


Abstract
The oxidative and conjugative metabolism of sertraline was examined in vitro to identify the enzymes involved in the generation of N-desmethyl, deaminated, and N-carbamoyl-glucuronidated metabolites in humans. In human liver microsomes, sertraline was N-demethylated and deaminated by cytochrome P450 (P450) enzymes with overall Km values of 98 and 114 μM, respectively, but the intrinsic clearance for N-demethylation was approximately 20-fold greater than for deamination. Using P450 isoform-selective inhibitors and recombinant heterologously expressed enzymes, it was demonstrated that several P450 enzymes catalyzed sertraline N-demethylation, with CYP2B6 contributing the greatest extent, and lesser contributions from CYP2C19, CYP2C9, CYP3A4, and CYP2D6. For deamination, data supported a role for CYP3A4 and CYP2C19. Purified human monoamine oxidases A and B also catalyzed sertraline deamination with comparable Km values (230-270 μM). Monoamine oxidase B catalyzed the reaction approximately 3-fold faster than did monoamine oxidase A. Sertraline N-carbamoyl glucuronidation was measured in human liver microsomes in bicarbonate buffer and under a CO2 atmosphere (Km = 50 μM) and was catalyzed at the fastest rate by recombinant human UGT2B7. The observation that multiple enzymes appear to be involved in sertraline metabolism suggests that there should be no single agent that could substantially alter the pharmacokinetics of sertraline, nor should there be any single drug-metabolizing enzyme genetic polymorphism (e.g., CYP2D6, CYP2C19, CYP2C9, UGT1A1) that could profoundly impact the pharmacokinetics of sertraline.
Sertraline is an effective and highly utilized drug for the treatment of depression and mania. It is one example of a class of drugs referred to as selective serotonin reuptake inhibitors. The members of this class of drugs are predominantly cleared by oxidative metabolism by the cytochrome P450 (P450) enzymes. For example, fluoxetine is primarily metabolized via N-demethylation by CYP2D6, 2C9, and 3A (von Moltke et al., 1997; Margolis et al., 2000), paroxetine by demethylenation of its methylenedioxy group by CYP2D6 (Bloomer et al., 1992), venlafaxine by O-demethylation by CYP2D6 (Otton et al., 1996; Fogelman et al., 1999), and citalopram by CYP3A4-, 2D6-, and 2C19-catalyzed N-demethylation (Kobayashi et al., 1997; von Moltke et al., 1999, 2001; Olesen and Linnet, 1999). As such, these drugs have been shown to be subject to drug interactions by various inhibitors of cytochrome P450 enzymes such as quinidine and ketoconazole (Lessard et al., 1999; Eap et al., 2003; Lindh et al., 2003). Also, some of these agents have been shown to exhibit substantial differences in pharmacokinetics in subjects who lack CYP2D6 or CYP2C19, such as fluoxetine, paroxetine, and venlafaxine (Hamelin et al., 1996; Lessard et al., 1999; Liu et al., 2001; Charlier et al., 2003; Lindh et al., 2003; Yu et al., 2003).
Previous reports have attempted to address the identities of cytochrome P450 enzymes responsible for sertraline N-demethylation (Greenblatt et al., 1999; Kobayashi et al., 1999; Xu et al., 1999). N-Demethylation is a major route of sertraline metabolism (Fig. 1). In vivo, the predominant metabolites of sertraline in excreta include entities that could arise via the further oxidative metabolism of the N-desmethyl metabolite or via initial deamination of the methylamino substituent (data on file, Pfizer, Inc.). Also, sertraline undergoes N-carbamoyl glucuronidation, an unusual reaction in drug metabolism, albeit that this pathway appears to be minor in human but major in the dog (Tremaine et al., 1989). The reports on sertraline N-demethylation in vitro have been conflicting, likely due to a complex number of cytochrome P450 enzymes that can catalyze the reaction. In the report of Kobayashi et al. (1999), recombinant heterologously expressed human P450 enzymes were used in determining that five enzymes all appeared to be significantly involved in sertraline N-demethylation: CYP2B6, 2C9, 2C19, 2D6, and 3A4. The report of Greenblatt et al. (1999) also identified CYP2C9, 2C19, 3A4, and 2D6 each as being partially involved in sertraline N-demethylation, with a very minor role for CYP2B6. Alternately, the report of Xu et al. (1999) claimed that CYP2C19 and CYP2C9 were the major P450 enzymes responsible for sertraline metabolism. These investigators utilized human liver microsomes phenotyped for CYP2C19 as well as chemical inhibitors to support their conclusions. However, in the studies of Xu et al. (1999), a role for CYP2B6 was not addressed. Furthermore, sertraline has been shown to exhibit no differences in pharmacokinetics in CYP2D6 extensive versus poor metabolizers (Hamelin et al., 1996) and minor differences in CYP2C19 extensive versus poor metabolizers (Wang et al., 2001), suggesting that neither of these enzymes predominates in the metabolic clearance of sertraline.

The metabolism of sertraline.
The study of drug metabolism in vitro has emerged as a powerful approach to address the potential for drug-drug interactions in vivo as well as addressing the potential for interpatient variability in pharmacokinetics that are due to metabolism (Venkatakrishnan et al., 2003). Such data can be used both retrospectively, to provide a mechanistic understanding of pharmacokinetic observations, and prospectively, to predict drug interactions or pharmacokinetics that are highly variable because of metabolism by enzymes subject to genetic polymorphism. The objective of the study described in this report was to provide greater clarity to our understanding of the enzymes involved in sertraline metabolism, and to reconcile the apparent conflicting information in the previous reports (Greenblatt et al., 1999; Kobayashi et al., 1999; Xu et al., 1999). Additionally, since sertraline contains a benzylamine moiety, a structure associated with monoamine oxidase substrates, the potential for MAO-catalyzed oxidation was also explored. Finally, the unusual metabolic reaction of sertraline N-carbamoyl glucuronidation was explored in the first systematic and quantitative study of this type of drug metabolism reaction in vitro.
Materials and Methods
Materials. Sertraline, N-desmethylsertraline, sertraline ketone, PPP, (+)-N-3-benzylnirvanol, and CP-105,162 were obtained from Pfizer, Inc. (Groton, CT). Other reagents used were from the following sources: furafylline, quinidine, omeprazole, diethyldithiocarbamate, sulfaphenazole, UDPGA, and quercetin from Sigma-Aldrich (St. Louis, MO); ketoconazole and NADPH, ICN (Aurora, OH). (+)-N-3-Benzylnirvanol was prepared according to the method of Suzuki et al. (2002). Sertraline N-carbamoyl glucuronide was biosynthesized using dog liver microsomes and isolated by sequential liquid extraction, solid phase extraction, and preparative HPLC. Human liver microsomes were prepared under contract to Pfizer by BD Gentest (Woburn, MA), and recombinant heterologously expressed human UGT enzymes were from this same source. HL-MIX-101 represents a pool of liver microsomes from 60 individual donors. Recombinant human cytochrome P450 enzymes heterologously expressed using a baculovirus expression system were obtained from PanVera Corp. (Madison, WI; for CYP1A2, 2B6, 2C9, 2C19, 2D6, and 3A4) or BD Gentest (CYP1A1, 2A6, 2C8, 2E1, and 3A5). Purified human MAO-A and -B were generously provided by Dr. Dale Edmonson, Emory University (Atlanta, GA).
