


Abstract
Momelotinib (MMB), a small-molecule inhibitor of Janus kinase (JAK)1/2 and of activin A receptor type 1 (ACVR1), is in clinical development for the treatment of myeloproliferative neoplasms. The pharmacokinetics and disposition of [14C]MMB were characterized in a single-dose, human mass-balance study. Metabolism and the pharmacologic activity of key metabolites were elucidated in multiple in vitro and in vivo experiments. MMB was rapidly absorbed following oral dosing with approximately 97% of the radioactivity recovered, primarily in feces with urine as a secondary route. Mean blood-to-plasma [14C] area under the plasma concentration-time curve ratio was 0.72, suggesting low association of MMB and metabolites with blood cells. [14C]MMB-derived radioactivity was detectable in blood for ≤48 hours, suggesting no irreversible binding of MMB or its metabolites. The major circulating human metabolite, M21 (a morpholino lactam), is a potent inhibitor of JAK1/2 and ACVR1 in vitro. Estimation of pharmacological activity index suggests M21 contributes significantly to the pharmacological activity of MMB for the inhibition of both JAK1/2 and ACVR1. M21 was observed in disproportionately higher amounts in human plasma than in rat or dog, the rodent and nonrodent species used for the general nonclinical safety assessment of this molecule. This discrepancy was resolved with additional nonclinical studies wherein the circulating metabolites and drug-drug interactions were further characterized. The human metabolism of MMB was mediated primarily by multiple cytochrome P450 enzymes, whereas M21 formation involved initial P450 oxidation of the morpholine ring followed by metabolism via aldehyde oxidase.
Introduction
Activation of alleles of the Janus kinase (JAK) 2 gene, such as the JAK2V617F allele, plays a central role in the pathogenesis of myeloproliferative neoplasms (MPNs) (Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Levine et al., 2007; Hasselbalch, 2012). The JAK2V617F mutation results in constitutive activation of JAK2/signal transducer and activator of transcription (STAT) signaling and cytokine hypersensitivity, and is present in >80% of patients with polycythemia vera (PV), ∼40% with essential thrombocytopenia (ET), and ∼70% with primary myelofibrosis (PMF) (Levine et al., 2007). The occurrence of JAK2 mutations in most patients with PV, ET, or PMF has reinforced the potential oncogenic role of abnormal JAK/STAT signaling in these disorders (Levine et al., 2007). Other mutations implicated in activation of the intracellular JAK/STAT pathway have also been identified in patients with MPNs, suggesting that dysregulation of the JAK/STAT pathway is a key component in their pathogenesis (Delhommeau et al., 2010). Accordingly, small-molecule JAK2 inhibitors have been developed to inhibit pathogenic JAK/STAT signaling in patients with MPNs and, as expected, have demonstrated a therapeutic benefit in patients with or without the V617F mutation (Pardanani, 2008; Verstovsek et al., 2012; Sonbol et al., 2013; Vannucchi et al., 2015).
Momelotinib (MMB, Fig. 1), previously known as CYT387 (Tyner et al., 2010; Sparidans et al., 2012; Durmus et al., 2013), is a potent and selective small-molecule inhibitor of JAK1/2 (Pardanani et al., 2009, 2013). This agent displays potent in vitro inhibitory activity against the JAK2V617F mutant and inhibits intracellular JAK1/2 signaling events (Winton and Kota, 2017). In phase 1 and 2 studies in patients with myelofibrosis, treatment with MMB resulted in significant improvement in splenomegaly and in nearly all patient-reported symptoms (Pardanani et al., 2011, 2013; Gupta et al., 2017). Two-year progression-free survival was reported in 74% of patients with long-term follow up (Gupta et al., 2017). In a phase 3 study evaluating the efficacy and safety of MMB compared with ruxolitinib in patients who had not received prior treatment with a JAK inhibitor, 24 weeks of MMB treatment was noninferior to ruxolitinib for spleen response but not for symptom response (Mesa et al., 2017). However, MMB treatment also alleviated anemia, increasing hemoglobin levels in transfusion-dependent patients, and was associated with a reduced transfusion requirement (Mesa et al., 2017). MMB also potently inhibits activin A receptor type 1 (ACVR1; Asshoff et al., 2017). Nonclinical studies suggest that the clinical anemia benefit of MMB may result from inhibition of ACVR1-mediated expression of hepcidin in the liver. Inhibiting hepcidin increases the release of iron from sequestered cellular stores, iron uptake from the gut, and iron availability for erythropoiesis (Asshoff et al., 2015; Warr et al., 2016).

Proposed general scheme for MMB metabolism and M21 formation. Generation of M21 involves subsequent metabolism by AOX, which is sensitive to inhibition by hydralazine.
This report presents the characterization of the pharmacokinetics (PK) and disposition of MMB in human and nonclinical species. MMB forms disproportionately higher amounts of a circulating metabolite (designated M21), a morpholino lactam, in humans relative to nonclinical species. The enzymology responsible for the formation of M21 was explored. On the basis of these experimental results, nonclinical repeat-dose studies were performed to characterize M21 to satisfy the regulatory requirements on safety testing of drug metabolites. In addition, MMB characteristics that may contribute to the reported anemia benefits of MMB were examined.
Materials and Methods
Materials
MMB and its metabolites (M8, M19, and M21) were synthesized at Gilead Sciences (Foster City, CA) (Brown et al., 2015; Sun et al., 2016; Zhu et al., 2017) (the physicochemical properties of MMB are described in the Supplemental Materials). [14C]MMB was synthesized at Moravek Biochemicals (Brea, CA). The [14C] in [14C]MMB was incorporated at the C-2 carbon on the substituted pyrimidine ring (Fig. 2) and was supplied in dimethyl sulfoxide solution. The [14C]MMB chemical purity and radiochemical purity were >99% in clinical and nonclinical mass-balance studies. All other chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO) or VWR International (West Chester, PA), and were of high-performance liquid chromatography or analytic grade.

