


Abstract
GDC-0834, a Bruton’s tyrosine kinase inhibitor investigated as a potential treatment of rheumatoid arthritis, was previously reported to be extensively metabolized by amide hydrolysis such that no measurable levels of this compound were detected in human circulation after oral administration. In vitro studies in human liver cytosol determined that GDC-0834 (R)-N-(3-(6-(4-(1,4-dimethyl-3-oxopiperazin-2-yl)phenylamino)-4-methyl-5-oxo- 4,5-dihydropyrazin-2-yl)-2-methylphenyl)-4,5,6,7-tetrahydrobenzo[b] thiophene-2-carboxamide) was rapidly hydrolyzed with a CLint of 0.511 ml/min per milligram of protein. Aldehyde oxidase (AO) and carboxylesterase (CES) were putatively identified as the enzymes responsible after cytosolic fractionation and mass spectrometry-proteomics analysis of the enzymatically active fractions. Results were confirmed by a series of kinetic experiments with inhibitors of AO, CES, and xanthine oxidase (XO), which implicated AO and CES, but not XO, as mediating GDC-0834 amide hydrolysis. Further supporting the interaction between GDC-0834 and AO, GDC-0834 was shown to be a potent reversible inhibitor of six known AO substrates with IC50 values ranging from 0.86 to 1.87 μM. Additionally, in silico modeling studies suggest that GDC-0834 is capable of binding in the active site of AO with the amide bond of GDC-0834 near the molybdenum cofactor (MoCo), orientated in such a way to enable potential nucleophilic attack on the carbonyl of the amide bond by the hydroxyl of MoCo. Together, the in vitro and in silico results suggest the involvement of AO in the amide hydrolysis of GDC-0834.
Introduction
GDC-0834 is a potent, selective, and reversible ATP-competitive small-molecule inhibitor of Bruton’s tyrosine kinase (BTK) that was under consideration as a therapeutic agent for rheumatoid arthritis (Liu et al., 2011a,b). One liability for GDC-0834 was the low confidence in the human pharmacokinetic prediction based on the species-dependent metabolism, with amide hydrolysis being more predominant in humans than in other preclinical species. Despite the uncertainty in the human clearance prediction, however, there was a high level of interest in a BTK inhibitor for clinical evaluation. An investigational new drug strategy was initiated in which GDC-0834 was rapidly advanced to a single-dose human clinical trial study. Exploratory clinical studies established that, after oral administration of 35 and 105 mg GDC-0834 to healthy volunteers, limited exposure of this drug was observed in circulation (<1 ng/ml in most plasma samples). This was attributed primarily to metabolism, with most drug-related circulating material being the aniline metabolite M1 (Fig. 1). At 35 mg, the mean highest observed plasma concentration (Cmax) of M1 was 142 ng/ml; at 105 mg, the mean Cmax of M1 was 390 ng/ml (Liu et al., 2011b).