Incubation Conditions.Desmethylsertraline Formation. Incubations containing sertraline (at various concentrations), human liver microsomes (0.1 mg/ml), NADPH (1.3 mM), and MgCl2 (3.3 mM) in a total volume of 0.2 ml of 25 mM potassium phosphate buffer (pH 7.4) were conducted at 37°C in a shaking water bath. Initial reaction velocity conditions were established such that product formation was linear over time to 20 min; thus, all subsequent incubations were conducted for this time period. The incubations were terminated by addition of 0.2 ml of 1 M NaOH. All incubations were conducted in triplicate.
Enzyme kinetic experiments in pooled human liver microsomes (MIX 101; combination of 60 individual livers) were conducted at sertraline concentrations ranging from 0.25 to 200 μM. In cases in which human recombinant heterologously expressed cytochrome P450 enzymes were examined, a total P450 concentration of 20 pmol of P450/ml was utilized, with protein concentrations of 0.5 mg/ml maintained by addition of microsomes from nontransfected cells. Sertraline concentrations were 0.5 or 50 μM. In incubations in which the effects of chemical inhibitors in human liver microsomes were examined, the sertraline concentration examined was 0.5 μM and the inhibitors were added in H2O/CH3CN (50:50) such that the final solvent concentration was 0.5%. Control incubations had solvent without inhibitor. For furafylline, methyl phenethyl piperidine, and diethyldithiocarbamate, the inhibitor was preincubated with microsomes and NADPH for 15 min at 37°C before addition of sertraline and additional NADPH.
Sertraline Ketone Formation by P450. Incubations containing sertraline (at various concentrations), human liver microsomes (1.0 mg/ml), NADPH (1.3 mM), and MgCl2 (3.3 mM) in a total volume of 1.0 ml of 25 mM potassium phosphate buffer (pH 7.4) were conducted at 37°C in a shaking water bath. Initial reaction velocity conditions were established such that product formation was linear over time to 10 min; thus, all subsequent incubations were conducted for this time period. The incubations were terminated by addition of 0.5 ml of 1 M HCl. All incubations were conducted in triplicate.
Enzyme kinetic experiments in pooled human liver microsomes (HL-MIX 101) were conducted at sertraline concentrations ranging from 5 to 500 μM. In cases in which human recombinant heterologously expressed cytochrome P450 enzymes were examined, a total P450 concentration of 40 pmol of P450/ml was utilized, with protein concentrations of 1.0 mg/ml maintained by addition of microsomes from sham transfected cells. Sertraline concentrations were 50 μM. Chemical inhibitors were utilized in the same manner as described above. Identical incubation conditions were applied when examining N-desmethylsertraline as a substrate, with the substrate concentration set at 50 μM for all experiments.
Sertraline Ketone Formation by MAO. Incubations containing sertraline (at various concentrations) and purified MAO (0.1 nmol/ml) in a total volume of 1.0 ml of 25 mM potassium phosphate buffer (pH 7.4) were conducted at 37°C in a shaking water bath. Initial reaction velocity conditions were established such that product formation was linear over time to 4 h; thus, all subsequent incubations were conducted for this time period. The incubations were terminated by addition of 0.5 ml of 1 M HCl. All incubations were conducted in triplicate. Enzyme kinetic experiments were conducted at sertraline concentrations ranging from 10 to 1000 μM. Identical incubation conditions were applied when examining N-desmethylsertraline as a substrate, with the substrate concentration set at 50 μM for all experiments.
Sertraline N-Carbamoyl Glucuronidation. Microsomes were first incubated with alamethacin, MgCl2, and sodium bicarbonate (pH adjusted to 7.5) on ice for 15 min in a total volume of 0.145 ml. This was followed by addition of sertraline and saccharolactone and warming to 37°C over 5 min under CO2. Incubations were commenced by addition of UDPGA in a final incubation volume of 0.5 ml. Final assay concentrations were: microsomes, 0.02 mg/ml; alamethacin, 0.05 mg/ml; NaHCO3, 100 mM; MgCl2, 5 mM; saccharolactone, 5 mM; sertraline, 2 to 200 μM; and UDPGA, 5 mM. Incubations were conducted in a Lucite container submerged in a heated water bath that permitted the continuous flow of CO2. The CO2 was passed through a gas warmer to prevent cooling of the incubations. After 45 min, incubations were terminated by the addition of 0.5 ml of HCl (1 M). The incubation time and protein concentration used were determined to be linear in initial experiments.
Analysis of N-Desmethylsertraline. To terminated incubation mixtures was added internal standard (20 μl of a 0.5 μg/ml solution of CP-105,162, the monochloro analog of desmethylsertraline), followed by 3 ml of methyl t-butyl ether and extraction by agitation on a multi-tube vortex mixer. The mixtures were spun in a centrifuge (2500g), after which the aqueous layer was frozen in a dry ice-acetone bath and the organic layer was decanted into a fresh test tube. The solvent was removed by evaporation under N2 at 35°C and reconstituted in 0.1 ml of water/acetonitrile (75:25) containing 0.1% formic acid.
The reconstituted extracts were analyzed by HPLC-tandem mass spectrometry. The HPLC system consisted of two Shimadzu LC-10ADvp pumps, DGU-14 solvent degasser, and SCL-10ADvp controller (Shimadzu, Columbia, MD), a CTC PAL autosampler (LEAP Technologies Inc., Carrboro, NC), and a Micromass Quattro Ultima tandem quadrupole mass spectrometer with ionspray interface (Waters, Milford, MA). Samples were injected (75 μl) onto a Metachem Polaris C18 column (4.6 × 250 mm; 5-μm particle size) equilibrated in a mobile phase consisting of 55% H2O/45% CH3CN containing 0.1% formic acid at a flow rate of 0.8 ml/min. This mobile phase composition was maintained for a run time of 14 min.
The effluent was introduced into the ionspray source of the mass spectrometer operated in the positive ion mode. The following settings on the mass spectrometer were established to optimize the signal for N-desmethylsertraline: capillary, 3.0 kV; cone, 20; source temperature, 135°C; desolvation temperature, 350°C; cone gas, 190 l/h; desolvation gas, 790 l/h; collision energy, 20 eV. Other potentials were adjusted to optimize the signal. Detection was accomplished in the multiple reaction monitoring mode following the transitions m/z 292→159 (N-desmethylsertraline) and m/z 241→125 (internal standard). Additionally, the mass transition m/z 275→159 was also monitored, which allowed visualization of both N-desmethylsertraline and sertraline. Under these conditions, the retention times for N-desmethylsertraline, sertraline, and internal standard were 9.8, 10.8, and 7.7 min, respectively. Quantitation was accomplished using a linear standard curve of N-desmethylsertraline ranging from 1.0 to 1000 ng/ml with 1/X2 weighting. In some experiments, concentrations of N-desmethylsertraline in incubation mixtures were anticipated to be well below 100 ng/ml, and in these cases, the standard curve was run from 1.0 to 100 ng/ml. In experiments in which high sertraline substrate concentrations were used (i.e., >20 μM), the incubation samples were diluted 10 times before analysis to avoid the excess of sertraline overwhelming the signal for N-desmethylsertraline.
