


Abstract
Intestinal cell lines are used as in vitro models for pharmacological and toxicological studies. However, a general report of the gene expression spectrum of proteins that are involved in the metabolism and the disposition of xenobiotics in these in vitro systems is not currently available. To fill this information gap, we systematically characterized the expression profile of 377 genes encoding xenobiotic-metabolizing enzymes, transporters, and nuclear receptors and transcription factors in intestinal mucosa (ileum, ascending colon, transverse colon, descending colon, and rectum) from five healthy subjects and in five commonly used intestinal cell lines (Caco-2, C2BBe1, HT29, T84, and FHC). For this, we performed a quantitative real-time reverse transcription-polymerase chain reaction analysis using TaqMan low-density arrays and analyzed the results by different statistical approaches: Spearman correlation coefficients, hierarchical clustering, and principal component analysis (PCA). A large variation in gene expression spectra was observed between intestinal cell lines and intestinal tissues. Both hierarchical clustering and PCA showed that two distinct clusters are visible, of which one corresponds to all cultured cell lines and the other to all intestinal biopsies. The best agreement between human tissue and the representative cell line was observed for human colonic tissues and HT29 and T84 cell lines. Altogether, these data demonstrated that gene expression profiling represents a new valuable tool for investigating in vitro and in vivo expression level correlation. This study has pointed out interesting expression profiles for various colon cell lines, which will be useful for choosing the appropriate in vitro model for pharmacological and toxicological studies.
Introduction
The intestine is the primary site for ingested xenobiotics, including toxicants, carcinogens, and drugs. Three different groups of proteins are involved in the metabolism and disposition of these compounds: enzymes, transporters, and nuclear factors. Phase I xenobiotic-metabolizing enzymes (XMEs) catalyze the first step of xenobiotic processing, referred to as xenobiotic functionalization; they catalyze oxidation, reduction, hydrolysis, cyclization, and decyclization reactions. The cytochrome P450 (P450) enzyme superfamily, for example, plays a dominant role in phase I biotransformation. Phase II XMEs conjugate xenobiotics or their phase I metabolites to glutathione, sulfate, glucuronide, methyl, or acetyl moieties, making molecules more hydrophilic and, consequently, more suitable for elimination. Transporters, mainly of the solute carrier (SLC) and of the ATP-binding cassette (ABC) families, mediate the influx of xenobiotics into cells or facilitate the efflux of xenobiotics or phase II metabolites from cells. Tight coupling between phase I XMEs, phase II XMEs, and transporters is ensured by nuclear receptors (NRs), which are ligand-activated transcription factors that control constitutive and inducible expression of numerous genes, including XME and transporter genes, in a coordinated manner (Nakata et al., 2006). Three major NRs (aryl hydrocarbon receptors, pregnane X receptors, and constitutive androgen receptors) are activated by xenobiotics and are therefore termed as xenosensors (Nakata et al., 2006).
To better understand xenobiotic metabolic pathways, efficacies, or toxicities, the establishment of a reliable research model system remains a key challenge. During past decades, primary intestinal cell cultures have been obtained from the small intestine (Perreault and Beaulieu, 1998; Aldhous et al., 2001) or from the colon (Baten et al., 1992; Fonti et al., 1994; Deveney et al., 1996; Grossmann et al., 2003; Mohammadpour, 2005), but a rapid loss of the differentiated characteristics of cells was observed. As a consequence, these primary cell models could be used only for short-term toxicity studies. Intestinal cell lines, such as Caco-2, C2BBe1, HT29, T84, and FHC, are the most widely used in vitro models for pharmacological and toxicological studies. These cell lines present several advantages including their availability, growth rate, homogeneous cell population, and reproducibility. However, they are not fully representative of the tissues. For example, a large variation in expression levels of drug-metabolizing enzymes is observed between cell lines and primary culture (Guo et al., 2011).
Many research groups have studied gene expression in human intestinal cells (Taipalensuu et al., 2001; Nakamura et al., 2002; Sun et al., 2002; Pfrunder et al., 2003; Anderle et al., 2004; van Erk et al., 2005; Seithel et al., 2006; Hilgendorf et al., 2007). To our knowledge, there has been no complete report comparing intestinal cell lines with both human ileum and colon, especially for systems involved in the metabolism and the disposition of xenobiotics. Such information would enhance our ability to predict potential pharmacological and toxicological consequences of xenobiotic exposure in the intestinal mucosa and would facilitate data translation to the in vivo situation.
