeptide
- P-gp
- P-glycoprotein
- PBPK
- physiologically based pharmacokinetic
- QconCAT
- quantification CONcatamer
- QTAP
- quantitative targeted absolute proteomic
- REF
- relative expression factor
- REFiBCRP
- relative expression factor for intestinal breast cancer resistance protein
- REFiNa/K-ATPase
- relative expression factor for intestinal sodium/potassium-ATPase
- REFiP-gp
- relative expression factor for intestinal P-glycoprotein
- SNP
- single-nucleotide polymorphism
- UoM
- The University of Manchester
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}