Analysis of Sertraline Ketone. To terminated incubation mixtures was added 3 ml of methyl t-butyl ether followed by extraction by agitation on a multitube vortex mixer. The mixtures were spun in a centrifuge (2500g), after which the organic layer was transferred into a fresh test tube. The solvent was removed by evaporation under N2 at 35°C and reconstituted in 0.04 ml of water/acetonitrile (50:50).
The reconstituted extracts were analyzed by HPLC-mass spectrometry. The HPLC system consisted of an Agilent 1100 quaternary pump with membrane degasser, a CTC PAL autosampler (LEAP Technologies, Inc.), and a PE Sciex API-100 single quadrupole mass spectrometer (PerkinElmerSciex Instruments, Boston, MA) with TurboIon spray interface. Samples were injected (30 μl) onto a Metasil AQ C18 column (2.0 × 50 mm; 5-μm particle size) equilibrated in a mobile phase consisting of 20 mM acetic acid (pH adjusted to 4.0 with NH4OH) containing 55% CH3CN at a flow rate of 0.5 ml/min. This mobile phase composition was maintained for a run time of 7 min.
The effluent was introduced into the ionspray source of the mass spectrometer operated in the positive ion mode. The following settings on the mass spectrometer were established to optimize the signal for sertraline ketone: ion spray voltage, 4500 V; orifice, 25 V; source temperature, 450°C; with other potentials and settings adjusted to optimize the signal. Detection was accomplished in the selected ion monitoring mode following m/z 291. Additionally, the effluent was monitored by UV at 245 nm, which allowed visualization of sertraline ketone. Under these conditions, the retention time for sertraline ketone was 3.4 min. Quantitation was accomplished using an external linear standard curve of sertraline ketone routinely ranging from 20 to 2000 ng/ml with 1/X weighting. In some cases, the limit of quantitation was extended down to 6.32 ng/ml.
Analysis of Sertraline N-Carbamoyl Glucuronide. Terminated incubation samples (0.2 ml) were extracted with methyl t-butyl ether (3 ml), and the organic fraction was collected by freezing the aqueous layer in dry ice/acetone and evaporated under N2. The residue was reconstituted in 0.04 ml of water/CH3CN (50:50) and injected (0.01 ml) onto the aforementioned HPLC-tandem mass spectrometry system. The column was a Varian Basic 2.0 × 50 mm narrow bore column (Varian, Inc., Palo Alto, CA) with material of 3-μm particle size. The mobile phase consisted of 0.1% formic acid containing 2 mM NH4OH at 65% and CH3CN at 35%, at a flow rate of 0.4 ml/min. After 2 min, a linear gradient to 65% CH3CN at 8 min was applied, followed by reequilibration. The effluent was introduced into the source of a Micromass Ultima tandem quadrupole mass spectrometer operated in the positive ion mode. Detection was accomplished by monitoring the mass transition m/z 543 (ammoniated molecular ion of sertraline N-carbamoyl glucuronide) to m/z 350. The following settings on the mass spectrometer were established to optimize the signal for sertraline N-carbamoyl glucuronide: capillary, 2.0 kV; cone, 30; source temperature, 100°C; desolvation temperature. 350°C; cone gas. 182 l/h; desolvation gas, 774 l/h; collision energy, 15. The retention time was 5.1 min. Quantitation was accomplished by extrapolation from a linear standard curve of N-carbamoyl glucuronide ranging from 10 to 1000 ng/ml (human liver microsomes) or 1.0 to 100 ng/ml (recombinant UGT enzymes).
Data Analysis. Enzyme kinetic data were fit using the Sigma Plot Enzyme Kinetics Module (v 1.0; SPSS Inc., Chicago, IL). The sertraline N-demethylation and N-carbamoyl glucuronidation data were best fit using the substrate inhibition equation:  For sertraline deamination, the data were best fit to the standard Michaelis-Menten equation:
For sertraline deamination, the data were best fit to the standard Michaelis-Menten equation: 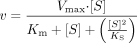 Inhibition data were fit to the equation:
Inhibition data were fit to the equation: 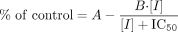 in which 100 - (A - B) represents the maximum inhibition and the IC50 is the inflection point of the curve on a plot of percentage of control versus log[I] (representing the potency of the inhibitor).
in which 100 - (A - B) represents the maximum inhibition and the IC50 is the inflection point of the curve on a plot of percentage of control versus log[I] (representing the potency of the inhibitor).
Percentage contribution by P450s using the relative activity approach (RAF; Venkatakrishnan et al., 2001) was determined using RAF values calculated from Vmax of rP450s and pooled human liver microsomes. RAF values for the pooled human liver microsomes used were 22 (CYP1A2; phenacetin O-deethylation), 32 (CYP2B6; bupropion hydroxylation), 23 (CYP2C9; tolbutamide methyl hydroxylation), 7 (CYP2C19; mephenytoin 4′-hydroxylation), 6 (CYP2D6; bufuralol 1′-hydroxylation), 86 (CYP2E1; chlorzoxazone 6-hydroxylation), and 48 (CYP3A4; average of testosterone 6β-hydroxylation, midazolam 1′-hydroxylation, and felodipine dehydrogenation).
Results
Sertraline N-Demethylation.Enzyme Kinetics in Liver Microsomes. The enzyme kinetics of sertraline N-demethylation in pooled human liver microsomes were measured to ascertain the Michaelis constant and ensure that subsequent reaction phenotyping experiments were conducted at a substrate concentration below Km. A plot of the substrate saturation curve is shown in Fig. 2A. The data analysis best fit a model of substrate inhibition occurring at high substrate concentrations. The enzyme kinetic parameters were: Km = 98 μM, Vmax = 1920 pmol/min/mg microsomal protein, and KS = 63 μM. Thus, all subsequent phenotyping experiments could be conducted at substrate concentrations below 98 μM. A decrease in sertraline N-demethylase activity at high sertraline concentrations was also observed previously (Greenblatt et al., 1999), and the Km value reported (84 μM) was similar to the one measured in the present report.

Substrate saturation plots for sertraline metabolism in pooled human liver microsomes and by purified human monoamine oxidases. Plot A, N-demethylation in HLM; plot B, deamination in human liver microsomes; plot C, N-deamination by MAO-A; plot D, N-deamination by MAO-B. Each point represents the mean from three determinations.