The aim of the present study was to investigate, using TaqMan low-density arrays, the expression profile of 377 genes encoding proteins that are involved in the metabolism and the disposition of xenobiotics in five cell lines (Caco-2 and HT29 differentiated or not, C2BBe1, T84, and FHC) and in intestinal mucosa from five different segments (ileum, ascending colon, transverse colon, descending colon, and rectum).
Materials and Methods
Tissues.
Human tissues were obtained after written informed consent, in agreement with local ethics committee. Fresh biopsy samples were obtained from five healthy subjects of either gender (aged 32–55 years) who were undergoing endoscopy. These patients were referred to the endoscopy center because of abdominal cramps and diarrhea of unknown cause, irritable bowel syndrome, polyp surveillance, obstipation, and anemia of unknown cause. They had no mucosal inflammation upon endoscopic and histological examination. Biopsy samples were taken from five different intestinal areas (ileum, ascending colon, transverse colon, descending colon, and rectum) using a standard biopsy forceps. Information on age, sex, and location of biopsy samples is listed in Table 1. To preserve the RNA, intestinal specimens were immediately placed in RNAlater (Ambion, Austin, TX), at 4°C for 16 h and then were stored at −20°C until used.
Description and origin of human intestinal biopsies
Cell Cultures.
Culture media and additives were obtained from Invitrogen (Cergy-Pontoise, France). Caco-2, C2BBe1, HT29, T84, and FHC cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA) (Table 2). Human colon carcinoma cell lines Caco-2, C2BBe1, and HT29 were cultured in DMEM supplemented with 10% fetal calf serum, 2 mM l-glutamine, 100 units/ml penicillin G, and 100 μg/ml streptomycin, as well as 1% nonessential amino acid solution for the Caco-2 cell line and 10 μg/ml transferrin for the C2BBe1 cell line. The human carcinoma cell line T84 derived from a lung metastasis of a colon carcinoma was cultivated in DMEM-F12 (1:1) supplemented with 5% fetal calf serum, 2 mM l-glutamine, 100 units/ml penicillin G, and 100 μg/ml streptomycin. The fetal human colon cell line FHC was cultured in DMEM-F12 (1:1) supplemented with 10% fetal calf serum, 10 ng/ml cholera toxin, 5 μg/ml insulin-transferrin-selenium, 100 ng/ml hydrocortisone, 10 mM HEPES, 100 units/ml penicillin G, and 100 μg/ml streptomycin. All cultures were maintained at 37°C in 5% CO2. The passage number was 4 for all experiments performed in each cell type (starting passage was determined as passage 1 after receipt from ATCC). Cells were seeded into six-well plates at 5 × 105 cells/well and grown to confluence (for five cell lines) or to 14 days postconfluence (for differentiation of Caco-2 and HT29 cells) before the RNA was isolated.
Description and origin of human colon cell lines
RNA Isolation.
Before extraction, intestinal biopsy samples were homogenized in Buffer RLT Plus (QIAGEN, Courtaboeuf, France) supplemented with 1% β-mercaptoethanol, using a gentleMACS Dissociator (Miltenyi Biotec, Paris, France). Cell lines were washed two times with phosphate-buffered saline and lysed in an appropriate volume of Buffer RLT Plus supplemented with 1% β-mercaptoethanol. Total RNAs from biopsy samples or cell lines were extracted using an RNeasy Plus Mini Kit (QIAGEN), according to the manufacturer's instructions. Elution was performed with 50 μl of RNase-free water. RNAs were then precipitated for 12 h at −20°C by addition of 3 M sodium acetate-ice-cold 100% ethanol (0.1 V/2.5 V). After centrifugation and washing with 70% ethanol (200 μl), pellets were resuspended in 10 μl of RNase-free water. RNA concentration was determined using the BioSpec-nano (Shimadzu, Champs-sur-Marne, France). The quality of RNA was evaluated using Experion RNA StdSens chips on an Experion electrophoresis system (Bio-Rad Laboratories, Hercules, CA).
cDNA Synthesis.
Two micrograms of total RNAs were retrotranscribed into single-stranded cDNAs using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Courtaboeuf, France) according to the manufacturer's recommendations.
Quantitative Real-Time PCR.
Gene expression data were obtained using TaqMan low-density arrays (TLDA) (Applied Biosystems), which are 384-well micro fluidic cards preloaded with sets of primers and 6-FAM-labeled TaqMan MGB probes that enable performance of 384 simultaneous real-time PCR runs and that have been used for gene expression profiling in several studies (Leclerc et al., 2010; Kodani et al., 2011). We chose a configuration with 379 different assays loaded in simplicate (listed in Tables 3 and 4 and Supplemental Tables 1, 2, and 3), the last five wells being dedicated to other assays not detailed in this study.
