


Abstract
Dabigatran etexilate (DABE) is an oral prodrug that is rapidly converted to the active thrombin inhibitor, dabigatran (DAB), by serine esterases. The aims of the present study were to investigate the in vitro kinetics and pathway of DABE hydrolysis by human carboxylesterase enzymes, and the effect of alcohol on these transformations. The kinetics of DABE hydrolysis in two human recombinant carboxylesterase enzymes (CES1 and CES2) and in human intestinal microsomes and human liver S9 fractions were determined. The effects of alcohol (a known CES1 inhibitor) on the formation of DABE metabolites in carboxylesterase enzymes and human liver S9 fractions were also examined. The inhibitory effect of bis(4-nitrophenyl) phosphate on the carboxylesterase-mediated metabolism of DABE and the effect of alcohol on the hydrolysis of a classic carboxylesterase substrate (cocaine) were studied to validate the in vitro model. The ethyl ester of DABE was hydrolyzed exclusively by CES1 to M1 (Km 24.9 ± 2.9 μM, Vmax 676 ± 26 pmol/min per milligram protein) and the carbamate ester of DABE was exclusively hydrolyzed by CES2 to M2 (Km 5.5 ± 0.8 μM; Vmax 71.1 ± 2.4 pmol/min per milligram protein). Sequential hydrolysis of DABE in human intestinal microsomes followed by hydrolysis in human liver S9 fractions resulted in complete conversion to DAB. These results suggest that after oral administration of DABE to humans, DABE is hydrolyzed by intestinal CES2 to the intermediate M2 metabolite followed by hydrolysis of M2 to DAB in the liver by CES1. Carboxylesterase-mediated hydrolysis of DABE was not inhibited by alcohol.
Introduction
The direct thrombin inhibitor dabigatran etexilate (DABE) is a new oral anticoagulant approved in the United States to prevent stroke and systemic embolism in patients with nonvalvular atrial fibrillation (Connolly et al., 2009). Dabigatran (DAB), the active moiety, is not orally bioavailable, so it is administered as a double prodrug (DABE) to improve absorption. The DABE prodrug is quickly converted to the active compound because DAB plasma concentrations peak rapidly after an oral dose and the two primary intermediate metabolites, as well as the DABE plasma concentrations, are very low (Stangier et al., 2007, 2008). This finding indicates that DABE undergoes a high first-pass metabolism prior to reaching the systemic circulation.
The conversion of DABE to DAB is a two-step process involving hydrolysis of an ethyl ester and a carbamate ester producing the active moiety (Blech et al., 2008). Experiments conducted in vitro using human liver microsomes confirm that enzymatic hydrolysis by serine hydrolases is the primary pathway for the formation of DAB from DABE and that the cytochrome P450 system plays no significant role in forming the active metabolite (Blech et al., 2008). Although the specific esterases involved in DABE metabolism have not been identified, the structure of DABE would suggest that human carboxylesterase-1 (CES1) and human carboxylesterase-2 (CES2) are likely to play a role in the formation of the DAB active metabolite (Satoh et al., 2002; Imai et al., 2006). Furthermore, based on the known substrate specificity of these enzymes, it would be predicted that CES1 hydrolyzes the DABE ethyl ester, whereas CES2 would metabolize the carbamate ester (Imai, 2006; Hu et al., 2013).
However, the specific hydrolytic pathways and the relative contributions of CES1 and CES2 to the formation of the DAB active metabolite have not been studied. Identification of the specific in vivo metabolic pathway is essential for understanding potential factors affecting the safety and efficacy of DABE, given the concentration-dependent anticoagulant effect of the DAB active metabolite and the correlation between DAB plasma concentrations and risk of stroke and bleeding (Stangier et al., 2007, 2008; Harper et al., 2012, Reilly et al., 2013). A growing body of evidence suggests that a number of factors can affect the catalytic activity of CES1 and CES2 and result in changes in drug disposition (Laizure et al., 2013). One such factor is drug interactions that inhibit carboxylesterase function (Parker and Laizure, 2010; Zhu et al., 2010; Rhoades et al., 2012). It is well established that alcohol is an inhibitor of carboxylesterase-mediated cocaine hydrolysis (Farre et al., 1997; Cami et al., 1998; Song et al., 1999; Laizure et al., 2003; Parker and Laizure, 2010). Whether this effect of alcohol is specific for cocaine or is more broadly applicable to other CES1 substrates is uncertain. However, recent work showing that alcohol inhibits the hydrolysis of the CES1 substrate drug methylphenidate in humans suggests that the hydrolysis of other CES1 substrate drugs might also be inhibited (Patrick et al., 2007; Bell et al., 2011). Given the large number of individuals who consume alcohol and its known effects on CES1 hydrolysis, understanding the effect of alcohol on carboxylesterase-mediated DABE hydrolysis could have important implications for the safety and efficacy of this agent.
Therefore, the objectives of this study are to characterize the human carboxylesterase-mediated DABE metabolic pathway and to determine the effect of alcohol on carboxylesterase-mediated hydrolysis of DABE.
Materials and Methods
DABE was purchased from TLC PharmaChem Inc. (Mississauga, ON, Canada). DAB and dabigatran-d3 (DAB-d3) were the products of Toronto Research Chemicals Inc. (North York, ON, Canada). Bis(4-nitrophenyl)phosphate (BNPP), fluorescein diacetate, cocaine (COC), benzoylecgonine (BE), and ecgonine methyl ester (EME) were purchased from Sigma-Aldrich (St. Louis, MO). Absolute alcohol (200 proof) was from Decon Laboratories (King of Prussia, PA). High-performance liquid chromatography (HPLC)–grade acetonitrile and methanol were purchased from Fisher Scientific (Pittsburgh, PA). Liquid chromatography (LC)-mass spectrometry–grade formic acid was purchased from Sigma-Aldrich (St. Louis, MO). HPLC-grade water was prepared with an in-house Milli-Q Advantage A10 Ultrapure water purification system (Bedford, MA). Recombinant human carboxylesterase 1b (named as CES1 hereafter) and 2 (CES2) are from baculovirus transfected insect cells (BD Supersomes); human liver S9 (HLS9) fractions (150 pooled donors of mixed sex) and human intestinal microsomes (HIMs, 7 pooled donors of mixed sex) were obtained from BD Gentest (San Jose, CA). Human liver cytosol has significant CES1 and CES2 activity; therefore, HLS9, which contains both cytosolic and microsomal enzymes, was used for in vitro metabolism studies instead of microsomes (Takahashi et al., 2009).