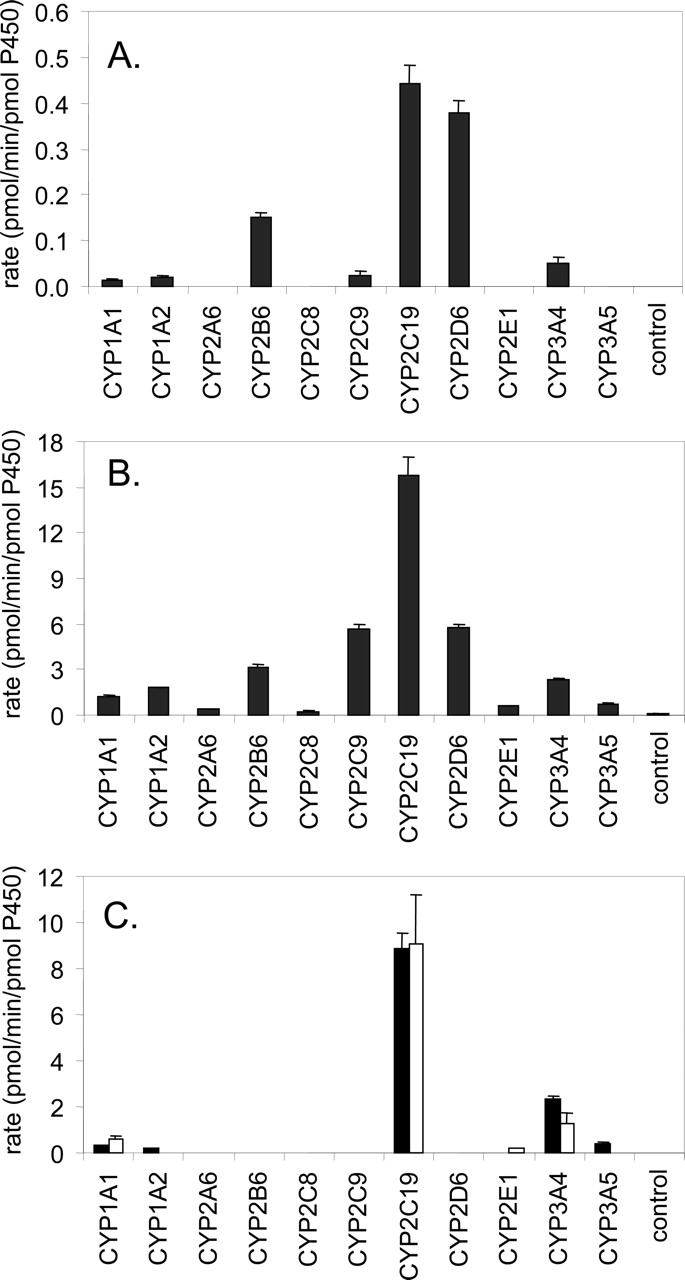
Incubations with rP450 Enzymes. Sertraline was incubated with recombinant human cytochrome P450s 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4, and 3A5, at substrate concentrations of 0.5 and 50 μM. Of the enzymes tested, N-desmethylsertraline was detected in most (Fig. 3, A and B). At [S] = 0.5 μM, the fastest rate was observed for CYP2C19, followed by CYP2D6, 2B6, 3A4, and 2C9. At [S] = 50 μM, the rank order was CYP2C19, 2D6, 2C9, 2B6, and 3A4. Trace quantities of N-desmethylsertraline were observed in incubations with CYP1A1, 1A2, 2A6, 2C8, 2E1, and 3A5. Application of relative activity factors (Venkatakrishnan et al., 2001) for the P450 activities in recombinant P450s and human liver microsomes suggest that at low concentrations, CYP2B6 > CYP2C19 ≈ CYP2D6 ≈ CYP3A4 > CYP2C9, whereas at high concentrations, the rank order changes to CYP2C9 ≈ CYP3A4 ≈ CYP2B6 ≈ CYP2C19 > CYP2D6 (Table 1).

Metabolism of sertraline and N-desmethylsertraline in human rP450 enzymes. A, sertraline N-demethylation at [Sertraline] = 0.5 μM; B, sertraline N-demethylation at [Sertraline] = 50 μM; C, sertraline (solid bars) and N-desmethylsertraline (open bars) N-deamination at substrate concentrations of 50 μM. Each bar represents the mean ± S.D. for three determinations.
Estimations of percentage contribution of human cytochrome P450 enzymes to sertraline N-demethylation
P450-Specific Inhibitors. The activity of human liver microsomes to catalyze sertraline N-demethylation was examined in the presence of chemical inhibitors and inactivators that are selective for specific cytochrome P450 enzymes. Of those tested, 2-phenyl-2-(1-piperdinyl)-propane (CYP2B6-selective inactivator) demonstrated the greatest inhibition at a sertraline concentration of 0.5 μM (Fig. 4). Only mild inhibition (ca. 10% or less) was observed for ketoconazole (CYP3A), sulfaphenazole (CYP2C9), and N-benzylnirvanol (CYP2C19).
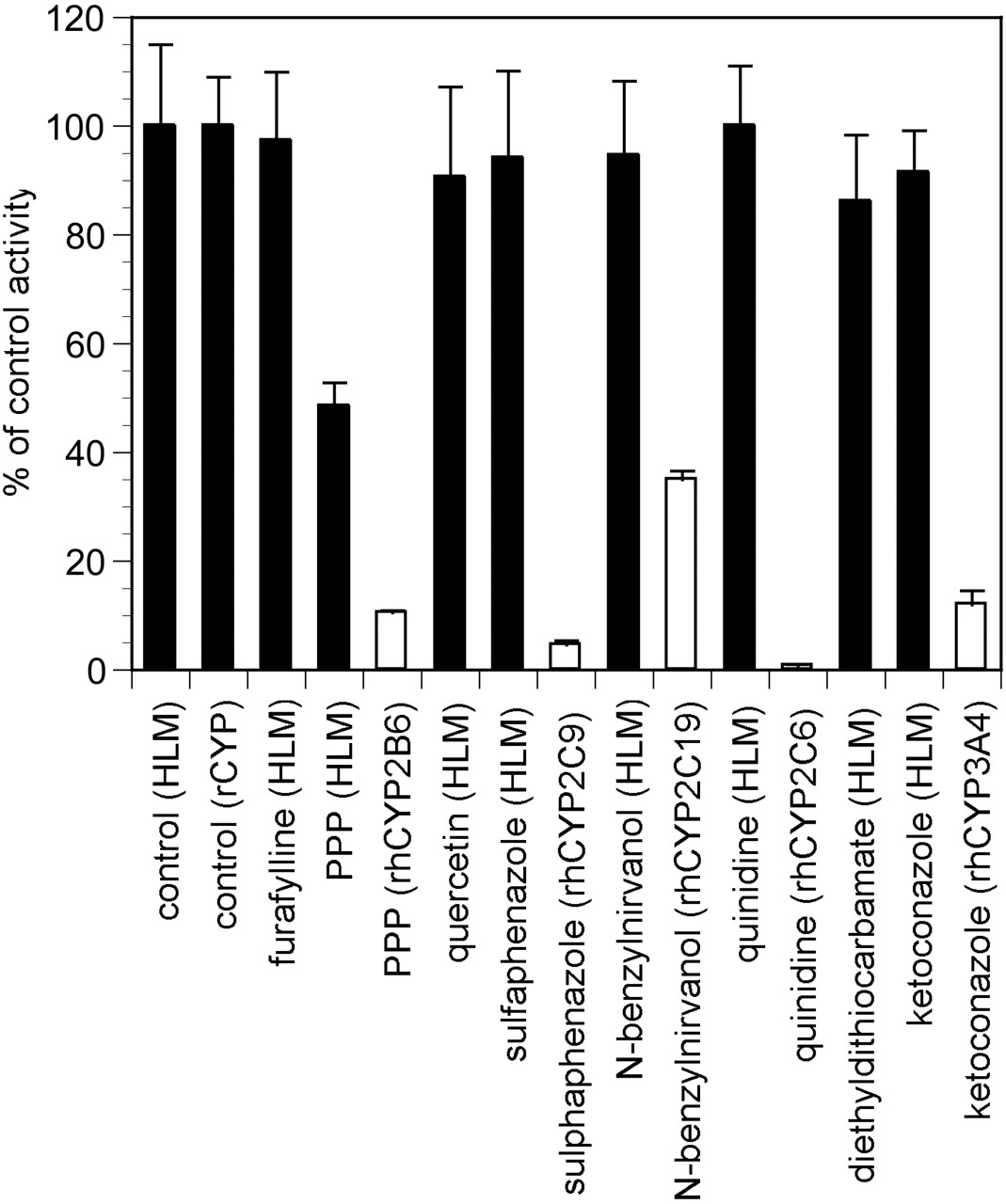
Inhibition of sertraline N-demethylase in pooled human liver microsomes and recombinant human P450 enzymes. Solid bars, human liver microsomes; open bars, recombinant human P450s. Concentrations of inhibitors used were: furafylline, 10 μM; PPP, 10 μM; quercetin, 10 μM; sulfaphenazole, 10 μM; N-benzylnirvanol, 10 μM; quinidine, 1 μM; diethyldithiocarbamate, 30 μM; ketoconazole, 1 μM. Each bar represents the mean ± S.D. for three determinations.