Relative abundance of XMEs, transporters, nuclear receptors, and transcription factors whose expression levels vary in ileum or along the colon
Percentages are expressed compared with transverse colon expression value [for each gene transverse colon expression is baseline (= 100%)].
Relative abundance of XMEs, transporters, nuclear receptors, and transcription factors expressed in colon cell lines and transverse colon biopsy samples
Percentages are expressed compared with transverse colon expression value [for each gene transverse colon expression is baseline (= 100%)]. Not represented in this table are 106 genes that are not expressed in transverse colon biopsy samples (for further details, see Supplemental Table 3).
The TLDAs were loaded with each cDNA template mixed with 2× TaqMan Gene Expression Master Mix (Applied Biosystems), according to the manufacturer's instructions. After centrifugation (two 1-min cycles at 1200 rpm), each reaction well contained 1 μl of reaction mixture corresponding to 1 ng of total RNA. The wells were immediately sealed with a TLDA sealer (Applied Biosystems) to prevent cross-contamination. The quantitative real-time PCR amplification was performed using an Applied Biosystems Prism 7900HT sequence detection system with the following thermal cycler conditions: 2 min at 50°C and 10 min at 94.5°C, followed by 40 cycles of 30 s at 97°C and 1 min at 59.7°C.
Relative Expression Analysis.
The detection threshold was set at 0.3 for all genes, except for seven genes for which a threshold at 0.3 would have led to inaccurate quantification (AKR7L, 0.2; ALDH16A1, 0.2; ALDH5A1, 0.1; CES2, 0.1; GSTM1, 0.1; PNMT, 0.2; and PON1, 0.1). The threshold cycle (Ct) values were determined with RQ Manager 1.2 software (Applied Biosystems). We chose Ct >35 as the cutoff for nonexpressed genes. Each Ct value was normalized to the average Ct of the two endogenous controls, 60S acidic ribosomal protein P0 (RPLP0) and peptidylpropyl isomerase A (PPIA), to correct for any tube-to-tube variations in reverse transcription efficiencies and amount of total RNA added to each reaction. The comparative ΔΔCt method was used to calculate relative quantification of gene expression.
Data Analysis.
Statistical analyses were performed using SPSS 17.0 (SPSS, Inc., Chicago, IL). Spearman correlation coefficients of all the ΔCt mean values were calculated between cell lines and intestinal segments. After ΔCt computation, unsupervised hierarchical clustering was performed with the Euclidean distance as an input parameter in the clustering algorithm.
Principal component analysis (PCA), a standard, nonparametric tool that reduces a complex data set to a lower dimension, was used for comparisons between different tissues and cell lines. This analysis was performed using ΔCt values. Two-dimensional plots were made, using principal component 1 and 2 as axes.
An arbitrary classification system designating expression levels as high (ΔCt <6), moderate (6 ≤ ΔCt <10), and low (ΔCt ≥10) was applied to the data throughout this article.
Results
In the current study, gene expression of 206 XMEs (137 phase I and 69 phase II enzymes), 102 transporters (including 30 ABC and 62 SLC transporters), 48 nuclear receptors and transcription factors (including coactivators and corepressors), and 21 miscellaneous genes (including 9 metallothioneins) was quantified in RNA samples from intestinal mucosa and intestinal cell lines, using custom TLDAs. Genes from each cell lines were evaluated in comparison with expression levels of biopsy samples from five donors.
Expression Level of Systems Involved in the Metabolism and the Disposition of Xenobiotics in Intestinal Biopsies.
Gene expression profiles of intestinal biopsy samples (Table 1) were analyzed using TLDAs. Of the genes, 21.7% were not detected (Ct >35) in RNA preparations from ileum and approximately 28% were not detected in colon. Some families of genes were poorly expressed in the intestine. For example, 29 of 54 (53.7%) P450 genes and 21 of 62 (33.9%) SLC genes were not detected in transverse colon (Supplemental Table 1). Of genes encoding P450s, 9.3% were expressed at low levels (10 ≤ ΔCt <14), 22.2% at moderate levels (6 ≤ ΔCt <10), and 14.8% at high levels (ΔCt <6) in transverse colon. We next calculated the relative abundance of each gene using gene expression levels measured in transverse colon biopsy samples as references (Table 3; Supplemental Table 2). Approximate abundance of genes expressed in transverse colon biopsy samples is listed as “Expression Value” in Table 3 and Supplemental Table 2, using housekeeping genes RPLP0 and PPIA as references and the expression value of each genes was defined using eqs. 1 and 2:
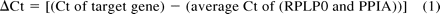

The expression value implies the relative mRNA expression abundance of a gene, arbitrarily assuming an expression level of the housekeeping genes RPLP0 and PPIA of 10,000 copies. Table 3 and Supplemental Table 2 are intended to provide very general information about gene expression in human transverse colon biopsy samples, in which the large interindividual variability of gene expression levels in human populations is certainly underrepresented.