In Vitro Metabolic Stability.
The metabolic stability of DABE in incubations containing recombinant human CES1, CES2, CES1/CES2 mixture, and HLS9 were performed. Assays were conducted in duplicate (triplicate for HLS9) in 96-well cluster tubes with a total assay volume of 100 μl in each well (at 37°C). The assay buffer was 0.1 M potassium phosphate, pH 7.4. Incubation times were 0, 5, 15, 30, and 60 minutes. Final protein concentrations were 0.025, 0.025, 0.025/0.025, and 0.25 mg/ml for CES1, CES2, CES1/CES2 mixture, and HLS9, respectively. Substrate concentrations in the incubations were 200 nM, which were similar to the human plasma concentrations of DAB (Stangier, 2008). The final acetonitrile concentration was not greater than 1% for all assays. Assays were initiated by adding the substrate/buffer mix to the enzyme/buffer mix. The metabolic depletion of fluorescein diacetate by each carboxylesterase enzyme was tested as a positive control to validate the enzyme activity (data not shown). Chemical stability of substrate in assay buffer was examined as the negative control. The reactions were terminated by the addition of an equal volume of ice-cold acetonitrile containing 200 nM internal standard (DAB-d3). After centrifugation at 16,000g for 5 minutes, 10 µl supernatant was injected into the liquid chromatography/triple quadrupole mass spectrometry (LC-MS/MS) instrument.
The sequential metabolism of DABE in HIMs (step 1) and HLS9 fractions (step 2) was also conducted. The incubation volume was 100 μl for each well in the first step. Three independent incubations were prepared for 0-, 5-, 15-, 30-, and 60-minute incubations (step 1). For the 60-minute incubations (step 1), 15 independent incubation samples were performed. The incubations in three of these samples were terminated after 60 minutes. For the remaining 12 samples (step 2), an equal volume of HLS9 (100 μl) was added and further incubated for 5, 15, 30, or 60 minutes (three samples for each time point). The final protein concentration was 0.25 mg/ml for HIMs in the first step, whereas the concentration was 0.50 mg/ml for HLS9 fractions in the second step. All of the reactions were terminated by the addition of an equal volume of ice-cold acetonitrile and then analyzed by LC-MS/MS.
The stability of DABE (1000 nM) in human plasma was tested with and without BNPP (400 µM), a known specific carboxylesterase inhibitor. The experimental procedure was similar to that of the stability studies with human carboxylesterases except the final acetonitrile concentration was 1% for all assays. The reaction was terminated by the addition of a 3-fold volume of ice-cold acetonitrile.
In Vitro Enzyme Kinetics.
To determine enzyme kinetics (Km, Vmax, and CLint), DABE was incubated with recombinant CES1, CES2, HLS9, and HIM under linear metabolite formation conditions. The experimental procedure was similar to that of the metabolic stability studies except the final substrate concentrations were 0.1, 0.2, 0.5, 1, 5, 10, 50, and 100 μM in the incubation system. The final acetonitrile concentration was 1% for all assays. The optimal incubation time was 5 minutes. The final protein concentration was 0.25 mg/ml for recombinant CES1, CES2, HLS9, and HIM. All reactions were run in triplicate.
In Vitro Inhibition.
Procedures to determine the effect of alcohol on the carboxylesterase-mediated hydrolysis of DABE were similar to the enzyme kinetics study. However, DABE was first mixed with alcohol at increasing concentrations (0, 12.5, 25, 50, and 100 mM) in the incubation tubes and then recombinant enzyme or HLS9 was added. The alcohol concentration was selected based on the reported human exposure levels (Umulis et al., 2005). Inhibition was tested under two conditions to determine the effect of alcohol on the formation of the intermediate M1 and M2 metabolites (condition A) and DAB (condition B). For condition A (low DABE depletion), the incubation time was 5 minutes and the protein concentrations in the incubations were 0.01, 0.025, and 0.025 mg/ml for CES1, CES2, and HLS9, respectively. For condition B (high DABE depletion), the incubation time was 10 and 5 minutes for the carboxylesterase enzymes and HLS9 fractions, respectively. The protein concentrations were 0.025 mg/ml for the carboxylesterase enzymes and 1.0 mg/ml for HLS9 fractions. The inhibitory effect of BNPP on the carboxylesterase-mediated metabolism of DABE was tested as a positive control. The concentrations of BNPP were 0, 1, 5, 25, and 125 μM.
The effects of alcohol on cocaine hydrolysis were determined to validate the in vitro model for alcohol-mediated inhibition of carboxylesterase activity. Cocaine metabolic stability when incubated with recombinant human CES1 and CES2 was evaluated for determination of linear metabolite formation conditions with respect to the incubation time. Recombinant CES1 and CES2 protein and cocaine concentrations in the incubation were 0.25 mg/ml and 10 µM, respectively. Incubation times were 0, 5, 15, 30, and 60 minutes. On the basis of the metabolic stability results, a 60-minute incubation time was used in the alcohol inhibition study. Final alcohol concentrations were 0, 12.5, 25, 50, 100, and 200 mM.
LC-MS/MS Analyses.