To better delineate the effects of these inhibitors and to ensure that concentrations used were adequate, they were also tested using their respective recombinant P450 enzymes (Fig. 4). For quinidine, sulfaphenazole, and ketoconaozle, inhibitor concentrations used in human liver microsomes (1.0, 10, and 1.0 μM, respectively) demonstrated potent inhibition in rP450s for sertraline N-demethylation, indicating that the inhibitor concentrations used for liver microsomes were appropriate. For N-benzylnirvanol, 10 μM yielded 65% inhibition of CYP2C19-catalyzed sertraline N-demethylation. For PPP, preincubation of 10 μM inhibitor yielded potent inhibition of CYP2B6 (89%) consistent with this inhibitor acting as a mechanism-based inactivator of CYP2B6 (Chun et al., 2000).
A full inhibitor concentration range was tested for N-benzylnirvanol and PPP, to aid in better delineating the relative contributions of CYP2B6 and CYP2C19 to sertraline N-demethylation. Inhibitor concentrations were tested ranging from 0.1 to 100 μM (with PPP undergoing preincubation with microsomes). The results are plotted in Fig. 5 and show that PPP yields a maximum inhibition of approximately 50%, indicating that in pooled human liver microsomes, CYP2B6 contributes approximately half the sertraline N-demethylation observed. N-Benzylnirvanol yielded a maximum inhibition of approximately 30%. Omeprazole was also tested. Omeprazole has been claimed as a CYP2C19 inhibitor, and a previous investigation had used this compound as an inhibitor to claim a role for CYP2C19 in sertraline N-demethylation (Xu et al., 1999). The data obtained with pooled human liver microsomes suggest that omeprazole does not cause a substantial amount of inhibition of sertraline N-demethylase except for when high inhibitor concentrations are tested (100 μM) (Fig. 5).

Inhibition of sertraline N-demethylation by PPP (CYP2B6-selective inactivator), N-benzylnirvanol (CYP2C19-selective inhibitor), and omeprazole (a CYP2C19-nonselective inhibitor) in pooled human liver microsomes. Squares, PPP; circles, (+)-N-3-benzylnirvanol; triangles, omeprazole. Each point represents the mean ± S.D. from three determinations. The data were fitted to the inhibition function described under Materials and Methods, “Data Analysis.”
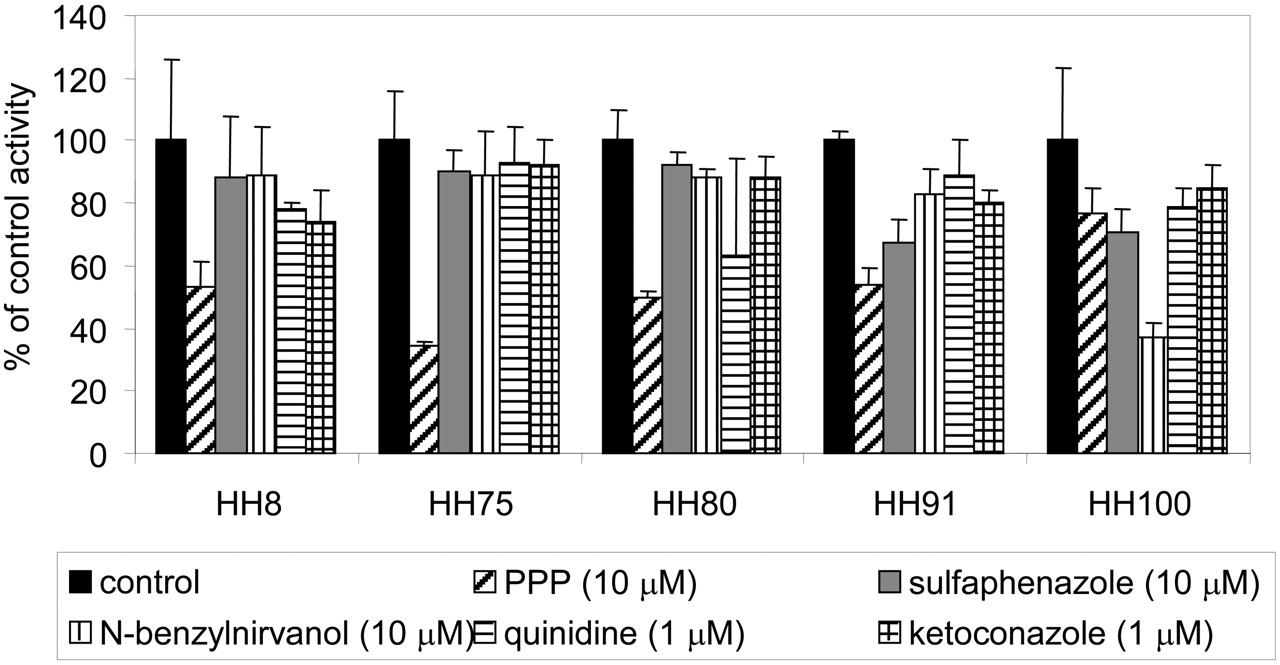
To further delineate the contributions of CYP2B6, 2C9, 2C19, 2D6, and 3A to sertraline N-demethylation in human liver microsomes, specific inhibitors of these five enzymes were tested in five lots of microsomes from individual donors. These lots were selected because each represented high activity for one of the enzymes, with moderate or low activity for the other four. The results are presented in Fig. 6. PPP demonstrated a range of inhibition from 23 to 66%. The lot of microsomes with the greatest CYP2B6 activity (HH75; as assessed with S-mephenytoin N-demethylase activity) was the one most affected by PPP (66% inhibition). The lot with the greatest CYP2C19 activity (HH100; assessed with S-mephenytoin 4-hydroxylase activity) demonstrated the greatest sensitivity to N-benzylnirvanol, with 63% inhibition. The sensitivity of the five lots to benzylnirvanol inhibition ranged from 11 to 63%. Corresponding results were found for the other three lots of human liver microsomes: ketoconazole had the greatest effect on HH8 (highest of the five lots for CYP3A activity), sulfaphenazole had the greatest effect on HH91 (highest of the five lots for CYP2C9 activity), and quinidine had the greatest effect on HH80 (highest of the five lots for CYP2D6 activity). Relative contributions of the P450 enzymes estimated from the inhibition data in these individual donors are listed in Table 1.