Expression of 46 genes was significantly higher (at least 4-fold higher) and expression of 6 genes (CYP2E1, CYP2W1, DHRS9, UGT2B10, SLC16A1, and THRB) was significantly lower (at least 4-fold lower) in ileum than in transverse colon biopsy samples (Table 3). In descending colon biopsy samples, only the CYP3A4 gene was significantly more expressed than in transverse colon biopsy samples, and SLC6A4 gene was significantly less expressed. Expression of 7 genes (ALDH1A2, CYP2W1, CYP3A4, GSTT1, SULT1C2, SLC15A1, and SLC28A2) was significantly higher in rectal biopsy samples than in transverse colon biopsy samples (Table 3; Supplemental Table 1) and expression of 12 genes (CYP2B6, CYP2C9, SLC6A4, UGT2B17, and others) was significantly lower in rectal biopsy samples than in transverse colon biopsy samples (Table 3). The most abundantly expressed genes in intestinal tissues were XMEs such as UGT2B17, MAOA, GSTP1, MGST1, and CES2 (Table 3).
Expression Level of Systems Involved in the Metabolism and the Disposition of Xenobiotics in Intestinal Cell Lines.
Gene expression profiles of five intestinal cell lines are described in Table 4 and Supplemental Table 3. The relative abundance of each gene detected in each cell line was calculated, using gene expression levels measured in transverse colon biopsy samples as references (Table 4).
A higher proportion of genes was not detected in each cell line, with 31.8, 29.2, 32.4, 40.8, 41.1, 42.4, and 42.4% of genes in Caco-2, differentiated Caco-2, C2BBe1, FHC, HT29, differentiated HT29, and T84 cell lines, respectively.
It is striking that several critical phase I enzymes, such as CYP3A4, CYP7B1, ADH1B, and BCHE were not detected in any of the cell lines. Nevertheless, ADH1B and CYP3A4 were found to be weakly expressed in differentiated Caco-2 and HT29 cells, respectively (Table 4). Among the genes expressed, between 18 and 44 genes, depending on the cell line, were barely detectable with a relative abundance of less than 5% of those in colon biopsy samples. CES2, one of key phase I enzymes, falls into this category. In all cell lines, abundance for the majority of expressed genes was at a level (25–400%) similar to that in colon biopsy samples. In addition, a few genes have much higher abundance (∼4–591 times higher) in cell lines. For example, CYP26B1 was expressed more than 250 times in FHC cells than in colon biopsy samples. Of note, although not detected in colon biopsy samples, several genes were found to be expressed in different cell lines, such as CYP1A1 in Caco-2 (differentiated or not), C2BBe1, FHC, and T84, making these cell lines potential surrogate tools for investigation of related XMEs (Supplemental Table 3). Likewise, the relative abundance for phase II XMEs, transporters, and NRs are listed for different intestinal cell lines compared with colon biopsy samples in Table 4.
Differences and Similarities between Intestinal Biopsies and Intestinal Cell Lines.
Differences and similarities in gene expression patterns among intestinal cell lines and intestinal biopsy samples are pronounced as indicated by the relative abundance of drug-metabolizing genes in different cells. To reveal similarities of gene expression patterns among these cells, a similarity matrix was evaluated by a pairwise comparison of the samples (Table 5), in which the Spearman correlation coefficient (rs) was calculated on the basis of the ΔCt obtained for each gene. In general, similarities among intestinal biopsy samples isolated from the different donors (with rs values between 0.914 and 0.992) were higher than the similarities among the different intestinal cell lines (with rs values between 0.703 and 0.980).
Spearman correlation coefficient between colon cell lines and intestinal biopsy samples
The correlation matrix was calculated on the basis of the averaged ΔCt of two technical replicates for colon cell lines and three biological replicates for intestinal biopsies. The numbers are the pairwise Spearman correlation coefficient (rs) values that represent the strengths of the linear relationship between any two sets of comparative components (rs = 0, no correlation; rs = 1, perfect correlation).