LC-MS/MS–based assays were used to measure the substrates and their metabolites. The LC-MS/MS system consisted of a Shimadzu HPLC separation module (Milford, MA) and an AB Sciex 3000 triple quadrupole mass spectrometer (Toronto, Canada) with a turbo ion spray (electrospray ionization) source. The LC separation for all of the analytes was achieved on a 5.0-μm Agilent Eclipse Plus C18 column (50 mm × 2.1 mm inside diameter; Santa Clara, CA) at 24°C. Mobile phases were as follows: methanol/water, 1:99 (v/v), containing 2.5 mM formic acid, for phase A; and methanol/water, 99:1 (v/v), modified with the same electrolyte, for phase B. A fast gradient chromatographic method was used, which we have employed successfully for the sensitive analysis of other drugs (Hu et al., 2011). This method is as follows: B 5% from 0.00 to 0.50 minutes, B 100% from 0.51 to 2.00 minutes, and B 5% from 2.01 to 4.50 minutes.
The precursor-product ion pairs (singly protonated species) used for multiple reaction monitoring of DABE, DAB, DAB-d3, COC, BE, EME, and CE were m/z 628.3→289.1, 472.2→289.1, 475.3→292.2, 304.3→182.1, 290.3→168.1, 200.3→182.1, and 318.3→196.1, respectively. The LC eluent was introduced to the electrospray ionization source at a flow rate of 0.40 ml/min over the period of 0.3–2.2 minutes. One internal standard, DAB-d3, was used for quantification of all of the analytes. Matrix-matched standard curves of the analyte/internal standard peak area ratio of a given analyte versus the nominal concentration in nanomoles were linear with correlation coefficients >0.99. The lower limit of quantification was 1.37 nM for all of the analytes except for EME, which was 12.3 nM. The within-run and between-run assay accuracies ranged from 93% to 109% and from 95% to 108%, respectively, whereas the ranges of precision values for the assays were from 1.8% to 12.5% and from 1.5% to 14.4%, respectively. The two intermediate metabolites (M1 and M2) in the study samples were quantified by our recently developed assay (Hu et al., 2013).
Data Analysis.
Michaelis constant (Km) and maximum velocity (Vmax) values were determined by nonlinear regression analysis of rates of metabolite formation as a function of substrate concentration using GraphPad Prism software (version 5.0; GraphPad Software Inc., San Diego, CA). In vitro intrinsic clearance (CLint) was calculated from the ratio of Vmax to Km. All data presented in the figures are the mean ± standard deviation.
Results
In Vitro Metabolic Stability.
To identify the specific enzymes responsible for DABE hydrolysis, separate incubations using recombinant CES1 and CES2 were conducted. Incubations using a mixture of recombinant CES1 and CES2 were also performed to assess the combined effect of these enzymes. The results of these experiments are summarized in Fig. 1 and show that CES1 converts DABE to the intermediate metabolite M1, whereas CES2 mediates the formation of intermediate metabolite M2. Furthermore, only a small quantity of the DAB active metabolite is formed in individual CES1 or CES2 incubations (Fig. 1). In contrast, the formation of DAB in incubations containing both CES1 and CES2 was approximately 4- and 12-fold higher compared with CES1 or CES2 alone, respectively. The metabolic profile of DABE in HLS9 fractions is shown in Fig. 2. Both M1 (major form) and M2 (minor form) were formed in HLS9 fractions. A moderate amount of DAB was also formed (Fig. 2).

DABE (200 nM) metabolite formation in recombinant CES1, CES2, and CES1/CES2 mixture (60-minute incubation).
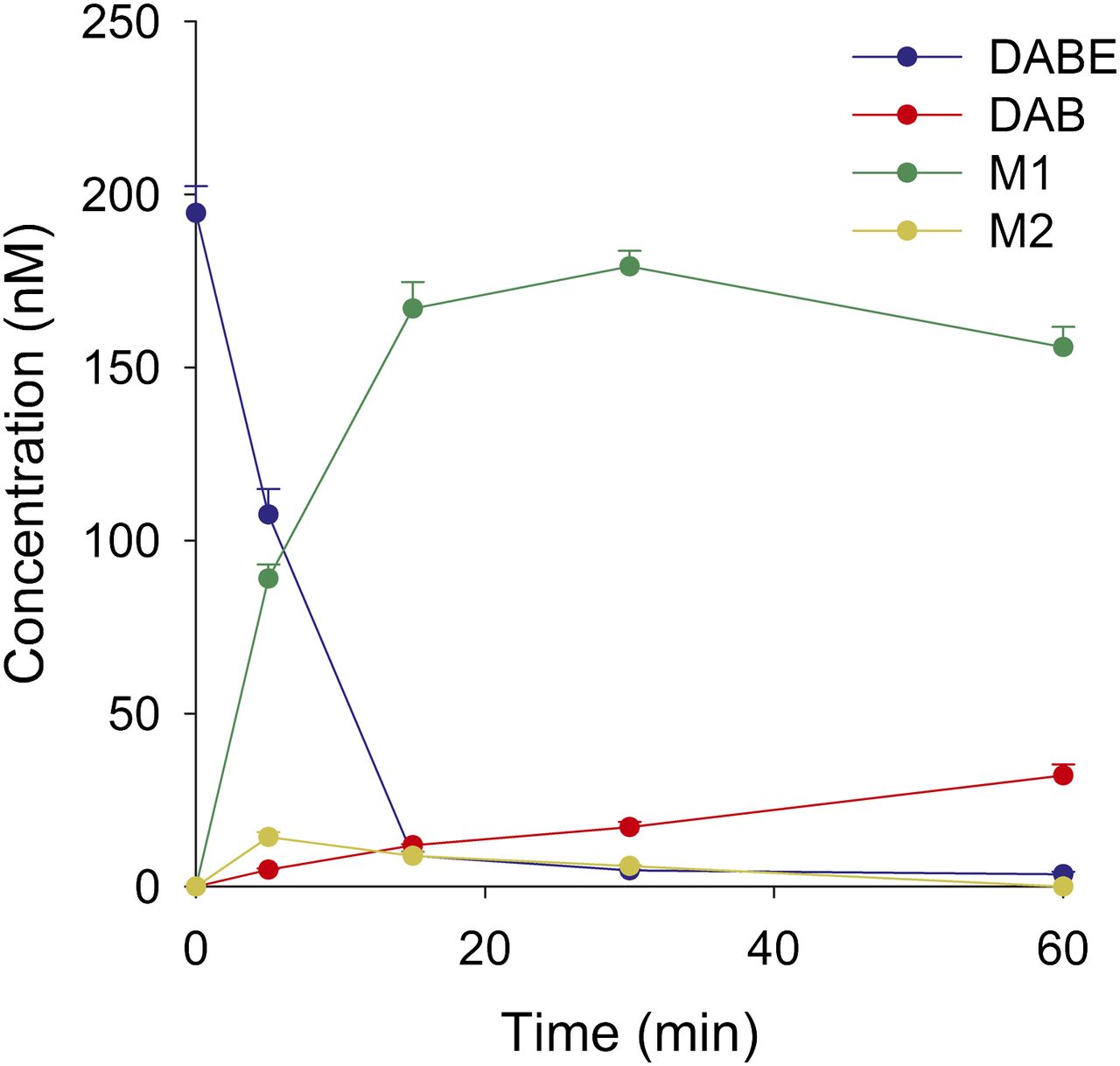
In vitro hydrolysis of DABE in HLS9.