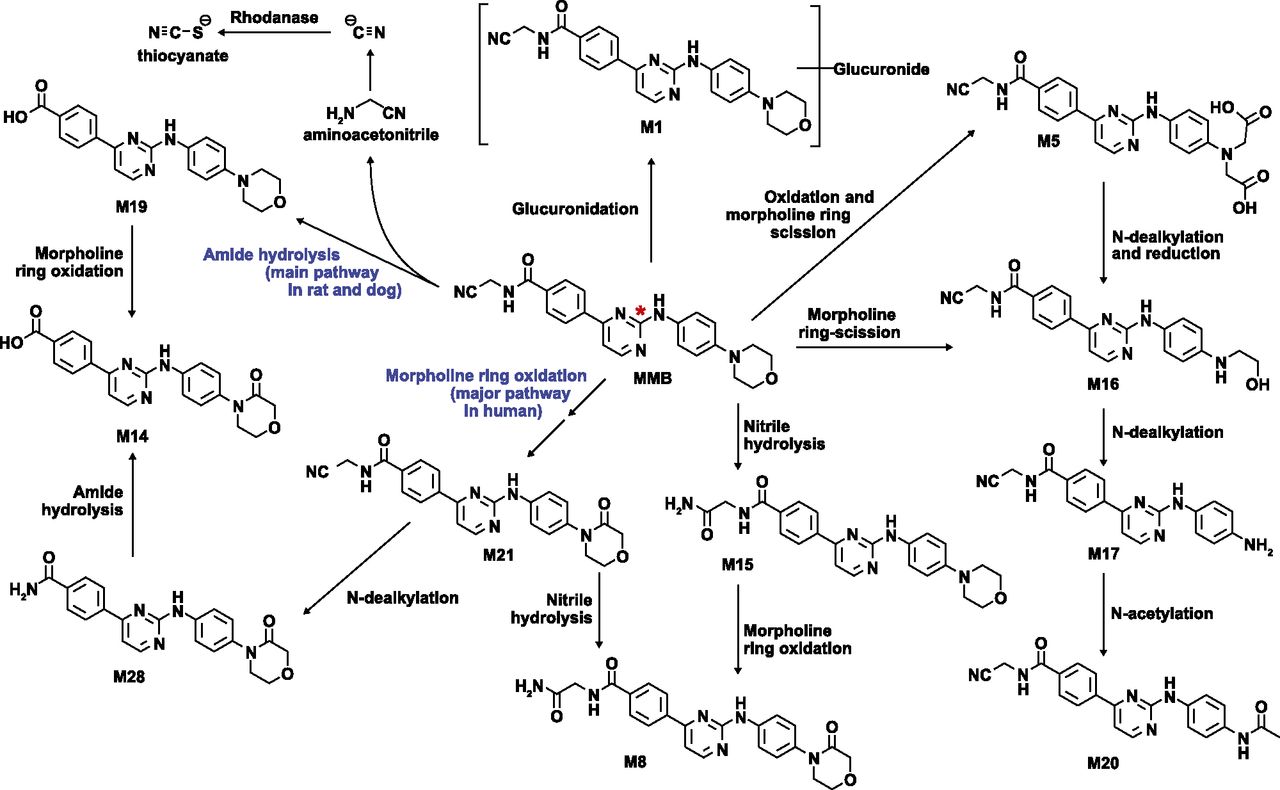
Proposed MMB biotransformation pathways (* denotes [14C] label position). Structures of MMB, M8, M19, and M21 were confirmed with synthetic standards. For all other metabolites, the proposed structures are consistent with available high-resolution mass spectrometry data and the biotransformation pathways leading to their formation.
Human whole blood was obtained from the South Bank Mobile Donor Centre (South Bank, Queensland, Australia). Pooled hepatic microsomal fractions, human cryopreserved hepatocytes, hepatocyte thawing medium, and Krebs-Henseleit buffer medium were procured from Bioreclamation IVT (Baltimore, MD). An NADPH-regenerating system, insect cell microsomal fraction containing baculovirus-expressed recombinant human cytochrome P450 (P450) or flavin-containing monooxygenase enzymes, and recombinant human monoamine oxidase enzymes and pooled human hepatic postmitochondrial supernatant fractions were purchased from BD Biosciences (Woburn, MA).
In Vitro Studies of MMB Metabolism and Physicochemical Properties
In vitro studies were performed using standard methods to characterize lipophilicity [distribution coefficient at pH 7.4 (logD7.4)], solubility, logarithmic of acid dissociation constant (pKa), permeability, blood-to-plasma ratio, binding in plasma, mixed-gender human liver microsomal fraction and pharmacodynamic assay media, stability in hepatocyte, and the enzymology of MMB metabolism, as well as formation of the MMB major circulating human metabolite. The analyte concentrations (MMB and metabolites, as applicable) were determined by high-performance liquid chromatography–tandem mass spectrometry (LC-MS/MS). In vitro study and bioanalysis methodologies are provided in Supplemental Table 1.
Human PK and Disposition Mass-balance Study
Study Design.
The PK and disposition of MMB following a single oral administration of [14C]MMB were evaluated in a phase 1, single-center, open-label, mass-balance study conducted in six healthy men. The study was conducted in accordance with recognized international scientific and ethical standards, including but not limited to the International Conference on Harmonization guideline for Good Clinical Practice and the principles embodied in the Declaration of Helsinki. Written informed consent was obtained from all participants. An overview of the purpose and design of this study is presented in Table 1.
Summary of nonclinical and clinical in vivo study design following oral administration of MMB
Screening for eligibility was performed within 28 days prior to the day-1 visit. On enrollment, eligible subjects were confined at the study center starting on day 1, received a single dose of study drug on day 1 and remained confined for a minimum of 5 days and up to 22 days until completion of protocol-specified assessments. Telephone follow up was conducted 7 (±1) days after discharge from the study center.
Subjects.
Subjects were healthy, nonsmoking men (aged 18–45 years) with a body mass index of 19–30 kg/m2 and confirmed to have normal kidney and liver function. All subjects were human immunodeficiency virus and hepatitis B/C negative and had not engaged in blood donation or other activities that might alter absorption, distribution, metabolism, and excretion (ADME) assessments.
Subjects were excluded if they engaged in alcohol or substance use that might interfere with compliance or safety, had taken a strong CYP3A4 inhibitor or inducer within 2 weeks prior to enrollment, or had taken any prescription or over-the-counter medication other than vitamins, acetaminophen, or ibuprofen within 28 days of study-drug administration.
Treatment and Sampling.
On the basis of pharmacokinetic and dosimetry data from a rat quantitative whole-body autoradiography study, administration of a single oral 100-μCi dose of [14C]MMB was not expected to represent a significant radiation exposure risk in humans. MMB (200 mg; ∼100 μCi) was formulated as a 200-ml solution that included 0.11% dimethyl sulfoxide, 0.38% hydrochloric acid (10% w/v), 5.62% purified water, and 93.8% cranberry juice. MMB was administered orally following overnight fasting on study day 1. Whole-blood and plasma samples were collected at prespecified time points. After the 96-hour postdose time point, additional blood samples were collected at 24-hour intervals up to the morning of day 22 (504 hours postdose) or until one of the following two conditions were met: Liquid scintillation counting (LSC) indicated that radioactivity levels in two consecutive whole-blood or plasma samples decreased to levels below the limit of quantitation, or both urine and fecal collections were discontinued.
All voided urine and feces were collected and pooled, beginning predose (within 12 hours for urine and within 24 hours for feces prior to the day-1 dose) and continuing over the prespecified collection intervals up to 120 hours postdose. After the 120-hour postdose collection, all voided urine and feces continued to be collected over successive 24-hour collection intervals up to the morning of day 22 (504 hours postdose), or until LSC indicated that radioactivity levels in samples from two consecutive 24-hour collection intervals were ≤1% of the administered radioactivity and the cumulative radioactivity recovered in urine plus feces was ≥90% of the administered radioactivity.
Determination of Radioactivity by LSC and Quantitation of MMB and Metabolites by LC-MS/MS.