Among these five intestinal cell lines, the highest rs value of 0.980 was observed between Caco-2 and C2BBe1, whereas the lowest rs value of 0.703 was found between differentiated Caco-2 and HT29. More importantly, the similarities between intestinal cell lines and the intestinal biopsy samples were lower, with rs values between 0.620 and 0.783. The highest similarity (rs = 0.783) was observed between T84 cells and ascending colon biopsy samples, whereas the lowest similarity (rs = 0.620) was observed between FHC cells and ileal biopsy samples (Table 5). Of interest, in terms of gene expression levels, the most often used intestinal cell lines HT29 and Caco-2 were relatively different from the intestinal biopsy samples with rs values between 0.672 and 0.747 (Table 5).
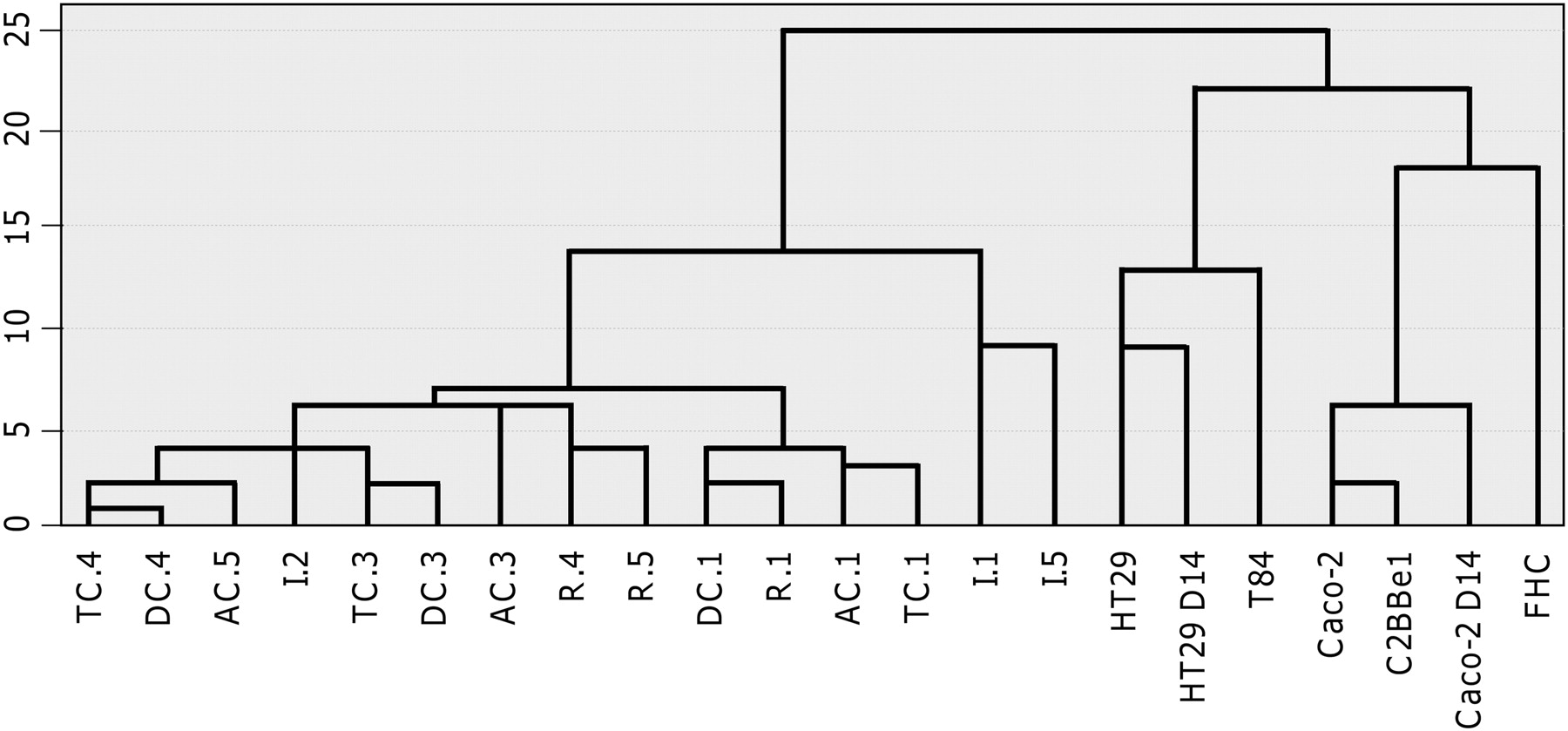
To visualize directly the distances of gene expression patterns among different intestinal cell lines and intestinal biopsy samples, a hierarchical cluster analysis was performed. Figure 1 shows a dendrogram of four clusters based on their gene expression levels. Two major clusters were clearly separated; one consisted of five intestinal cell lines and the other consisted of intestinal biopsy samples. In the cell line cluster, HT29 and T84 profiles were separated from those of other cells (Caco-2, C2BBe1, and FHC). In the intestinal biopsy cluster, profiles of ileal biopsy samples were separated from profiles of colon biopsy samples, except for the ileal biopsy sample from patient 2.