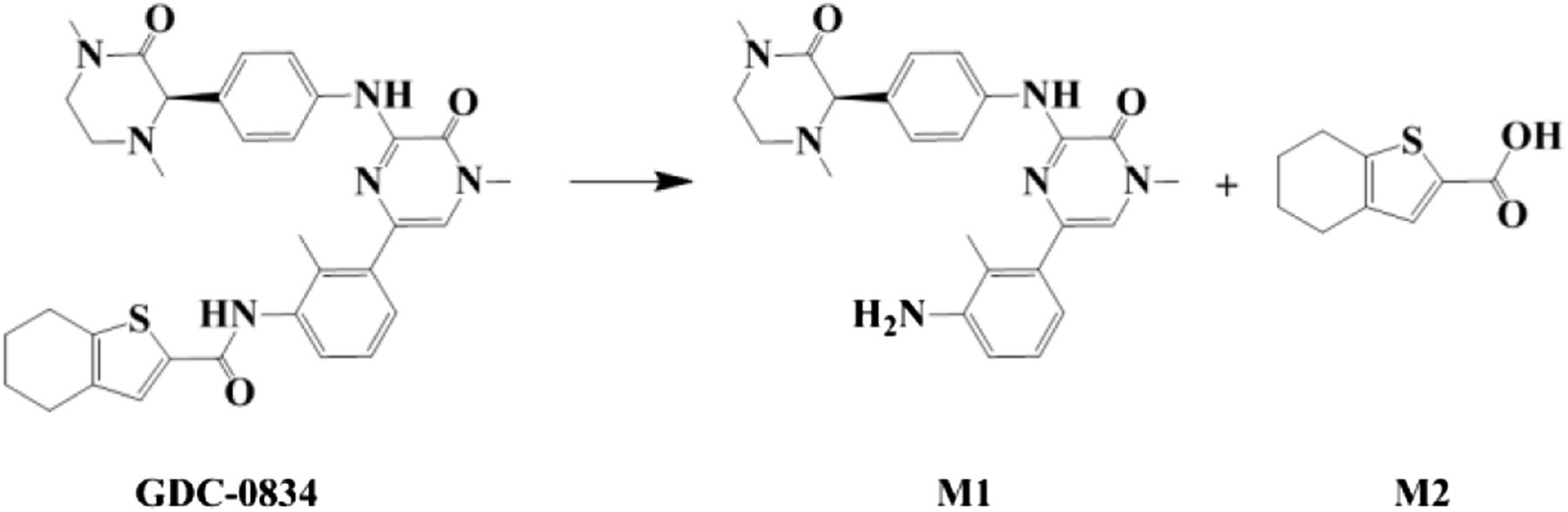
Amide hydrolysis pathway of GDC-0834 to form M1 and M2.
In contrast to humans, in preclinical species, the extensive metabolism of GDC-0834 was not evident as GDC-0834 was orally bioavailable after oral administration to mice, rats, dogs, and monkeys. In plasma of preclinical species, M1 was minor, and the percent exposure ratios of M1 area under the curve/parent area under the curve were 9.3% (severe combined immunodeficiency, or SCID, mice), 1.5% (rats), 26% (dogs), and negligible (monkeys). It is interesting that in PXB mice with humanized livers, the ratio increased to 74.1% (Liu et al., 2011b). In vitro metabolic stability studies of GDC-0834 in liver microsomes (in the presence and absence of NADPH) and hepatocytes predicted hydrolysis as the route of metabolism in humans and indicated significant differences in amide hydrolysis rates between humans and other preclinical species (Liu et al., 2011b). This specific GDC-0834 to M1 biotransformation was much more pronounced in human liver fractions than those of preclinical species.
Here we investigated the enzyme(s) involved in the amide hydrolysis of GDC-0834. Preliminary in vitro metabolism experiments using various liver fractions revealed that soluble enzyme(s) present in human liver cytosol (HLC) mediated the amide hydrolysis of GDC-0834. Therefore, HLC was chosen over human liver microsomes and hepatocytes since it contained the soluble enzyme(s) to facilitate fractionation and mass spectrometry (MS)–proteomics analysis. A series of kinetic experiments with probe substrates and chemical inhibitors of aldehyde oxidase (AO), carboxylesterase (CES), and xanthine oxidase (XO), as well as in silico modeling studies of GDC-0834, were conducted.
Materials and Methods
Chemicals.