The amount of radioactivity present in blood, plasma, urine, and feces was measured using LSC. Concentrations of MMB and metabolites (M19 and M21) were determined using an LC-MS/MS method (for additional details, see the metabolic profiling methodology in the Supplemental Materials and Methods). PK parameters for total radioactivity in plasma and blood, as well as nonradiolabeled MMB and metabolites (M19 and M21), were estimated using noncompartmental analyses by Phoenix WinNonlin 6.4 (Pharsight Corporation, Princeton, NJ), including maximal concentration (Cmax), clearance (CL) of the drug from plasma after oral administration (CL/F), the area under the plasma concentration-time curve (AUC) from time 0 to infinity (AUC∞), time to Cmax (Tmax), and half-life (t1/2).
Plasma metabolite profiling was done with AUC-pooled samples (Hamilton et al., 1981). An individual-subject AUC pooled sample was prepared from samples collected from 1 to 24 hours from each of the six subjects. The individual time-pooled plasma samples were then further pooled to create a single AUC-pooled sample. Individual subject urine and feces metabolite profiles were obtained from pooled samples that were prepared by combining a weight-proportional aliquot from each time interval. The pooled sample represented ∼90% of the total radioactivity excreted over the cumulative collection intervals up to 24 and 72 hours in urine and feces, respectively. Radiochromatograms were generated and MMB-derived structures were assigned by liquid chromatography–high-resolution mass spectrometry (HRMS). Identification of metabolites (M8, M19 and M21), for which reference standards were available, was also confirmed with coelution experiments.
Nonclinical In Vivo PK Studies
An overview of the purpose and design of nonclinical studies is provided in Table 1. All nonclinical PK and disposition studies were conducted at a contract research organization in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited facility, and all protocols were approved by an Institutional Animal Care and Use Committee.
Cross-Species Comparison of MMB and Metabolite Exposure in Steady-State Plasma.
Relative steady-state plasma exposures of MMB and key metabolites (M19 and M21) were compared in AUC-pooled samples (Hamilton et al., 1981) collected from rats, dogs, and humans at PK steady state (Table 1).
The rat AUC pool (0–24 hours) was prepared from day-91 samples from animals given a 20-mg/kg per day dose of MMB. The dog AUC pool (0–24 hours) was derived from day-91 samples from animals given a 60-mg/kg per day dose of MMB. Dose levels of 20 and 60 mg/kg per day were selected for rat and dog samples, respectively, because they represented the no-observed-adverse-effect level (NOAEL) doses for these species. The human AUC pool (0–24 hours) was prepared from day-28 samples from subjects given a 300-mg/d dose of MMB in capsules. The 300-mg human dose in capsules was equivalent in exposure to the 200-mg tablets evaluated in the phase 3 studies (Xin et al., 2017). All AUC pools contained plasma from equal numbers of males and females. The rat control pool was derived from plasma samples taken 3 hours after dosing of the vehicle alone and the predose samples of selected animals on day 1. The dog and human control pools were derived from day-1 predose samples.
For each pairwise comparison (human-to-rat and human-to-dog), two samples were prepared by mixing equal volumes of the AUC pool of each species with a control pool of the comparator species to ensure that both samples would have exactly the same matrix (Gao et al., 2010). Specifically, for the human-to-rat comparison, a human AUC pool was mixed with a rat control pool, and a rat AUC pool was mixed with a human control pool; for the human-to-dog comparison, a human AUC pool was mixed with a dog control pool, and a dog AUC pool was mixed with a human control pool. The plasma samples were analyzed by LC-MS/MS.
PK of MMB and M21 following Single Oral Coadministration to Rats.
Given limited M21 formation in rats (see Table 4), it was necessary to determine whether coadministration of MMB and M21 in rats would produce sufficient M21 systemic AUC exposure to allow for M21 safety assessment, as per FDA guidelines (https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm074916.pdf; https://www.fda.gov/downloads/Drugs/.../Guidances/ucm079266.pdf; http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S1B/Step4/S1B_Guideline.pdf). An overview of the purpose and design of this study is provided in Table 1. MMB and M21 were coformulated with 0.05 M hydrochloric acid in sterile water and were dosed by oral gavage. Under this condition, MMB and M21 existed as a solution and suspension, respectively. Plasma PK samples were collected at specific time points and analyzed by LC-MS/MS.
In Vitro Cellular Pharmacodynamic Assays and Pharmacologic Activity Index
Cellular assays to determine the 50% effective concentration (EC50) for the inhibition of IL6-stimulated, JAK1/2-mediated phosphorylation of STAT3 in primary human peripheral blood mononuclear cells (PBMCs), and ACVR1 inhibition by hepcidin gene expression analysis on HepG2 cells (a hepatoma cell line) have been previously described (Asshoff et al., 2017).
In the present series of experiments, the pharmacologic activity index (PAI) was used as a framework for evaluating the potential contributions of individual metabolites relative to the pharmacologic activity of MMB in PBMCs and HepG2 cells. A PAI value >25% was indicative of a pharmacologically active metabolite (Leclercq et al., 2009). The PAI index was calculated as: Where AUCm and AUCp are the AUC values for metabolite and parent, respectively, fum and fup are the fraction unbound values for metabolite and parent in plasma, and EC50, p and EC50, m are the EC50 values for parent and metabolite in the cellular pharmacodynamic assays.
Where AUCm and AUCp are the AUC values for metabolite and parent, respectively, fum and fup are the fraction unbound values for metabolite and parent in plasma, and EC50, p and EC50, m are the EC50 values for parent and metabolite in the cellular pharmacodynamic assays.
Results
Permeability.
MMB at 4.15 μg/ml (10 μM) showed high passive permeability (16.4 × 10−6 cm/s) through human colonic adenocarcinoma (Caco-2) monolayers with an efflux ratio of 2.1. MMB assayed at a 3-fold higher concentration showed an increased passive permeability (30.4 × 10−6 cm/s) and decreased efflux ratio (0.9). On the basis of these findings, MMB free base is considered a Biopharmaceutics Classification System Class II compound (Amidon et al., 1995; https://www.fda.gov/downloads/Drugs/Guidances/ucm070246.pdf).
Human ADME: Single-Dose Mass-Balance Study in Healthy Subjects
Study Population.
Six male subjects, five white and one African-American, were enrolled in the study, received a single dose of [14C]-radiolabeled MMB oral solution and 200 mg/100 μCi, and completed all assessments. The mean age was 29 years (range 25–36) and the mean body mass index was 25.2 (S.D., 2.4) kg/m2. Mean (S.D.) creatinine clearance was 130 (28) ml/min.
Pharmacokinetics and Disposition Results.
Overall recovery of radioactivity was 96.7%, with more of the drug-related materials (DRM) recovered in feces (69.3% of the DRM) than in urine (27.5% of the DRM). MMB metabolism involved oxidation and scission of the morpholine ring, amide hydrolysis, N-dealkylation, nitrile hydrolysis, nitrile oxidation, and glucuronidation (Fig. 2). A summary of MMB and its metabolites observed in excreta is provided in Table 2. The HRMS data and structure assignments for MMB and its metabolites are provided in Supplemental Figs. 1–10. The primary metabolite excreted in feces was the M14 metabolite (21.4% of the DRM), followed by M21 (12.7% of the DRM), unchanged MMB (12.6% of the DRM), and coeluted M19 and M33 (7.1% of the DRM). An additional 10 metabolites each accounted for <4% of the DRM. In urine, M21 was the main component recovered (11.5% of the DRM), with low levels of unchanged parent MMB and minor metabolites observed.