Hierarchical clustering of colon cell lines and intestinal biopsy samples. The dendrogram represents the relationship of gene expression profiles. The lengths of the arms are proportional to the similarities of gene expression profiles, with shorter arms indicating closer relationships. After ΔCt computation, this clustering analysis identified two distinct clusters based on the similarity of their gene expression profiles, separating colon cell lines and intestinal biopsy samples.
Furthermore, similarities and differences in gene expression levels between intestinal biopsy samples and intestinal cell lines were further illustrated by PCA. The plot shown in Fig. 2 is a simple scatterplot of the first two principal components (PCs). PC1 and PC2 explain 42.2 and 16.49% of the total variation, respectively. Distance of separation between clusters is indicative of degree of similarity on a gene level; a greater distance is equivalent to reduced similarity. Along PC1, two distinct clusters were visible, one corresponding to all cultured cell lines, and the other to all intestinal biopsy samples. In the cluster of the cell lines, two subclusters exist along PC2, corresponding to profiles from HT29 together with T84 cells and Caco-2 together with C2BBe1 cells. PC1 was negatively correlated with the expression of SLC7A11, SLC7A5, AHRR, and CRABP2 and positively correlated with ADH1B, ADH5, AOC3, CES2, FMO5, NAT2, UGT2B17, ABCC8, SLC18A2, SLC22A5, NCOR1, RXRG, GZMA, and GZMB, for example. PC2 was negatively correlated with the expression of GSTT1, TBXAS1, SLC38A5, and SLCO5A1 and positively correlated with AKR1D1, AKR1E2, ALDH7A1, ABCA3, ABCC9, SLC1A3, SLC29A4, CRABP1, and AIP, for example.

Principal component analysis of gene expression profiles generated from five colon cell lines (Caco-2, C2BBe1, HT29, T84, and FHC) and intestinal biopsy samples from five segments (ileum, ascending colon, transverse colon, descending colon, and rectum). Samples are labeled with different symbols. Closed symbols represent colon cell lines, and open symbols represent intestinal biopsy samples. For the XME genes, transporter genes, and nuclear receptor and transcription factor genes, the relative contribution of the ΔCt variance is shown by two major principal components (PC1 and PC2) plotted in two dimensions.
Discussion
This study provides an overview of gene expression profiles of phase I and phase II XMEs, transporters, and NRs and transcription factors in human intestinal tissues, as well as in intestinal cell lines.
Expression analysis was performed using a real-time quantitative RT-PCR strategy based on TLDAs. This biotechnology allows simultaneous measurement of mRNA expression of 380 genes for a single sample and is considered to be one of the most robust, sensible, reproducible, and reliable techniques for high throughput screening in functional genomics. On the basis of a careful and extensive review of the literature and of human genome sequence databases, we identified 377 genes known or suspected to code for XME, xenobiotic transporters, or nuclear factors involved in xenobiotic cellular processing. Many genes, such as CYP, ADH, FMO, UGT, GST, and MT, are clustered in the same chromosomal regions, and their nucleotide and amino acid sequences show high homology. The high specificity of the TaqMan primers and probe sets enables discrimination among closely related members of gene families.
RNA expression data do not always correlate with the expression of encoded protein. To fully align mRNA data with protein expression profiles, a protein expression analysis of all the proteins would be necessary. However, quantification of all proteins remains a major technical challenge in proteomics, and, until this issue is solved, quantitative mRNA analysis is the most viable alternative. Consequently, we used mRNA profiles to estimate the expression patterns of the 377 proteins investigated in this study.
Quantification with RT-PCR technology uses housekeeping genes as a reference. There has recently been vigorous debate in the literature about how best to achieve this and about which housekeeping genes are suitable (Tricarico et al., 2002; Dydensborg et al., 2006; Said et al., 2009). The principle for choice of internal reference genes is that they should be unregulated in all samples of interest and their amplification behavior should be comparable to that of the genes of interest. If not all these prerequisites are taken into consideration, it could be impossible to build a relative expression analysis on one single reference gene.