The sequential hydrolysis of DABE in HIMs and HLS9 fractions is shown in Fig. 3. The metabolic depletion of DABE in HIMs showed that M2 was the major metabolite and only a small quantity of DAB was formed (Fig. 3A, step 1). After addition of HLS9 fractions, M2 was rapidly and completely hydrolyzed to DAB (Fig. 3B, step 2).

Sequential hydrolysis of DABE (200 nM) in HIMs (A) (step 1) and HLS9 fractions (B) (step 2). Because the incubations for step 2 (B) were diluted after the addition of HLS9, the resulting concentration of DABE and its metabolites in (B) are normalized to 200 nM.
The stability study of DABE in human plasma showed that less than 25% of DABE was converted to M1 after a 60-minute incubation (the amounts of M2 and DAB formed were very low; data shown in Supplemental Figure 1). The addition of the carboxylesterase inhibitor BNPP did not affect this process, suggesting that the slow hydrolysis of DABE in human plasma was spontaneous or mediated by other enzymes.
In Vitro Enzyme Kinetics.
The enzyme kinetics results are shown in Table 1 and Supplemental Figure 2. The CLint values for the formation of M1 in CES1 and M2 in CES2 were 27.2 and 12.9 µl/min per milligram protein, respectively. In contrast, CLint values were ≤0.3 µl/min per milligram protein for formation of M2 in CES1 and M1 in CES2. Although the Vmax for the formation of M1 by CES1 was 9.5-fold higher than the formation of M2 by CES2, the Km for the latter conversion was much lower (5.5 µM) than that of M1 formation (24.9 µM). The Km value for M1 formation in HLS9 fractions was comparable with that in the recombinant CES1 preparation (33.5 µM versus 24.9 µM). In addition, the kinetics for the formation