Mean percentage of the [14C]-radioactivity of the most abundant metabolites in AUC pooled human plasma (0–24 h) and their corresponding percentage of the DRM in pooled urine (0–24 h) and feces (0–72 h)
Plasma radioactivity consisted of MMB and six metabolites, as shown in Fig. 3 and Table 2. The M21 metabolite made up the largest percentage of DRM in plasma (64.2%), followed by MMB (17.3%), M8 (5.8%), M19 (5.2%), and M5, M28, and M20 (2.3–2.7% each). M21 was the only major circulating metabolite present at >10% of the circulating DRM, the threshold above which safety testing for metabolites is required (https://www.fda.gov/downloads/Drugs/.../Guidances/ucm079266.pdf). The three circulating metabolites present at >25% of the parent systemic exposure were M21 (371%), M8 (34%), and M19 (30%). Twenty-five percent exposure is the threshold above which the drug-drug interaction potential of metabolites should be evaluated (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf; https://www.fda.gov/downloads/Drugs/Guidances/ucm292362.pdf). In contrast, MMB and metabolite M19 were the two most abundant circulating DRM in a single-dose mass-balance study in rat and dog (data on file; Supplemental Fig. 11; Supplemental Table 2). The mean blood-to-plasma radioactivity AUC ratio of [14C] was 0.72, suggesting a low association of radioactivity with blood cells for MMB and metabolites, consistent with previous in vitro results in which the blood-to-plasma concentration ratios for MMB were close to 1 in rats and humans (data on file).

Representative radiochromatogram in AUC pooled human plasma from human subjects administered 200 mg [14C]MMB (100 μCi). Circulating radioactivity consisted of M21 (64.2%), MMB (17.3%), M8 (5.8%), M19 (5.2%), M5 (2.7%), M28 (2.5%), and M20 (2.3%). M21 is the most abundant circulating DRM and is the only major circulating metabolite of MMB in human. Radioprofiling was performed using chromatography and fraction collection followed by off-line radiocounting (see Supplemental Materials and Methods). The percentage of each identifiable metabolite used the actual counts obtained as a basis and may not appear as a distinct peak(s) in this reconstructed chromatogram. However, there was a clear baseline separation between M19 and M21 in the liquid chromatography–high resolution mass spectrometry and liquid chromatography-UV chromatograms of MMB and metabolites in AUC pooled plasma (data on file). M19 and M21 peak integration was also facilitated by the availability of individual reference standards. CPM, count per minute.
Plasma concentration-time profiles for total radioactivity and concentrations for nonradiolabeled MMB and its M19 and M21 metabolites following a single oral dose of [14C]MMB are shown in Fig. 4. Plasma PK parameters for total radioactivity as well as for nonradiolabeled MMB, M19 and M21 are summarized in Table 3. Plasma concentrations of M21 were higher than those of MMB and M19. MMB, M21, and M19 all declined in a monoexponential manner. Median nonradiolabeled MMB Cmax in plasma was observed 1.3 hours following a single dose of MMB, and median t1/2 was 3.7 hours. Median Cmax of the nonradiolabeled M21 and M19 metabolites were observed 3.8 and 4.0 hours postdose, respectively, with median t1/2 of 3.6 and 9.3 hours.
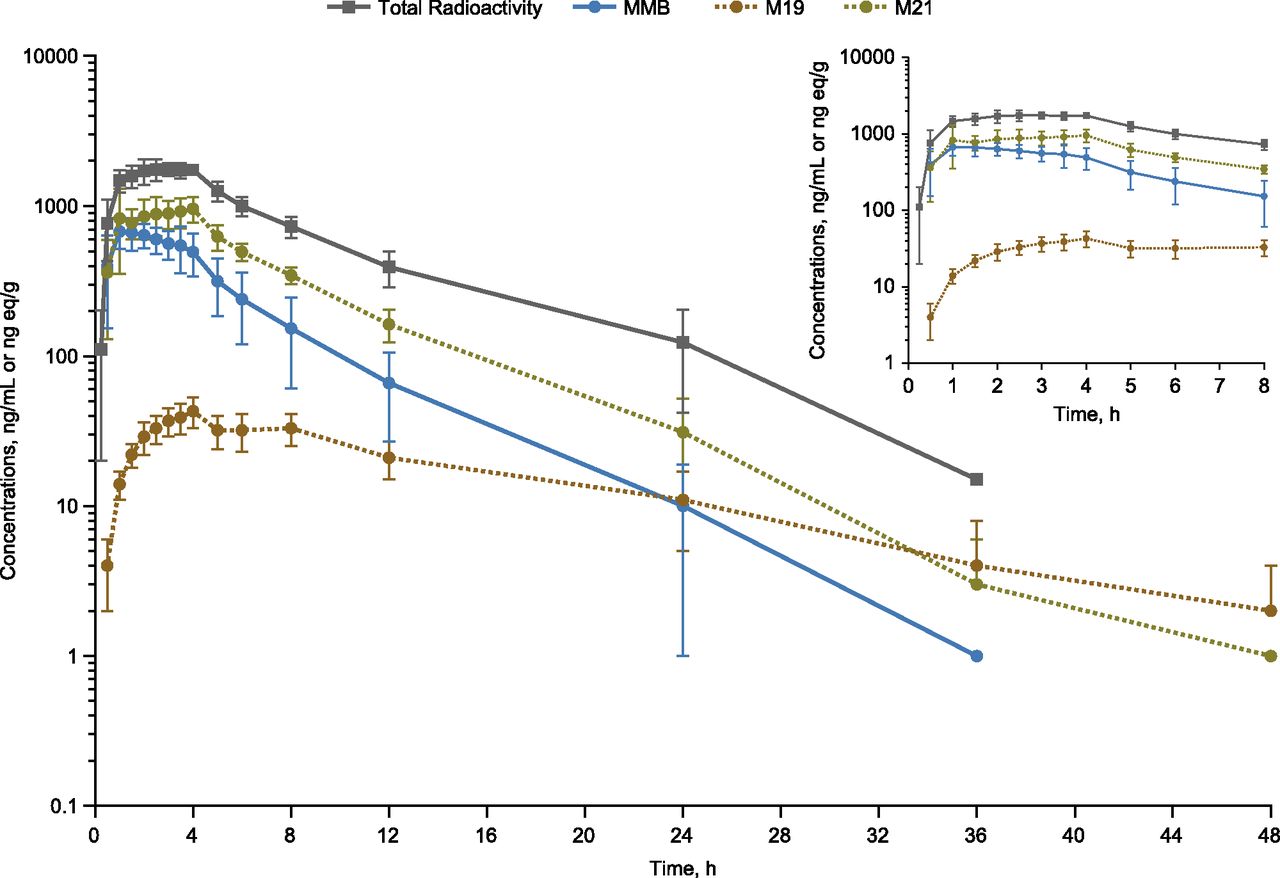
Plasma concentration (total radioactivity)-time curves for MMB and its M21 and M19 metabolites after a single 200-mg (100 μCi) oral dose in healthy subjects (n = 6, mean ± S.D.). Insert figure provides detailed view of values through hour 8.