The optimal internal reference gene for the comparison over all samples was determined using the geNorm algorithm software (Vandesompele et al., 2002). A panel of 16 potentially suitable housekeeping genes was analyzed for 8 cDNA samples (data not shown). Because RPLP0 and PPIA mRNA displayed the smallest coefficients of variation between tissues and between patients (<1.89% for RPLP0 and <2.1% for PPIA), they were selected as reference genes for normalization of target gene data in this study. Although the coefficient of variation looked best for 18S mRNA, it was decided to exclude 18S mRNA, because it had Ct values of approximately 10, making it clearly different from the target genes, which had Ct values between 20 and 40. The maximal fold intrinsic variance in the sample set was low, which allows quantitative comparison between all samples included in the analysis.
In this study, mRNA expression profiles of 377 genes encoding proteins that are involved in the metabolism and the disposition of xenobiotics were investigating for the first time in biopsy specimens from five different intestinal areas (ileum, ascending colon, transverse colon, descending colon, and rectum). Apart from data for cytochromes P450 (Obach et al., 2001; Bergheim et al., 2005; Zhang et al., 2007), available data on other genes expressed in human intestinal tissues are very limited (Kaminsky and Zhang, 2003; Anderle et al., 2004; Hilgendorf et al., 2007). Hierarchical clustering revealed that profiles of ileal biopsy samples were separated from profiles of colon biopsy samples. Many genes encoding XMEs (i.e., AADAC, CBR1, CYP2C9, CYP2D6, CYP3A4, FMO1, GSTA1, SULT1E1, SULT2A1, and others) and transporters (i.e., ABCC2, ABCG8, SLC10A2, SLC15A1, SLC28A1, SLC6A4, and others) were overexpressed in the small intestine compared with colon, indicating major differences in the metabolism and disposition of xenobiotics between these two tissues (Table 3). This finding can be explained by different functions of these two segments. Indeed, although the small intestine function comprises the major part of the digestion processes, and most of the food gets absorbed in the small intestine, the large intestine function in digestion mostly pertains to absorption of water and excretion of solid wastes through the anus.
Furthermore, hierarchical clustering revealed that the gene expression profile of the ileal biopsy sample from patient 2 was separated from profiles of ileal biopsy samples from patients 1 and 5. The variability of gene expression among humans is largely attributed to genetic and environmental factors. Genetic polymorphisms, including single nucleotide polymorphism, copy number variation, and insertion and deletion variations, contribute greatly to gene expression profiles, drug metabolism, and clinical impacts. In addition, environmental factors such as exogenous inducers, inhibitors, and/or intestinal disorders may produce more heterogeneous gene expression and xenobiotic responses. Wojtal et al. (2009) observed a statistically significant increase in expression of SLC15A1 mRNA in ileum and colon of patients with inflammatory bowel disease. The intestinal SLC mRNA levels are dysregulated in patients with inflammatory bowel disease, which may be linked to the inflammatory status of the affected tissue (Wojtal et al., 2009).
In the current study, similarities and differences in gene expression levels between intestinal biopsy samples and five intestinal cell lines were observed using Spearman rank correlation coefficients, principal component analysis, and hierarchical clustering analysis. These similarity comparison analyses suggest that intestinal cell lines only partially reflect the gene expression characteristics of intestinal tissues, indicating their limitations as surrogate cell models for human intestinal epithelial cells in toxicological and pharmacological studies.
This result could be partly explained by the tumoral origin of the cell lines. Four of the five cell lines directly originate from colon cancer (Caco-2, C2BBe1, HT29, and T84), and the FHC cell line, which has been established from fetal colonic mucosa, exhibits a tumorigenic phenotype (Soucek et al., 2010). Chromosomal aberrations including gene amplification, gene deletion, and heteroploidy are common events in carcinogenesis, which introduces gene dosage differences between normal cells and transformed cell lines.
Furthermore, in comparison with the in vivo situation with influences from different neighboring cells, (neuro)endocrine regulators, and blood flow, the in vitro model is relatively simple. Although ideal for specific research questions, this factor complicates the translatability of in vitro results to in vivo situations. In addition, cell culture environments, such as the composition of the culture medium and the oxygen concentration, can alter gene expression profiles in cell lines (Warabi et al., 2004; Brunet De La Grange et al., 2006).
Above all, it is not clear whether cells in culture retain gene expression profiles of their in vivo counterparts. A study of the gene expression patterns in 60 human cancer cell lines, including colon cancer cell lines, revealed that the tissue from which the cells are derived is the main factor accounting for the variation in gene expression (Ross et al., 2000).