Inhibition of sertraline N-demethylase in human liver microsomes from five individual donors. Each bar represents the mean ± S.D. for three determinations.
Enzyme Kinetics in Liver Microsomes. The enzyme kinetics of sertraline N-deamination in pooled human liver microsomes were determined. The data demonstrated inhibition at the highest sertraline concentration, but inclusion of the 500 μM data point did not allow for fitting of the kinetics. Exclusion of this point yielded enzyme kinetic parameters of Km = 114 μM and Vmax = 106 pmol/min/mg microsomal protein. Thus, all subsequent phenotyping experiments could be conducted at substrate concentrations below 114 μM. A plot of the substrate saturation curve is shown in Fig. 2B.
Incubations with rP450 Enzymes. Sertraline N-deamination was measured in rP450 at a substrate concentration of 50 μM. (Due to assay sensitivity limitations, a substrate concentration of 0.5 μM could not be examined as it was for the N-demethylation reaction.) Of the 11 rP450s tested, measurable activity was detected for CYP1A1, 1A2, 2C19, 3A4, and 3A5 (Fig. 3C). Application of RAF as described above, yielded estimates of 63%, 35%, and 2% contribution from CYP3A4, 2C19, and 1A2, respectively (Table 2).
Estimations of percentage contribution of human cytochrome P450 enzymes to sertraline N-deamination
P450-Specific Inhibitors. Following demonstration that sertraline deamination was catalyzed by rCYP1A2, 2C19, and 3A4, incubations were conducted in HLM-101 using furafylline, N-benzylnirvanol, and ketoconazole as selective inhibitors for these three enzymes. Whereas furafylline had no effect on the activity, N-benzylnirvanol (10 μM) and ketoconazole (1 μM) demonstrated 9% and 53% inhibition, respectively (Fig. 7). This is consistent with a role for CYP2C19 and CYP3A in this metabolic reaction.
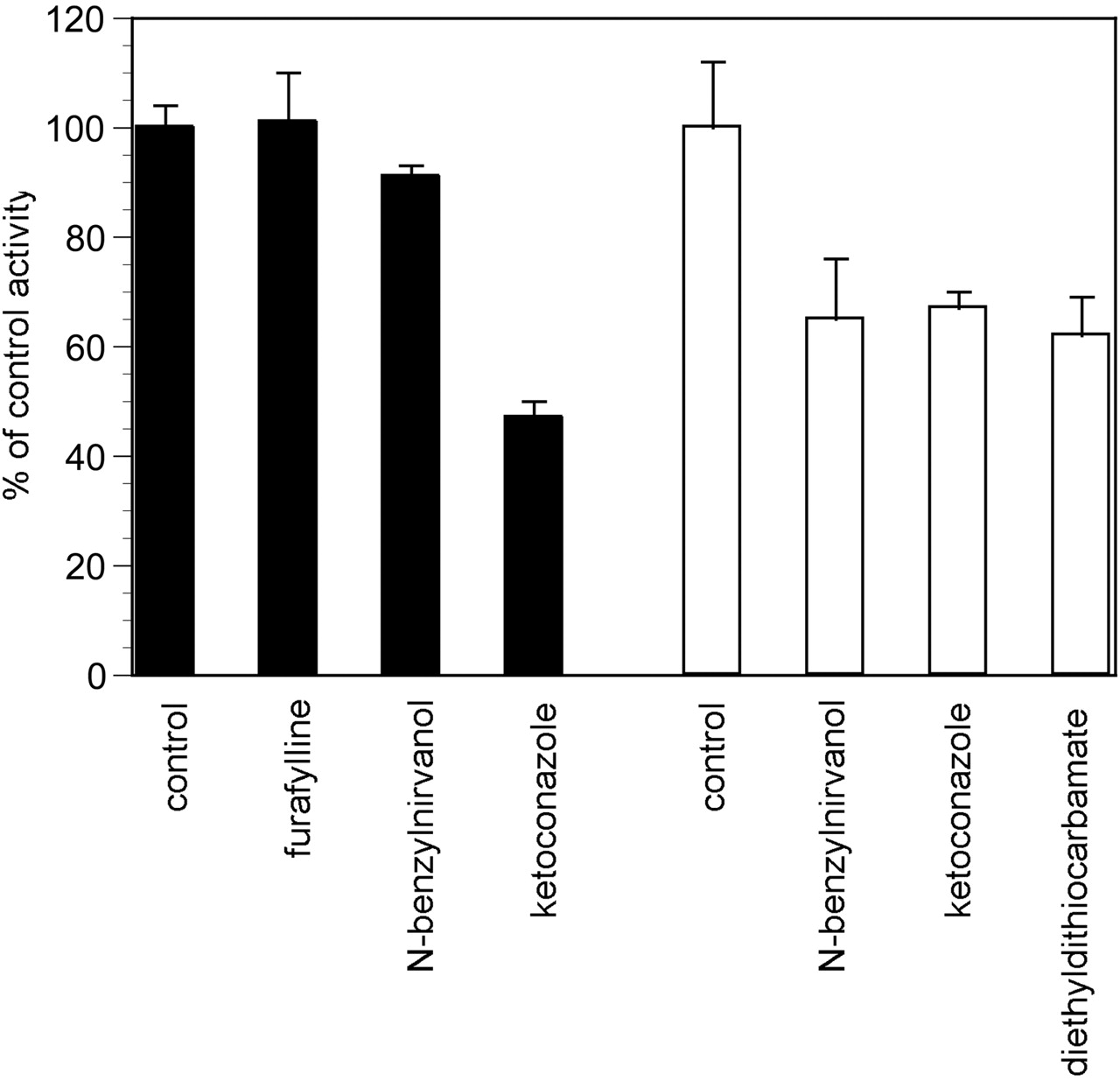
Inhibition of sertraline and N-desmethylsertraline N-deamination in pooled human liver microsomes. Solid bars, sertraline; open bars, N-desmethylsertraline. Concentrations of inhibitors used were: furafylline, 10 μM; N-benzylnirvanol, 10 μM; diethyldithiocarbamate, 30 μM; ketoconazole, 1 μM. Each bar represents the mean ± S.D. for three determinations.
Sertraline N-Deamination by MAO. Sertraline N-deamination was catalyzed by purified human MAO-A and MAO-B. Enzyme kinetics were determined (Fig. 2, C and D) with Km values of 230 and 270 μM, and Vmax values of 39.9 and 120 pmol/min/nmol MAO for MAO-A and MAO-B, respectively. Thus, the intrinsic clearance for MAO-B is approximately 2.7 times greater than that for MAO-A, when normalized on a per nanomole of enzyme basis. The relative contributions of these enzymes to the N-deamination of sertraline, and relative to the P450 contribution to this reaction, will depend on the relative expression levels of these enzymes in various human tissues.
N-Desmethylsertraline N-Deamination.N-Desmethylsertraline was also demonstrated to generate sertraline ketone when incubated with purified MAO-A and MAO-B, as well as with human liver microsomes. At a substrate concentration of 50 μM, rates of N-desmethylsertraline N-deamination were lower than those for sertraline N-deamination for all three enzyme systems (Table 3).