Blood and plasma PK summary of total [14C] radioactivity by liquid scintillation count and plasma PK of MMB and metabolites by LC-MS/MS following a single 200-mg dose in healthy subjects (n = 6, mean ± S.D.)
Safety.
All six subjects had one or more treatment-emergent adverse events (AE) that were considered potentially related to treatment with MMB. The most frequently reported AEs by preferred term were dizziness (100%), headache (83%), and nausea (33%), all of which were reported as possibly treatment related. All other AEs were reported in single subjects. All AEs were Grade 1, except for a single occurrence of Grade 2 dizziness that resolved on the same day. No serious AEs (≥Grade 3), AEs leading to discontinuation, or deaths were reported.
Cross-Species Comparison of MMB and Metabolite Exposure in Plasma
Relative plasma AUC exposures of MMB and its metabolites (M19 and M21) in humans, rats, and dogs are presented in Table 4. M19 was measured because it was the most abundant circulating metabolite in animal studies, had a longer t1/2 than MMB or M21 in human plasma, and therefore was considered to have the potential for accumulation at steady state. The steady-state plasma AUC of intact MMB was similar in dogs and humans, but plasma AUC was ∼13-fold more abundant in rats than in humans. At steady state, M19 was more abundant in rats and dogs than in humans. M21 plasma AUC was 4.5-fold more abundant in humans than in rats. M21 was not detectable in dogs.
Relative AUC exposure levels of MMB and metabolites in rat, dog, and human AUC pooled plasma samples at steady state at the corresponding NOAEL dose level in rat and dog
Assessment of Pharmacodynamic Activity
The MMB EC50 for inhibition of interleukin-6-stimulated, JAK1/2-mediated phosphorylation of STAT3 in human PBMCs, and the EC50 for ACVR1 inhibition by hepcidin gene expression analysis on HepG2 cells were previously reported (Asshoff et al., 2017).
In the present analysis, the in vitro pharmacologic activity of the M21 metabolite was assessed and compared with MMB within the same experiment. The mean EC50 values for inhibition of interleukin-stimulated, JAK1/2-mediated, phosphorylation of STAT3 in human PBMCs were 259 and 689 nM for MMB and M21, respectively (Table 5). MMB and M21 resulted in dose-dependent reductions in hepcidin mRNA levels, with mean EC50 values of 652 and 1420 nM, respectively. MMB and its metabolites (M19 and M21) were highly bound in rat plasma but moderately bound in dog and human plasma (Table 6). Free fractions of MMB and M21 were moderate-high in cell culture media. The PAI values of 47% and 57% for M21 were well above the 25% threshold, indicating that M21 is pharmacologically active for both JAK1/2 and ACVR1 inhibition, respectively. M19 was not active in either assay.
In vitro JAK1/2 and ACVR1 inhibition by MMB and M21 and the corresponding pharmacologic activity index values
Protein binding of MMB, M19, and M21 in plasma, hepatic microsomal fraction, and cell culture media
Metabolism and Enzymology
MMB exhibited variable rates of metabolism in cryopreserved hepatocytes, with a predicted low hepatic extraction ratio of 0.22 in rats, high EH of 0.73 in dogs, and intermediate EH of 0.63 in humans [EH, defined as the ratio of the predicted hepatic clearance of a drug to the corresponding liver blood flow (Qh) in vitro, and calculated as 1 – AUC∞ from the femoral vein over AUC∞ from the portal vein following oral administration in vivo]. The in vitro hepatocyte-predicted EH in humans indicates that in vivo, MMB exposure entering the portal vein would be 2.7-fold that in systemic blood vessels. In human hepatic microsomes, the average free fractions for MMB and M21 were high (Table 6). M21 was predicted to be more stable than MMB in human hepatocytes, with a predicted low EH of 0.16.
In vitro experiments were conducted to characterize the enzymes responsible for the metabolism of MMB and the formation of its major, active circulating M21 metabolite. These experiments demonstrated low turnover by individual recombinant enzymes. There was detectable metabolism by major P450s, CYP1A2, 2C8, 2C9, 2C19 and 3A4, as well as minor CYPs, 1A1, 2J2 and 4F2. Rates of metabolism by CYP2A6, 2B6, 2D6, 2E1, 4F3A, 4F3B and 4F12 were below the limit of quantification.
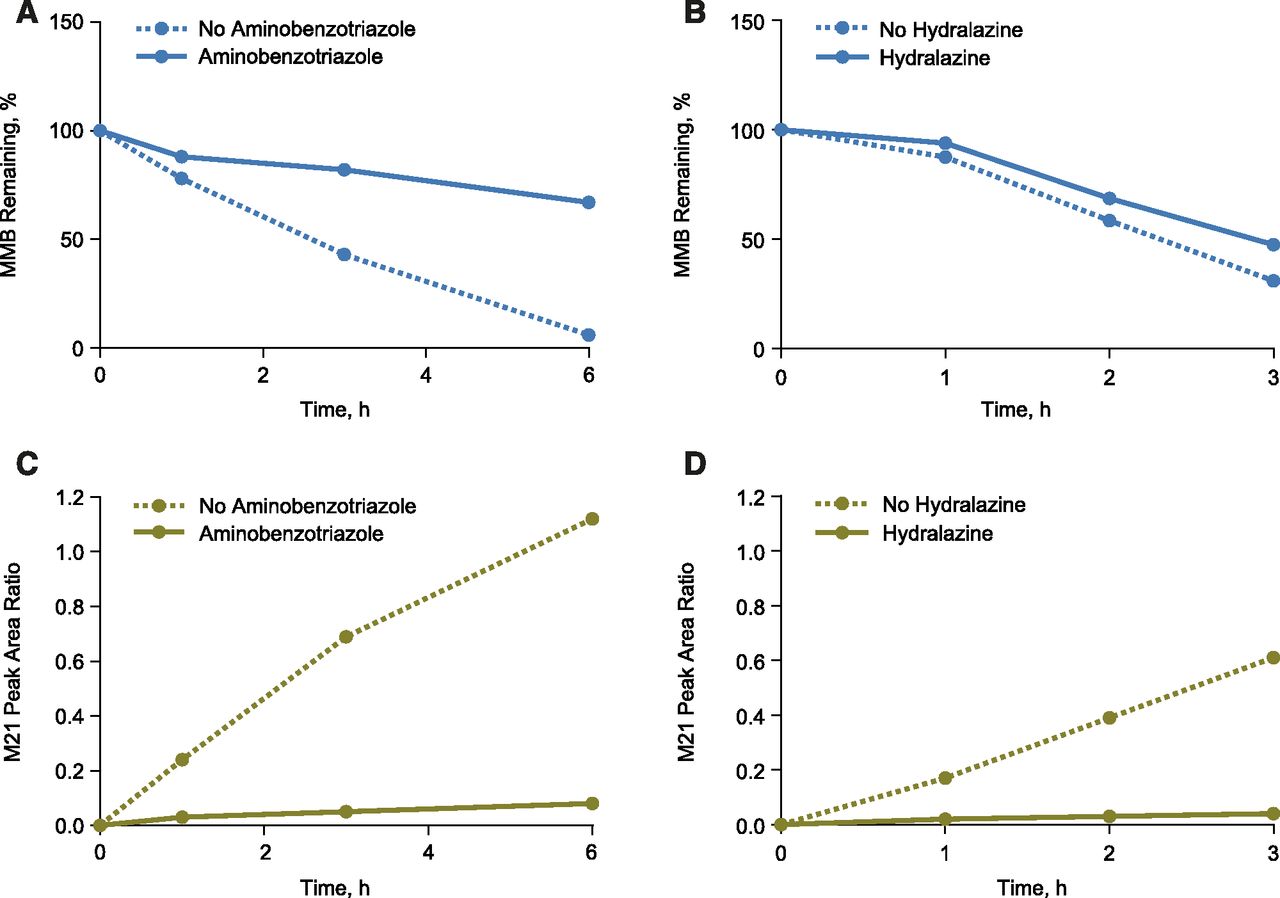
To determine the quantitative contributions of the P450 enzymes responsible for the metabolism of MMB, and to identify the enzyme(s) responsible for the generation of M21, initial studies were attempted with human hepatic S9 (a fraction which contains both cytosol and microsomes). Although some loss of MMB was detected under these conditions, there was little formation of M21. Subsequent enzymology studies were carried out in cryopreserved human hepatocytes. The rate of MMB metabolism in hepatocytes was reduced by 87% in the presence of 1-aminobenzotriazole (ABT; 1 mM) (Fig. 5A). Similar results were obtained with the pan-P450 inhibitors 1-benzylimidazole (100 μM) and 1-phenylimidazole (100 μM), confirming that NADPH-dependent MMB metabolism was mediated primarily by P450 enzymes.