of M2 was comparable between HIMs and recombinant CES2 (Km 8.6 µM versus 5.5 µM). The CLint for M2 formation in HLS9 fractions was much less than that for the formation of M1 in HLS9 fractions (2.0 µl/min per milligram protein versus 35.0 µl/min per milligram protein).
Enzyme kinetic parameters for DABE hydrolysis in recombinant CES1, CES2, pooled HLS9 fractions, and pooled HIMs
All reactions were run in triplicate and results are presented as mean ± S.D.
In Vitro Alcohol Inhibition.
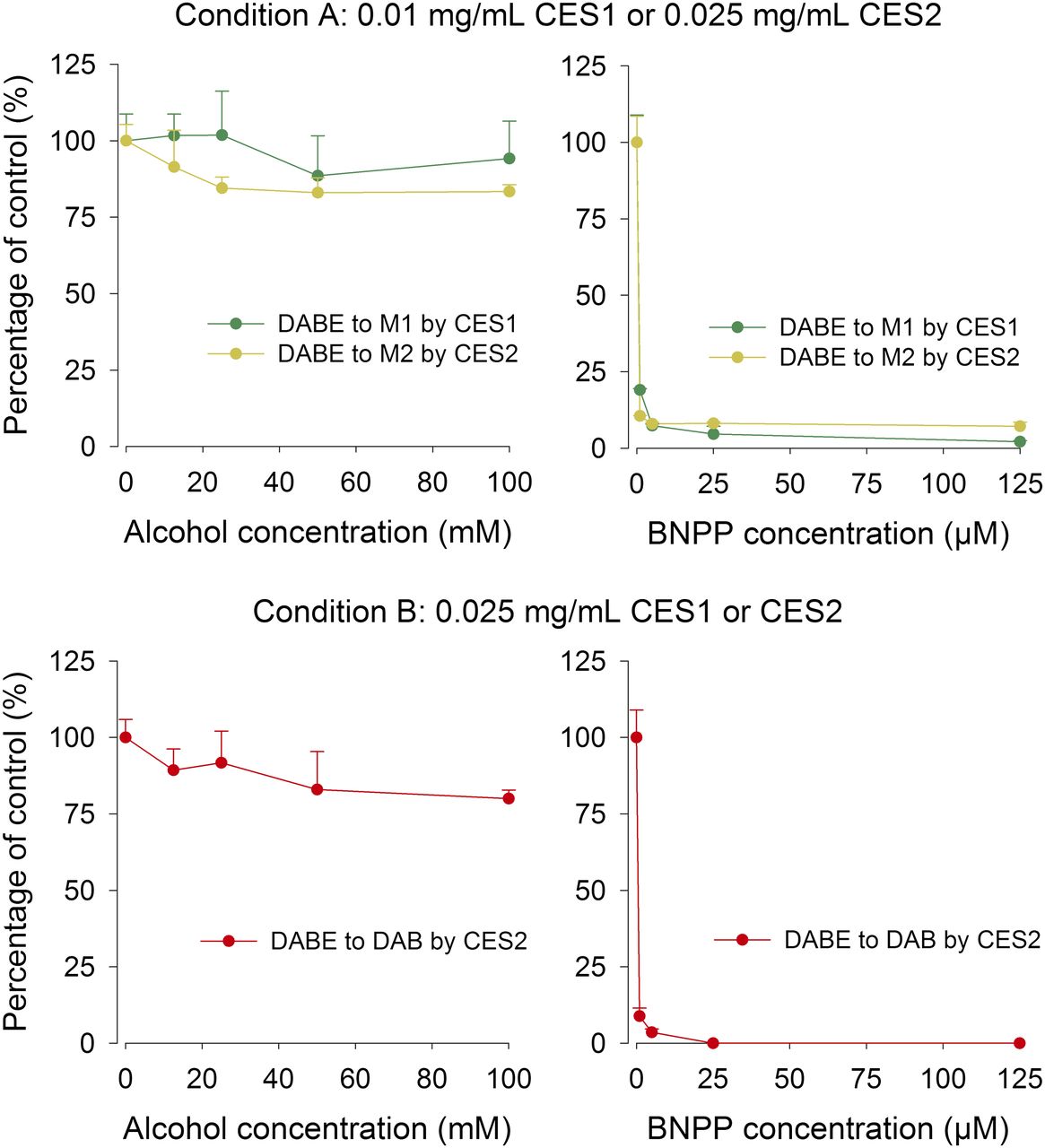
The effects of alcohol and BNPP on the hydrolysis of DABE by recombinant CES1 and CES2 are shown in Fig. 4. Alcohol showed no significant inhibitory effect on the hydrolysis of DABE in CES1 or CES2. However, carboxylesterase-mediated hydrolysis of DABE was almost completely inhibited by BNPP (Fig. 4). The effects of alcohol and BNPP on the hydrolysis of DABE in HLS9 fractions are shown in Fig. 5. The results are similar to those observed with the recombinant carboxylesterase enzymes. DAB can be quantified in both conditions A and B using HLS9.
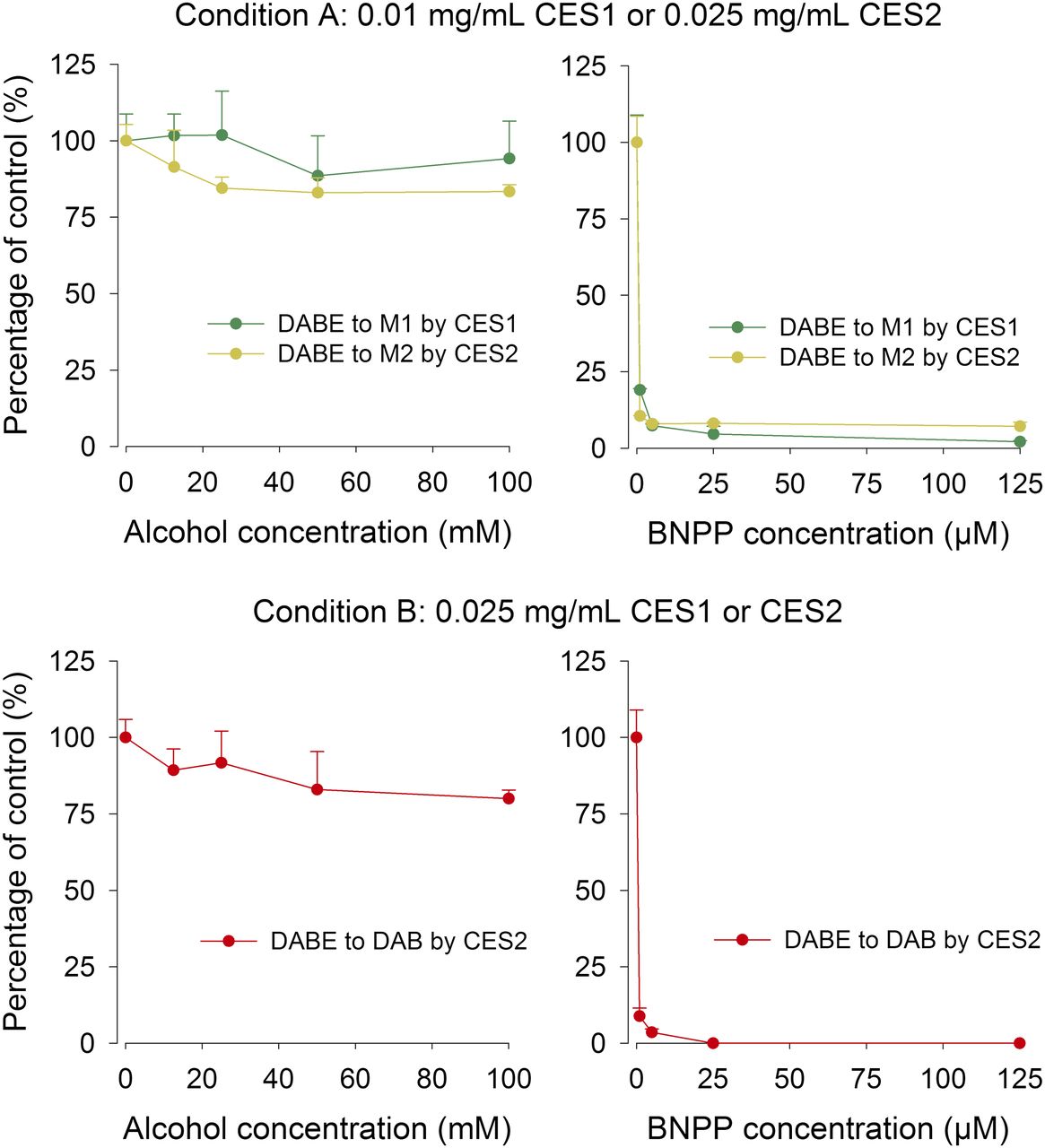
The effect of alcohol (left) and BNPP (right) on the carboxylesterase-mediated metabolism of DABE in recombinant CES1 or CES2 enzymes. The concentrations of the resulting metabolites without inhibitors were set as 100%. Condition A (5-minute incubation, low DABE depletion) and condition B (10-minute incubation, high DABE depletion) were used to test the effect of alcohol on the formation of intermediate metabolites (M1 and M2) and the final metabolite (DAB), respectively. The concentration of DAB in the incubation with CES2 was too low to be detected under condition B.