To avoid all these changes in the expression profile, it would be preferable to use primary cultures for toxicological studies. Primary cultures have been described in other tissues, such as liver (LeCluyse, 2001) and kidney (Brown et al., 2008), as having a genetic profile similar to that of native tissue. However, primary human cells have high variability and short life spans, and availability is limited. Moreover, primary cultures of human intestinal epithelial cells have been difficult to obtain to date. Therefore, intestinal cell lines, which are relatively easy to maintain in culture, are widely used for transport and toxicity studies of xenobiotics and therapeutic agents.
Caco-2 cells were derived from a human colon adenocarcinoma, and they differentiate spontaneously in vitro under standard culture conditions, thereby exhibiting enterocyte-like structural and functional characteristics. In the differentiated state, they mimic typical characteristics of the human small intestinal epithelium, like a well developed brush border with associated enzymes such as alkaline phosphatase and sucrase isomaltase. Nevertheless, the Caco-2 cell model is different from the small intestine in several aspects, and the phenotype of Caco-2 cells is dependent on the time in culture (Mehran et al., 1997; Engle et al., 1998). Of interest, our results reveal a relatively good correlation between Caco-2 cells and human colon with a Spearman correlation coefficient of 0.7.
The HT29 cell line is also of human colon adenocarcinoma origin and is described as an in vitro model of normal epithelial cells because HT29 cells can reversibly display some structural and functional features resembling those of human normal mature intestinal epithelial cells (Andoh et al., 2001). Comalada et al. (2006) had observed that a confluent culture of HT29 cells (differentiated culture) showed characteristics of normal epithelial cells. For the expression of our 377 genes, differentiated HT29 cells and human colonic tissues do not appear to be significantly different (rs = 0.75).
Up to now, the Caco-2 and HT29 cell lines have proved to be the best models for studies of intestinal absorption and toxicity of xenobiotics. Nevertheless, studies have shown that several proteins, including transporters, are over- or underexpressed in those cell lines, in comparison with human intestinal tissues (Calcagno et al., 2006; Seithel et al., 2006; Hilgendorf et al., 2007; Lenaerts et al., 2007). This fact needs to be considered by scientists using these cell lines to prevent misinterpretation of findings obtained in vitro that are being translated to the in vivo situation. Our results showed that the best agreement between human tissue and the representative cell line was observed for human colonic tissues and HT29 and T84 cells for expression of 206 XMEs, 102 transporters, 48 nuclear receptors and transcription factors, and 21 miscellaneous genes. In addition, the large gap observed in the PCA between the HT29 cluster and the Caco-2 cluster suggests that the latter cell line seems not to be the best in vitro model for toxicological and pharmacological studies.
In conclusion, this study was designed to compare expression profiles of widely used intestinal cell models with those of their in vivo counterparts. PCA and hierarchical clustering analysis revealed that gene expression of intestinal biopsy samples differed considerably from that of intestinal epithelial cell lines. Expression profiles of colon cell lines will be useful for choosing the appropriate model system for toxicological and pharmacological studies.
Authorship Contributions
Participated in research design: Lo-Guidice and Broly.
Conducted experiments: Bourgine, Billaut-Laden, and Happillon.
Contributed new reagents or analytic tools: Lo-Guidice, Maunoury, and Broly.
Performed data analysis: Bourgine and Billaut-Laden.
Wrote or contributed to the writing of the manuscript: Bourgine, Billaut-Laden, Lo-Guidice, and Imbenotte.
Acknowledgments
We thank Dr. Julie Leclerc for scientific and technical contribution. We are grateful to Dr. Bruno Lefebvre for final proofreading assistance.
Footnotes
This work was supported by the Centre Hospitalier Régional et Universitaire de Lille; Université de Lille 2; Fonds Européen de Développement Régional; Institut de Recherche en Environnement Industriel; and Conseil Régional du Nord-Pas de Calais.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.ABBREVIATIONS:
- XME
- xenobiotic-metabolizing enzyme
- P450
- cytochrome P450
- SLC
- solute carrier
- ABC
- ATP-binding cassette
- NR
- nuclear receptor
- DMEM
- Dulbecco's modified Eagle's medium
- PCR
- polymerase chain reaction
- TLDA
- TaqMan low-density array
- PCA
- principal component analysis
- PC
- principal component
- RT
- reverse transcription.

- Received August 19, 2011.
- Accepted January 4, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}