Effects of ABT and hydralazine on metabolic stability of MMB in human hepatocytes. (A) Metabolism of MMB was markedly reduced in the presence of the pan-P450 inhibitor ABT, indicating that MMB undergoes P450-mediated metabolism. (B) Metabolism of MMB was not altered in the presence of the AOX inhibitor, hydralazine, indicating AOX has no direct role in MMB metabolism. (C and D) M21 formation was markedly reduced in the presence of either pan-P450 inhibitor ABT or the AOX inhibitor hydralazine, indicating that M21 formation requires involvement of both P450s and AOX and that MMB metabolism undergoes P450-mediate metabolism followed by AOX metabolism (mean, n = 2).
The rate of MMB metabolism was not significantly altered by incubation with hydralazine at 50 μM (rate reduced by 13%, Fig. 5B) in human hepatocytes, indicating no direct role of aldehyde oxidase (AOX). Although hydralazine is considered a specific AOX inhibitor, the 13% rate reduction would be considered consistent with the reported inhibitory effect of hydralazine at 50 μM on individual human P450 enzymes (Strelevitz et al., 2012). ABT and hydralazine reduced the rate of M21 formation by 95% (Fig. 5C) and 94% (Fig. 5D) in human hepatocytes, respectively, indicating that both P450 and AOX enzymes were necessary for its formation. Simultaneous addition of ABT and hydralazine yielded a result similar to that for ABT alone (results not shown). The differential effects of ABT and hydralazine on MMB stability in human hepatocytes indicate that P450 enzymes are responsible for the primary metabolic event, with AOX being responsible for a subsequent step, and that M21 formation may not depend solely on sequential metabolism by P450s and AOX. The proposed scheme of MMB general metabolism is summarized in Fig. 1.
In human hepatocytes, MMB was also not metabolized by flavin-containing monooxygenase or monoamine oxidase (data on file), further supporting the hypothesis that P450 enzymes are mainly responsible for the primary oxidation of MMB. The effects of enzyme-selective inhibitors on the metabolism of MMB were tested in human hepatocytes. The effects of the inhibitors on both the rates of MMB metabolism and M21 formation were tested in the same assay. The P450 isoforms responsible for the metabolism of MMB, assessed by the loss of parent drug, ranked in order of high to low fraction of metabolism (fm), were CYP3A4 (36%), 2C8 (19%), 2C19 (19%), 2C9 (17%), and 1A2 (9%) (Table 7). No turnover was observed for MMB when incubated with P450s CYP2B6, 2D6, 2E1, or 3A5. In human hepatocytes, MMB was efficiently metabolized by multiple P450s without a single dominant metabolizing P450 enzyme.
Enzyme-selective inhibitors reactions used in enzymology studies in human hepatocytes to determine relative contributions of human P450 enzymes to the rates of MMB metabolism and M21 formation (mean ± S.D., n = 6–8)
No turnover was observed for MMB when incubated with CYP2B6, 2D6, 2E1, or 3A5. In parallel incubations, ABT (1 mM) reduced midazolam oxidation by 95%, and hydralazine (50 μM) reduced zoniporide oxidation by 87%, confirming the effectiveness of the inhibitors. Addition of ABT (1 mM) greatly attenuated the loss of MMB, with an 87% reduction in the rate of loss. Hydralazine (50 μM) had little effect on MMB metabolism (rate reduced by 13%). ABT and hydralazine reduced the rate of M21 formation by 95% and 94%, respectively, indicating that both P450 and AOX enzymes were necessary for its formation. Simultaneous addition of ABT and hydralazine yielded a result similar to that observed with ABT alone. Additionally, the combination of furafylline, montelukast, sulfaphenazole, benzylnirvanol, and ritonavir resulted in 84% inhibition, which is close to the effect observed with ABT, indicating that the majority of P450 enzymes generating M21 had been identified. Fraction of metabolism was calculated by normalizing each percent inhibition to the total reduction in rate for all inhibitors exhibiting an inhibitory effect.
Raloxifene is another potent inhibitor of human liver AOX (Obach, 2004), but is also a nonspecific inhibitor of P450 enzymes and therefore generally not recommended for use as a selective inhibitor for AOX (Nirogi et al., 2014). Nevertheless, a parallel experiment was conducted in human hepatocytes to assess the effects of raloxifene and ritonavir (RTV, a mechanism-based inhibitor of CYP3A4 in humans) on M21 formation. Whereas RTV (3 μM) did not inhibit M21 formation, raloxifene (3 μM) inhibited 50% of M21 formation with no synergy observed between the two inhibitors, further supporting the role of AOX on M21 formation.