Comparison of rates of N-deamination for sertraline and N-desmethylsertraline in MAO and human liver microsomes Substrate concentration used was 50 μM.
For P450 enzymes, CYP1A1, CYP2C19, CYP3A4, and, to a very small extent, CYP2E1 demonstrated generation of sertraline ketone from N-desmethylsertraline (Fig. 3C). In human liver microsomes, N-desmethylsertraline was inhibited by N-benzylnirvanol by 35%, ketoconazole by 33%, and diethyldithiocarbamate by 38%, suggesting roles for CYP2C19, CYP3A, and CYP2E1 in this reaction (Fig. 7).
Sertraline N-Carbamoyl Glucuronidation. Incubation of pooled human liver microsomes with sertraline, along with other reagents needed for observation of in vitro glucuronidation reactions, yielded sertraline N-carbamoyl glucuronide. The incubations were conducted in bicarbonate buffer and under a continuous flow of CO2 to generate the carbamoyl glucuronide. Under these conditions, the enzyme kinetics of this reaction showed substrate inhibition by sertraline at concentrations above 100 μM (Fig. 8), and values determined for Km, Vmax, and KS were 50 μM, 960 pmol/min/mg, and 58 μM, respectively. In incubations with recombinant heterologously expressed human UGT enzymes, sertraline N-carbamoyl glucuronidation was measurable in UGT1A3, 1A6, 2B4, and 2B7, with the greatest activity observed with the latter enzyme (Fig. 8). Product formation was under the limit of quantitation for UGT1A1, 1A4, 1A7, 1A8, 1A9, 1A10, and 2B15.
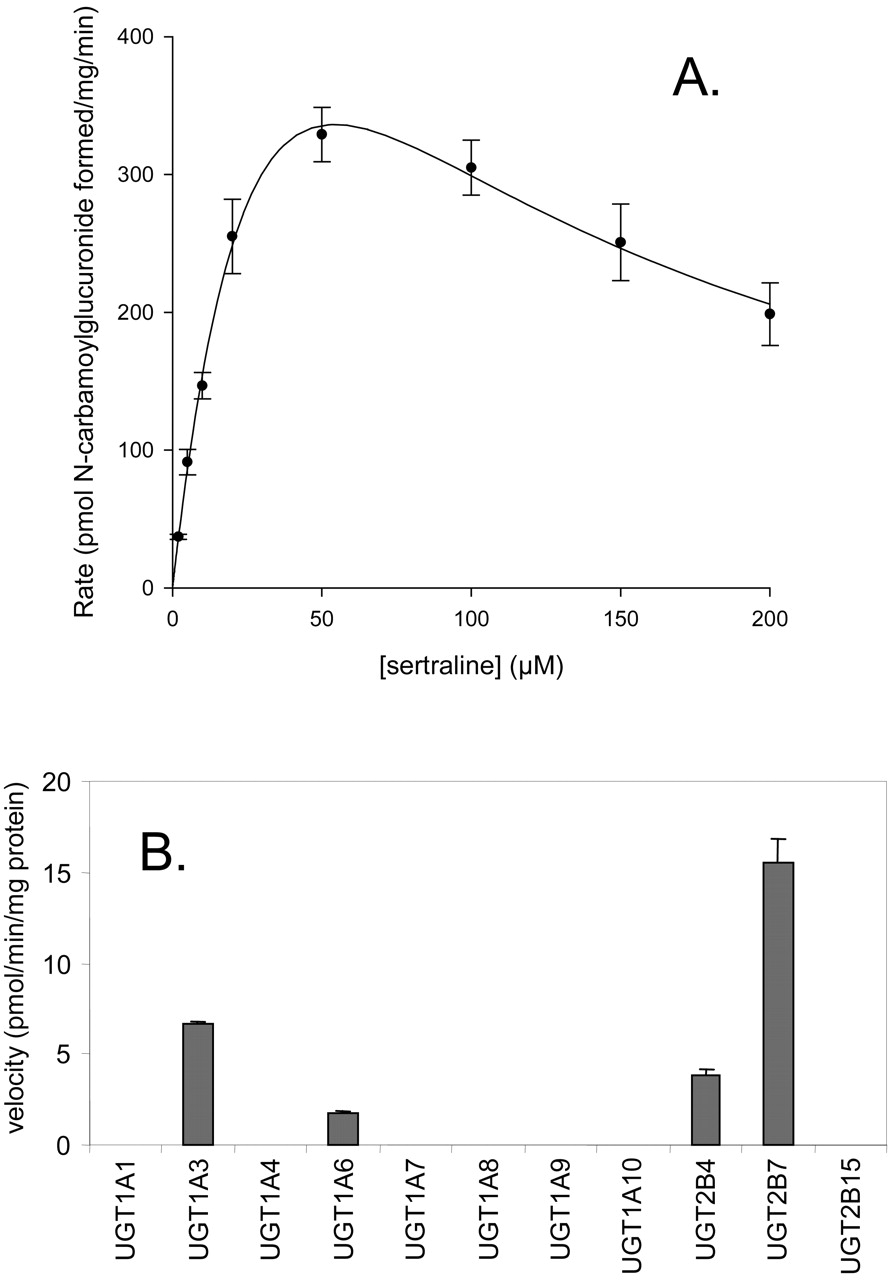
Sertraline N-carbamoyl glucuronidation in pooled human liver microsomes and human UGT enzymes. A, substrate saturation plot in pooled human liver microsomes; B, reaction velocities for sertraline N-carbamoyl glucuronidation in recombinant human UGT enzymes at a sertraline concentration of 10 μM.
Discussion
To this point, there have been three published reports of in vitro data that address identification of the cytochrome P450 enzymes involved in sertraline metabolism, with focus only on the N-demethylation reaction (Greenblatt et al., 1999; Kobayashi et al., 1999; Xu et al., 1999). In the report of Kobayashi et al. (1999), the enzyme kinetics of sertraline N-demethylation were examined in recombinant human P450s, and the conclusion made was that several P450s (CYP2B6, 2C9, 2C19, 2D6, and 3A4) were involved in this reaction. In the work of Greenblatt et al. (1999), four of these enzymes, 2C9, 2C19, 2D6, and 3A4, were stated to be nearly equally contributing to sertraline N-demethylation. However, in the report of Xu et al. (1999), a different conclusion was made using human liver microsomes and chemical inhibitors: CYP2C19 and 2C9 were claimed to be the enzymes involved. However, in this latter example, examination of CYP2B6 was not made. When it appears that multiple P450 enzymes are involved in a metabolic transformation, as is the case with sertraline N-demethylation, ascertaining the relative contributions of each is more challenging. The present work described in this report was undertaken to attempt to clarify this transformation and included the use of inhibitors selective for CYP2B6 and CYP2C19 that have been described since the earlier reports were published. The data in this report support the idea that sertraline N-demethylation is catalyzed by CYP2B6 along with lesser roles for CYP2C19, 2C9, 2D6, and 3A, and that different expression levels of these enzymes among individuals will contribute to different percentages of contribution of these enzymes in different subjects.