The effect of alcohol (left) and BNPP (right) on the carboxylesterase-mediated metabolism of DABE in HLS9. The concentrations of the resulting metabolites without inhibitors were set as 100%. Conditions A (5-minute incubation, low DABE depletion) and condition B (5-minute incubation, high DABE depletion) were used to test the effect of alcohol on the formation of intermediate metabolites (M1 and M2) and the final metabolite (DAB), respectively.
Alcohol-mediated inhibition of cocaine metabolism was used to validate the in vitro carboxylesterase substrate–alcohol interaction model. Only CES1 converted cocaine to BE (Supplemental Figure 3A) and the addition of alcohol resulted in the formation of the transesterification product cocaethylene (data not shown). In contrast, both CES1 and CES2 enzymes catalyzed the hydrolysis of COC to EME (Supplemental Figure 3A). Alcohol significantly inhibited the hydrolysis of COC to BE and EME (Supplemental Figure 3B).
Discussion
The primary findings of this study are that CES1 catalyzes the hydrolysis of DABE to M1 and CES2 catalyzes the hydrolysis of DABE to M2. There was little cross-reactivity of carboxylesterase hydrolysis, suggesting that both enzymes are required for the formation of the active metabolite, DAB. We also found that alcohol did not inhibit carboxylesterase-mediated DABE hydrolysis by CES1 or CES2.
The formation of DABE hydrolysis products shown in Table 1 and Fig. 1 is consistent with the reported substrate specificities of CES1 and CES2 with CES1 hydrolyzing DABE to the M1 metabolite (ester hydrolysis) and CES2 hydrolyzing DABE to the M2 metabolite (carbamate hydrolysis) (Imai, 2006). Carboxylesterases are considered relatively substrate nonspecific, so it is common for a hydrolysis site to be susceptible to both CES1 and CES2 (although usually one predominates). However, as Table 1 shows, recombinant CES1 and CES2 only produce M1 and M2, respectively, and fail to produce the alternate metabolite. It is also apparent from Table 1 that the inability of CES1 and CES2 to hydrolyze the carbamate and ester groups, respectively, also applies to the M2 and M1 metabolites because the individual incubations conducted with recombinant CES1 and CES2 produce only small quantities of the DAB active metabolite.
Both M1 and M2 metabolites were formed in HLS9 fractions (Fig. 2). The metabolite concentration pattern was as expected since CES1 accounts for 80%–95% of hydrolase activity in the liver (Imai et al., 2006). Thus, the high M1 concentrations are consistent with the high levels of hepatic CES1 activity. Similarly, since M2 formation is dependent on CES2 hydrolysis, the low M2 concentrations reflect the low level of hepatic CES2 expression. The incubation of DABE in HLS9 fractions also shows a low amount of DAB active metabolite formation. Compared with oral administration of DABE to humans, in vitro incubations in HLS9 fractions cannot account for the significant prehepatic formation of M2 from DABE by CES2 in the intestine (Imai et al., 2006; Imai and Ohura, 2010). This intestinal M2 formation is supported by sequential hydrolysis experiments showing formation of the M2 intermediate metabolite in HIMs followed by the subsequent complete conversion to DAB in HLS9 fractions (Fig. 3).