M21 is a morpholino lactam metabolite of MMB (Fig. 1), and its formation involved two oxidation steps: The first step involved oxidation to initially form a carbinolamine intermediate followed by a second oxidation of the intermediate to M21. The formation of an iminium intermediate by MMB was supported by a cyanide-trapping experiment (Argoti et al., 2005) in human liver microsomes (HLM), and compared with cyanide adduct formation when MMB was incubated with recombinant CYP3A4. In the presence of NADPH, formation of cyanide adduct was observed when MMB was incubated in HLM, as when MMB was incubated with recombinant CYP3A4 (Supplemental Fig. 12). No cyanide adduct was observed in the absence of NAPDH, suggesting the cyanide adduct formation is NADPH-dependent. A proposed mechanism of M21 formation is shown in Fig. 1. For the first oxidation step, the P450 enzymes responsible for formation of M21 were the same as those responsible for other MMB metabolism, with CYP1A2 playing a greater role and CYP2C9 playing a lesser role than for total MMB metabolism (Table 7). The fm for the P450 isoforms responsible for the formation of M21 were similar for 3A4 (24%), 2C8 (22%), 2C19 (23%), 1A2 (23%), but lower for 2C9 (8%). Though the possibility of two sequential oxidations via P450s, possibly even mitochondrial P450s, cannot be completely ruled out, the totality of data from in vitro inhibition, cyanide trapping experiments, and in vivo species differences collectively imply that the second oxidation step leading to formation of M21 involved AOX, and that the morpholine moiety was not a direct AOX substrate but rather the carbinolamine-imine intermediate.
PK of MMB and M21 following Single Oral Coadministration to Male Rats
Following oral coadministration of MMB at 5 mg/kg and M21 at 25 mg/kg to male Sprague Dawley rats, MMB AUC∞ was 34,895 ± 3053 ng·h/ml and the corresponding M21 AUC∞ was 2354 ± 344 ng·h/ml. The M21 AUC approached 50% of the overall mean M21 AUC exposure (AUC∞ = 4739 ± 38 ng·h/ml, n = 178) from all available human subjects at the time of study; although this value was approximately 33% of the mean M21 AUC observed in the single-dose clinical mass-balance study (Table 3).
Discussion
ADME studies enable a holistic understanding of drug disposition (Beumer et al., 2006; Roffey et al., 2007; Penner et al., 2009). The single [14C]MMB dose (200-mg; 100 μCi) administered to healthy human subjects was almost quantitatively recovered in this study. Radioactivity was mainly eliminated in feces (primary route) and urine (secondary route) as a combination of unchanged MMB and metabolites.
Following single-dose MMB administration, the most abundant circulating DRM in humans were the parent drug and M21 metabolite, representing 17.3% and 64.2% of the AUC of total radioactivity in plasma, respectively. In the single-dose mass-balance studies in rats and dogs, MMB was an abundant circulating DRM. Metabolite M19 was the most abundant and major circulating metabolite in dogs and the most abundant but not major (i.e., <10% of the circulating DRM) circulating metabolite in rats (Supplemental Fig. 11; Supplemental Table 3). This was consistently observed in comparisons of the relative plasma AUC exposures of MMB and its metabolites (M19 and M21) at steady state in rats, dogs, and humans. Overall, systemic exposure of M21 was disproportionately higher in humans than in rats and dogs. Given disproportionately higher M21 human systemic AUC exposure, a combination dosing approach (i.e., coadministration of both M21 and MMB) was employed to increase M21 systemic AUC exposure in rats. With single-dose oral coadministration of 5 mg/kg MMB and 25 mg/kg M21, the M21 AUC in rats reached 50% of the M21 overall mean AUC in humans, and was considered sufficient to allow for M21 safety assessment. This combination dosing approach was adopted in subsequent 104-week rat safety testing to meet regulatory guidelines (https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm074916.pdf; http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S1B/Step4/S1B_Guideline.pdf).
Metabolite M21 was 2- to 3-fold less active than MMB for inhibition of JAK1/2 and ACVR1 in vitro. However, on the basis of the PAI assessment, M21 is anticipated to contribute significantly to the pharmacological activity of MMB, not only as a JAK1/2 inhibitor but also as an ACVR1 inhibitor. ACVR1 inhibition is hypothesized to be responsible for the improvement of inflammatory anemia by MMB (Asshoff et al., 2017). This benefit was an unexpected finding for a JAK1/2 inhibitor because erythropoietin-mediated JAK2 signaling is essential for erythropoiesis, and because new-onset anemia is a major adverse event associated with ruxolitinib treatment (Neubauer et al., 1998; Parganas et al., 1998; Verstovsek et al., 2012).
Higher free liver exposure may also play a role in the anemia benefit observed with MMB in clinical trials (Pardanani et al., 2013; Gupta et al., 2017; Mesa et al., 2017). The in vitro predicted intermediate EH in humans suggests MMB exposure would be higher in portal versus systemic veins in vivo. In vitro, M21 was more stable than MMB in human hepatocytes. In vivo, the median t1/2 of M21 was similar to MMB in human plasma. In addition, mean free fractions of both MMB and M21 were significantly higher in human hepatic microsomes than in human plasma.
Metabolite M21 was undetectable in dog plasma, and M21 exposure was substantially higher in humans than in rats. This suggests either a species difference in the pattern of primary metabolites generated, or that M21 is a secondary metabolite that requires additional metabolism from the parent, for which dogs lack the necessary enzyme. AOX was a plausible candidate for this deficient enzyme, as it is absent in dogs (Beedham et al., 1987; Garattini et al., 2008; Pryde et al., 2010). Following a literature review (Brandänge and Lindblom, 1979; Pryde et al., 2010; Zientek et al., 2010; Garattini and Terao, 2012; Hutzler et al., 2012; Strelevitz et al., 2012; Sanoh et al., 2015), it was hypothesized that experiments in which the compound is incubated with a cytosolic or an S9 fraction in the presence of NADPH would be the best option to study the two-step process of M21 formation. However, this was unsuccessful because minimal MMB turnover was observed and M21 formation was very low, suggesting poor coupling between P450 and AOX reactions within the S9 fraction. The reason for poor reaction coupling observed for MMB to M21 in S9 is not clear but could be a result of insufficient subcellular fraction enzyme enrichment and/or involvement of nonendoplasmic reticulum/cytosolic drug metabolism enzymes. Low turnover of an AOX substrate in human hepatic S9 fraction that prevented enzymology assessment was recently reported by other investigators (Crouch et al., 2017). Higher MMB turnover rates and M21 formation were observed in cryopreserved human hepatocytes. As a result, subsequent AOX reaction phenotyping studies were carried out in cryopreserved human hepatocytes, an assay which was atypical and challenging (Zientek and Youdim, 2015).
The AOX-mediated production of M21 was not predicted by results from studies in rats or dogs in which M19 systemic exposure is significantly more abundant than M21. Increased M21 formation in humans compared with nonclinical species is consistent with reports that rats generally have low AOX activity and dogs are completely devoid of such activity. The molecular properties of substrate recognition by AOX do not correlate with any specific area of molecular space, such as polarity and lipophilicity, but rather reflect specific structural features (Pryde et al., 2010). Not surprisingly, among those heterocyclic motifs with a high risk of AOX turnover, morpholine was not noted (Garattini et al., 2008; Pryde et al., 2010).
Metabolism of MMB in humans was mediated by multiple P450 enzymes, whereas M21 formation involved initial P450-mediated biotransformation by the same enzymes followed by secondary metabolism via AOX. Drugs metabolized by multiple P450 enzymes have been reported (Vickers et al., 1999; Obach et al., 2005). The finding that multiple P450s catalyzed the oxidation of the morpholine ring of MMB without a single dominant metabolizing P450 enzyme is unusual. The initial involvement of multiple P450s and the sequential metabolism by AOX may be part of the reason why the phenotyping studies were challenging.