The enzyme kinetics of sertraline N-demethylation were consistent with a “single enzyme model,” i.e., that just a single Km value could be observed, in this case representing a hybrid of several enzymes with Km values that are kinetically indistinguishable. This is consistent with the values measured in rP450s by Kobayashi et al. (1999) and the value measured by Greenblatt et al. (1999), in human liver microsomes. These data are in contrast to those of Xu et al. (1999), in which a two-enzyme model was observed in liver microsome samples containing CYP2C19 and a single-enzyme model in microsomes deficient in CYP2C19 activity. The low Km value in CYP2C19 extensive metabolizer microsomes was reported to be approximately 1 to 2 μM (Xu et al., 1999). The data in the present report showed no sign of a low Km enzyme, even with sertraline substrate concentrations as low as 0.25 μM being tested in the substrate saturation experiment. Furthermore, in the reports of Kobayashi et al. (1999) and Greenblatt et al. (1999), Km values of 9 and 33 μM were measured for rCYP2C19, which are well above the value claimed to be attributed to CYP2C19 in the report of Xu et al. (1999).
2-Phenyl-2-(1-piperdinyl)propane and (+)-N-3-benzylnirvanol are recently reported inhibitors of CYP2B6 and CYP2C19, respectively (Chun et al., 2000; Suzuki et al., 2002). These inhibitors were not reported at the time of the previous publications on sertraline N-demethylation, and they were successfully applied in the present work. PPP was shown to inhibit sertraline N-demethylation 15 to 65%, depending on the activity of CYP2B6 in the liver microsome preparation. N-Benzylnirvanol demonstrated inhibition between 9 and 42%, again depending on the activity of CYP2C19 in the liver microsome preparation tested. The use of these inhibitors provides the most convincing in vitro evidence of the roles of CYP2B6 and CYP2C19 in sertraline N-demethylation. A minor contribution from CYP2C9, 2D6, and 3A4 is also apparent from these data.
The finding that sertraline N-demethylation is catalyzed by several P450 enzymes in humans is consistent with in vivo data. In clinical drug interaction studies, no drug has been identified that can cause a large increase in sertraline exposure by inhibiting sertraline metabolism (DeVane et al., 2002). Sertraline pharmacokinetics are not different in CYP2D6 extensive and poor metabolizers, suggesting that CYP2D6 does not contribute substantially to sertraline clearance in vivo (Hamelin et al., 1996). In CYP2C19 poor metabolizers, sertraline exposure was about 40% greater than in extensive metabolizers (Wang et al., 2001). This suggests a minor role for CYP2C19 in sertraline metabolism. If this enzyme contributed a major role, then the difference between CYP2C19 extensive and poor metabolizer subjects would be great, and a bimodal distribution of sertraline exposure values would have been observed in the population, as has been observed for drugs for which CYP2C19 plays a considerable role, such as omeprazole (Desta et al., 2002). CYP2B6 has not been studied as extensively as other human P450 enzymes with regard to clinical pharmacokinetics and drug interactions. In vitro studies have established a few drugs, such as bupropion, ifosphamide, and cyclophosphamide, as CYP2B6 substrates, but there have been no in vivo studies in which a profound increase in the exposure to these drugs has been demonstrated. CYP2B6 is also subject to genetic polymorphisms that can manifest themselves in a poor metabolizer phenotype, as well as observed gender differences in expression (Lang et al., 2001; Lamba et al., 2003). However, since sertraline appears to be metabolized by several enzymes, an effect of this variability in CYP2B6 activity among individuals on sertraline pharmacokinetics may not be observable.
In humans, the major excretory metabolite is hydroxyl sertraline ketone (data on file, Pfizer, Inc.). This metabolite could arise from three initial possible parallel pathways: N-demethylation followed by N-deamination and hydroxylation; N-deamination of the methylamine substituent followed by hydroxylation, and/or initial hydroxylation followed by oxidative metabolism of the methylamine moiety. The latter seems unlikely because hydroxylated sertraline was not observable in incubations of sertraline, liver microsomes, and NADPH (data not shown). In this report, we did demonstrate that sertraline can be directly N-deaminated to sertraline ketone. This reaction was catalyzed by both P450 enzymes (CYP3A4 and 2C19) as well as monoamine oxidases (MAO-A and MAO-B). Whereas P450 enzymes are predominantly expressed in the liver, MAO enzymes are abundantly expressed in numerous tissues. This difference complicates any attempt to determine a role for MAO versus P450 in sertraline metabolism and also complicates the determination of whether the main initial metabolic pathway for sertraline is N-demethylation or N-deamination. Some estimates of MAO expression levels have been made, and values range, depending on the tissue examined, from 1 to 30 pmol/mg tissue protein (O'Carroll et al., 1989; Riley and Denney, 1991; Saura et al., 1992, 1996). Nevertheless, quantitative comparison of the role of MAO versus P450 in sertraline metabolism would require further study, such as the measurement of sertraline metabolism in preparations of other tissues besides liver (e.g., brain, kidney, lung, etc.) and application of specific P450 versus MAO inhibitors, or in vivo studies in humans with concomitant administration of selective P450 versus MAO inhibitors.
N-Carbamoyl glucuronidation is an unusual metabolic reaction exhibited by some primary and secondary amines. Sertraline N-carbamoyl glucuronide, along with tocainide N-carbamoyl glucuronide, was one of the first examples of this type of metabolite (Ronfeld et al., 1982; Tremaine et al., 1989). Subsequent investigations of the N-carbamoyl glucuronidation of carvedilol described the first means by which such metabolism could be observed in an in vitro system and demonstrated that the incubation needed to be conducted under a CO2 atmosphere and in carbonate buffer (Schaefer, 1992). The data reported in this paper describe the first observations of the generation of sertraline N-carbamoyl glucuronide in vitro, and this is the first time that this type of reaction was characterized for its enzyme kinetics and the identity of UGT enzymes involved. In order for the reaction to occur, it is hypothesized that the amine spontaneously forms a transient carbamic acid intermediate with the dissolved CO2 followed by glucuronidation. However, proof that the carbamic acid is the actual UGT substrate and that UGT does not also catalyze the formation of the carbamic acid would require that this intermediate is stable and isolable. Sertraline N-carbamoyl glucuronidation was primarily formed by UGT2B7, which is also responsible for numerous acyl glucuronidation reactions of carboxylic acid drugs (Tephly and Green, 2000). This might suggest that sertraline N-carbamic acid is the actual UGT substrate; however, such a claim remains speculative without mechanistic proof. Exploring this metabolic reaction remains the object of an ongoing investigation.
In conclusion, because it appears that sertraline has multiple enzymes involved in its initial metabolic pathways, it would be difficult for any single agent to cause a meaningful drug interaction via inhibition of the metabolic clearance of sertraline. To date, there have been no agents identified that can give rise to a marked increase in sertraline exposure upon concomitant administration (DeVane et al., 2002). The findings in this study are consistent with these observations.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.002428.
-
ABBREVIATIONS: P450, cytochrome P450; MAO, monoamine oxidase; PPP, 2-phenyl-2-(1-piperdinyl)propane; RAF, relative activity factor; UDPGA, uridine diphosphoglucuronic acid; UGT, uridine diphosphoglucuronic acid transferase; HPLC, high-performance liquid chromatography; rP450, recombinant cytochrome P450; HLM, human liver microsome(s); CP-105,162, 4-(4-chlorophenyl)-1,2,3,4-tetrahydro-1-naphthalenamine.
- Received September 20, 2004.
- Accepted November 12, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}