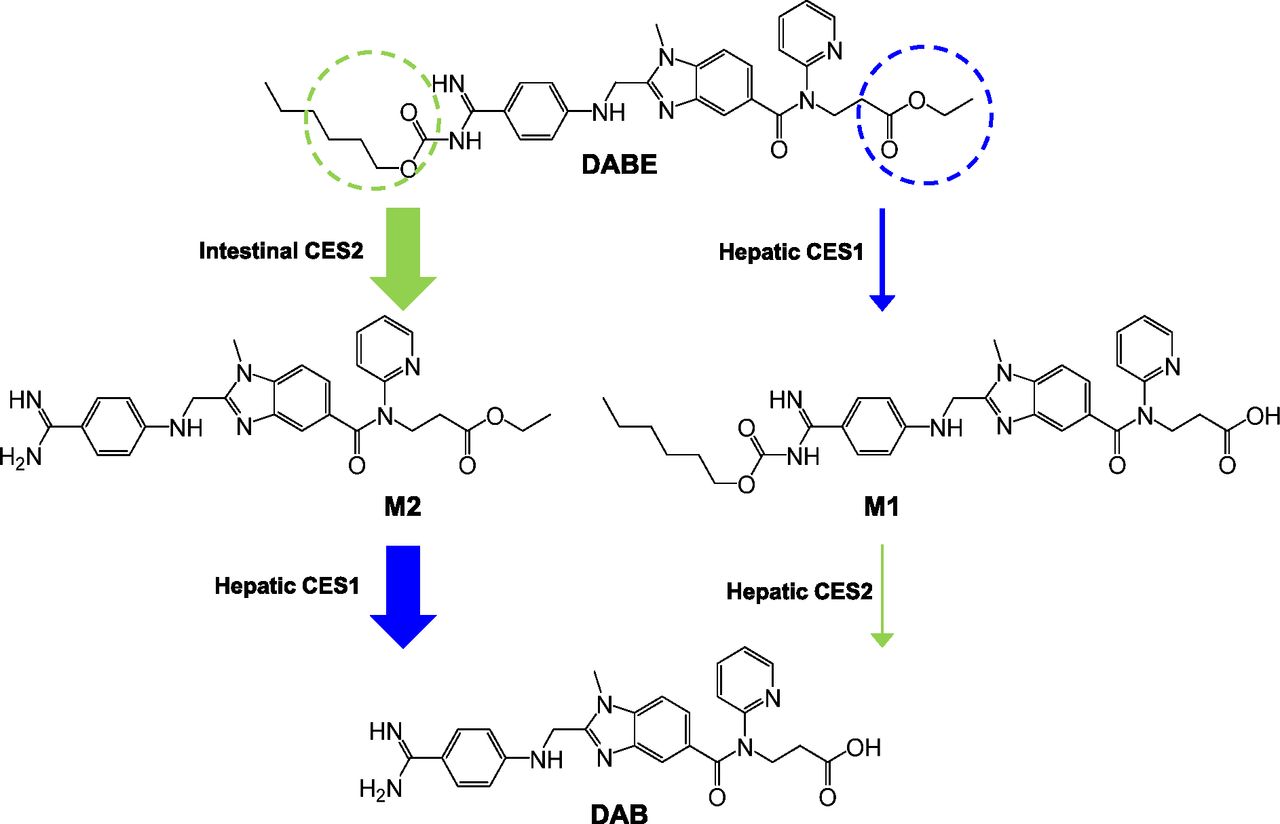
Collectively, these experiments demonstrate that hydrolysis of the DABE double prodrug by both CES1 (ethyl ester) and CES2 (carbamate ester) is required for formation of the DAB active moiety. When this drug is given to humans, DAB is rapidly formed with peak concentrations occurring within 2 hours after an oral dose, with negligible plasma concentrations of M1, M2, and DABE (Blech et al., 2008; Stangier et al., 2008). This indicates that the hydrolysis of both the ethyl and carbamate ester sites undergo high first-pass metabolism prior to reaching the systemic circulation. On the basis of these findings, a proposed metabolic pathway for DABE after oral administration to humans is shown in Fig. 6. Our data suggest that DABE undergoes extensive presystemic conversion to M2 by intestinal CES2. The M2 metabolite is then subject to further first-pass hydrolysis by hepatic CES1, resulting in formation of the DAB active metabolite. This proposed metabolic pathway is supported by the sequential hydrolysis of DABE by HIMs and HLS9 fractions shown in Fig. 3. In addition, this proposed metabolic pathway is consistent with the anatomic locations of CES1 (liver) and CES2 (intestine) expression (Satoh et al., 2002; Imai et al., 2006), the specific susceptibility of the ethyl and carbamate esters to hydrolysis by CES1 and CES2 (Table 1), and the disposition of DABE demonstrated in human studies (Blech et al., 2008; Stangier et al., 2008). If there were large first-pass formation of the M1 metabolite via CES1-mediated ester hydrolysis, then high plasma concentrations of M1 would be expected, because this metabolite would not be subsequently exposed to the high CES2 activity in the intestines. In this case, DAB formation would depend on the relatively low levels of hepatic CES2. Thus, as shown in Fig. 6, we propose that the conversion of DABE to M1 to DAB represents a minor metabolic pathway with the primary formation of the DAB active metabolite occurring via CES2-mediated hydrolysis of DABE to M2 that is subsequently hydrolyzed by hepatic CES1 to DAB.
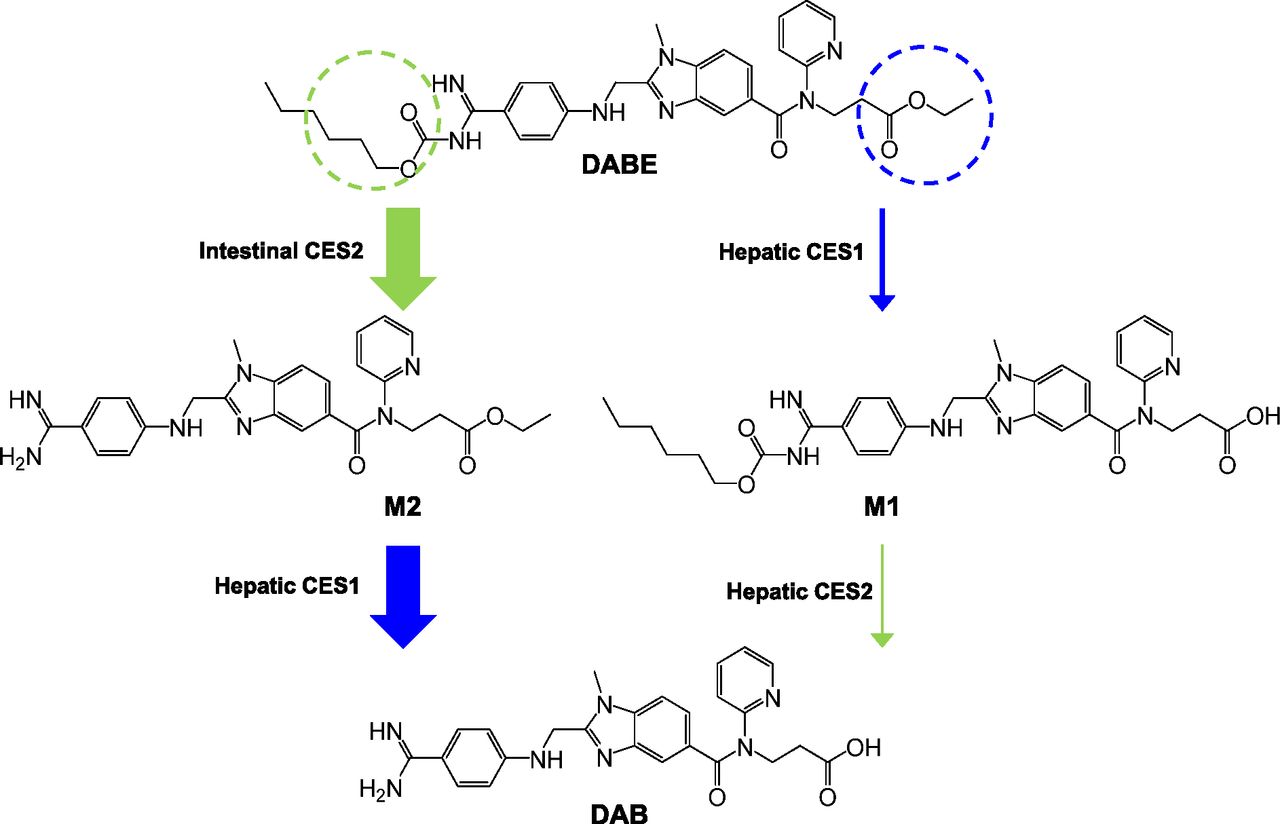
Proposed in vivo metabolic pathway of orally administered DABE in humans. The thickness of each arrow indicates the relative contribution of each transformation to the hydrolysis of DABE to DAB. Circles indicate the hydrolysis sites by CES1 (blue) and CES2 (green).