Although bioactivation of the morpholine ring is uncommon and generally considered undesirable (Bolleddula et al., 2014), formation of the pharmacologically active M21 metabolite contributed to the JAK1/2 and ACVR1 inhibitory effect of MMB observed in humans. A similar improvement in efficacy via bioactivation of the morpholine ring has been reported with the anticancer drug methoxymorpholinyl doxorubicin (a doxorubicin analog), but AOX was not involved (Bolleddula et al., 2014). Further, the metabolism of crizotinib on the piperidine ring leads to the formation of its active, major circulating metabolite crizotinib lactam without AOX involvement (Johnson et al., 2015). Involvement of AOX has been observed with the metabolism of the pyrrolidine ring of prolintane, and the morpholine rings of doxapram and phenmetrazine (Vickers and Polsky, 2000). Nicotine undergoes initial metabolism by hydroxylation on the pyrrolidine ring catalyzed by P450, followed by metabolism by AOX to form the inactive metabolite cotinine (Brandänge and Lindblom, 1979). It was demonstrated that morpholine hydroxylation by P450s occurs at the α-carbon next to nitrogen through a hydrogen atom abstraction and rebound mechanism facilitated by the presence of nitrogen in a morpholine heterocycle (Shaikh et al., 2009). In the case of MMB, however, bioactivation of the morpholine ring at the α-carbon next to nitrogen with nitrogen directly attached to phenyl via P450 followed by AOX appears to be a unique occurrence. Metabolism of the morpholine ring in drugs such as linezolid, gefitinib, and methoxymorpholinyl doxorubicin typically occurs via the P450 system (Lau et al., 1989; Wynalda et al., 2000; Quintieri et al., 2005; Bolleddula et al., 2014).
M19 formation was a minor metabolic pathway in humans but a main pathway in rats and dogs. Given the limited M19 systemic exposure (<10% of the circulating DRM) and the lack of pharmacological activity in humans, enzymology of M19 formation was not studied. Hydrolysis at the amide bond of MMB is, however, known to lead to the formation of M19 and the simultaneous formation of aminoacetonitrile, which can undergo further metabolism to form thiocyanate (Fleming et al., 2010; Takahashi et al., 2017). In addition, cyanide degradation via various enzymes and mechanisms was involved in MMB metabolism and led to the formation of metabolites M15 and M8 (Gupta et al., 2010). Plasma thiocyanate concentrations following MMB administration were quantified in a chronic rat toxicity study and in humans. In humans, MMB-related increases in systemic thiocyanate exposure were less than the S.D. at baseline and are unlikely to pose safety concerns (Le et al., 2016).
The enzymology and metabolite profiling for MMB have important implications with respect to its drug-drug interaction potential, safety, efficacy, and anemia benefit. Because multiple P450 enzymes are involved in its metabolism, MMB PK probably is not altered in a clinically meaningful way by an inhibitor or inducer of any single enzyme (Vickers et al., 1999; Obach et al., 2005). MMB has not been shown to be a strong inhibitor or inducer of P450 enzymes in vitro (data on file). Considering its moderate plasma protein binding, MMB is unlikely to be a clinically meaningful inhibitor or inducer of P450 enzymes at the typical plasma concentrations anticipated after oral administration. In general, AOX has not been found to play a substantial role in drug interactions (Foti and Dalvie, 2016). The potential for MMB to interact with AOX inhibitors has not been formally assessed and cannot be excluded.
Regulatory guidelines recommend evaluation of drug-drug interaction potential for metabolites present at ≥25% of the parent drug AUC (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf; https://www.fda.gov/downloads/Drugs/Guidances/ucm292362.pdf) and >10% of the circulating DRM (https://www.fda.gov/downloads/Drugs/.../Guidances/ucm079266.pdf). Given the extent of MMB metabolite formation, in vitro studies were conducted to assess the drug interaction potential of M21, M19, and M8; results from these experiments will be reported separately. Thus, accurate quantitation of MMB metabolite formation, including species differences in M21 formation, has proven essential in assessing the efficacy and safety of MMB in accordance with regulatory requirements.
In summary, MMB is metabolized via multiple pathways and eliminated as a combination of metabolites and unchanged parent drug. MMB is metabolized by multiple P450 enzymes, and formation of its major metabolite M21 in human requires P450-mediated oxidation on the morpholine substituent of the parent drug with subsequent oxidation by AOX to form a stable lactam. M21 systemic exposure is disproportionately higher in humans than in rats and dogs. Given this discrepancy, a combination dosing approach was implemented to increase M21 systemic exposure in rats for safety assessment. PAI estimation suggests M21 contributes significantly to the pharmacological activity of MMB for the inhibition of both JAK1/2 and ACVR1. ACVR1 inhibition, higher free liver exposure, the activity of M21, and the unique enzymology of M21 formation may all contribute to the observed anemia benefits by MMB.
Acknowledgments
The authors thank Dr. Adrian Ray, Dr. Chris Yang, Dr. Gerry Rhodes, and Dr. Srini Ramanathan for their scientific contributions; Kelly Wang for conducting the protein-binding assay; Covance Laboratories (Conshohocken, PA) for performing the [14C]MMB ADME and metabolite profiling in rats and dog; and Frontage Laboratories Co. Ltd. (Exton, PA) for performing the [14C]MMB ADME and metabolite profiling in humans. The authors thank all patients and volunteers who participated in these studies and acknowledge the expert technical and editorial assistance of Jeffrey Coleman of BioScience Communications, New York, NY, funded by Gilead.
Authorship Contributions
Participated in research design: Zheng, Silverman, Whitney, Warr, Xin, Murray, Ling, Smith.
Conducted experiments: Warr, Hemenway, Zhang.
Performed data analysis: Zheng, Moorehead, Xin, Subramanian, Murray.
Wrote or contributed to the writing of the manuscript: Zheng, Silverman, Kwan, Whitney, Warr, Xin, Subramanian, Ling, Smith.
Footnotes
- Received October 12, 2017.
- Accepted January 2, 2018.
This research was funded by Gilead Sciences, Inc.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ABT
- 1-aminobenzotriazole
- ACVR1
- activin A receptor type 1
- ADME
- absorption, distribution, metabolism, and excretion
- AE
- adverse event
- AOX
- aldehyde oxidase
- AUC
- area under time-concentration curve
- AUC∞
- AUC from time 0 to infinity
- Cmax
- maximum concentration
- DRM
- drug-related materials
- EH
- hepatic extraction
- HRMS
- high-resolution mass spectrometry
- JAK
- Janus kinase
- LSC
- liquid scintillation counting
- M21
- circulating metabolite of MMB
- MMB
- momelotinib
- MPN
- myeloproliferative neoplasms
- P450
- cytochrome P450
- PAI
- pharmacologic activity index
- PBMCs
- peripheral blood mononuclear cells
- PK
- pharmacokinetic(s)
- STAT
- signal transducer and activator of transcription
- t1/2
- terminal half-life
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}