Alcohol has been shown to inhibit cocaine hydrolysis catalyzed by CES1 and CES2 (Roberts et al., 1993; Song et al., 1999) and the hydrolysis of methylphenidate (Bourland et al., 1997) and clopidogrel (Tang et al., 2006) (all by CES1) in microsomes or HLS9 fractions. In human studies, cocaine and methylphenidate hydrolysis are significantly inhibited by the consumption of alcohol and the transesterification products cocaethylene and ethylphenidate are produced (Farre et al., 1997; Cami et al., 1998; Patrick et al., 2007). These studies show that the inhibition of carboxylesterase hydrolysis and the formation of transesterified metabolites are not unique to the cocaine-alcohol interaction and may occur with other CES1 substrates. However, unlike cocaine, dabigatran hydrolysis was not affected by alcohol and no transesterified product was formed (see Figs. 4 and 5). Although BNPP significantly inhibited the hydrolysis of DABE by CES1 and CES2, even alcohol concentrations up to 100 mM did not significantly affect the hydrolysis of DABE by CES1 or CES2. Thus, although past studies clearly demonstrate that the inhibition of CES1 by alcohol occurs with some CES1 substrates, the lack of effect on DABE hydrolysis indicates that the inhibitory effects of alcohol on CES1 cannot be generalized to all CES1 substrates. The potential mechanism(s) for the inability of alcohol to inhibit DABE hydrolysis are unclear. Although speculative, the lack of formation of transesterified metabolites and the absence of alcohol-mediated inhibition of DABE hydrolysis may be linked. Other CES1 substrate drugs that are inhibited by alcohol also undergo transesterification, including cocaine, methylphenidate, clopidogrel, and meperidine (Bourland et al., 1997; Farre et al., 1997; Song et al., 1999; Tang et al., 2006; Patrick et al., 2007). In addition, the molecular weight of DABE is 2- to 3-fold higher than these other CES1 substrate drugs that are susceptible to inhibition/transesterification with alcohol. This could potentially affect the conformational orientation or access to the CES1 active site pocket (Imai et al., 2006).
DABE is a unique prodrug requiring hydrolysis at two sites to form the active direct thrombin inhibitor. A recent analysis demonstrating that both stroke and bleeding risk in patients with atrial fibrillation are directly linked to DAB plasma concentrations suggests that understanding DABE’s metabolic pathway and factors affecting it are crucial for assessing benefits and risks of therapy (Reilly et al., 2013). We attempt to address this issue in this report with our results showing that both CES1 and CES2 are essential for DAB active metabolite formation. Characterizing this drug’s metabolic pathway is a crucial first step in identifying how factors affecting the activity of CES1 and CES2 may have important effects on this drug’s disposition, and in turn, efficacy and safety. The common assumption applied to DABE and other carboxylesterase substrate drugs is that these agents are not subject to significant variation in disposition. However, a growing body of evidence indicates that these enzymes may be affected by numerous factors including drug–drug interactions and genetic variability in activity (Laizure et al., 2013). Further investigation is warranted to understand the relationship between factors affecting DAB formation and therapeutic response and toxicity.
Authorship Contributions
Participated in research design: Laizure, Parker, Herring, Hu.
Conducted experiments: Hu.
Performed data analysis: Hu.
Wrote or contributed to the writing of the manuscript: Laizure, Parker, Herring, Hu.
Footnotes
- Received August 20, 2013.
- Accepted November 8, 2013.
This research was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant R15-GM096074].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- BE
- benzoylecgonine
- BNPP
- bis(4-nitrophenyl) phosphate
- CE
- cocaethylene
- CES1
- human carboxylesterase 1
- CES2
- human carboxylesterase 2
- CLint
- in vitro intrinsic clearance
- COC
- cocaine
- DAB
- dabigatran
- DAB-d3
- dabigatran-d3
- DABE
- dabigatran etexilate
- EME
- ecgonine methyl ester
- HIM
- human intestinal microsome
- HLS9
- human liver S9
- HPLC
- high-performance liquid chromatography
- LC
- liquid chromatography
- LC-MS/MS
- liquid chromatography/triple quadrupole mass spectrometry
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}