GDC-0834 ((R)-N-(3-(6-(4-(1,4-dimethyl-3-oxopiperazin-2-yl)phenylamino)-4-methyl-5-oxo- 4,5-dihydropyrazin-2-yl)-2-methylphenyl)-4,5,6,7-tetrahydrobenzo[b] thiophene-2-carboxamide), M1, M2 (acid metabolite; Fig. 1), and an internal standard (structural analog of GDC-0834 and a proprietary compound) were synthesized at Genentech, Inc. (South San Francisco, CA). Phosphate-buffered saline (pH 7.4), potassium phosphate buffer (100 mM; pH 7.4), and 4-morpholinepropanesulfonic acid (MOPS, 2.5 mM; pH 7.4) were provided by the Media Preparation Facility at Genentech, Inc. High-performance liquid chromatography–grade solvents (acetonitrile, methanol, and water) were purchased from EMD Chemicals, Inc. (Gibbstown, NJ). Formic acid (FA) was purchased from Mallinckrodt Baker, Inc. (Phillipsburg, NJ). Allopurinol, O6-benzylguanine, bis-(p-nitrophenyl) phosphate (BNPP), 2,6-dichlorophenolindophenol (DCPIP), β-estradiol, 7-ethyl-10-hydroxycamptothecin (SN-38), irinotecan (7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycampthothecin; CPT-11), loperamide, zaleplon, and zoniporide were purchased from Sigma-Aldrich (St. Louis, MO). Raloxifene, 4-hydroxycarbazeran, 8-oxobenzylguanine, 5-oxozaleplon, and 2-oxozoniporide were purchased from Toronto Research Chemicals Inc. (North York, ON, Canada). Phthalazine was purchased from Alfa Aesar (Ward Hill, MA). Menadione and phthalazinone were purchased from Acros Organics (Geel, Belgium). Carbazeran was purchased from Chemoraga, Inc. (Oakland, CA). HLC from mixed male and female donor pools (n = 150 donors; protein content of 20 mg/ml) and dog liver cytosol (DLC) from a pool of male beagle dogs (n = 4 dogs; protein content of 20 mg/ml) were purchased from BD Biosciences (San Jose, CA) and stored at −80°C. Fresh whole blood and plasma collected from male humans, male Sprague-Dawley rats, female CD-1 mice, male beagle dogs, and male cynomologus monkeys were purchased from BioreclamationIVT (Westbury, NY). The synthesis of N-[(2-dimethylamino) ethyl]acridine-4-carboxamide (DACA) was performed as described by Barr and Jones (2013).
Protein Separation and Liquid Chromatography–Tandem Mass Spectometry Proteomic Analysis.
HLC was fractionated using an Agilent 1100 Series pump and a Varian 701 fraction collector. HLC (50 µl; 20 mg/ml) was manually injected via a 100-µl loop onto a Xenix SEC-300, 3 µm, 300 Å size-exclusion column from Sepax Technologies (Neward, DE) and separated at a flow rate of 0.5 ml/min using phosphate-buffered saline (pH 7.4) as the mobile phase. The eluent was collected as 500-µl fractions every minute for 1 hour into glass test tubes. A portion of each fraction (45 µl) was assessed for enzymatic activity by incubating with GDC-0834 (0.8 µM) for 30 minutes. Reactions were terminated using acetonitrile containing the internal standard (100 µl) and centrifuged for 10 minutes at 2000g. The supernatants (100 µl) were removed, combined with water (200 µl), and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) to quantify the relative abundance of M1 formed from GDC-0834.
MS Proteomics and Correlation Profiling.
MS proteomics and correlation profiling were used to help identify potential candidate enzymes responsible for conversion of GDC-0834 to M1. A series of seven fractions (fractions 25–31) containing measurable amide hydrolytic activities and one fraction (fraction 24) lacking activity were loaded onto a series of lanes on an SDS-PAGE 4–12% Bis/Tris gradient gel run in MOPS buffer. Proteins were partially separated and subjected to in-gel trypsin digestion, and peptides were analyzed by LC-MS/MS on an LTQ-Orbitrap XL mass spectrometer (ThermoFisher Scientific, San Jose, CA) as described previously (Phu et al., 2011; Sheng et al., 2012). Precursor ions were analyzed in high resolution (resolving power of 60K) in the Orbitrap and MS/MS spectra were collected in the ion trap using data-dependent acquisition. MS/MS were searched using Mascot (Matrix Science, London, UK) against a concatenated target-decoy database comprising forward and reverse sequence of human proteins from UniProt and common contaminants with a 50 ppm precursor ion tolerance. Peptide spectral matches were filtered to a 1% false discovery rate using linear discriminant analysis. Pearson correlations were determined for each protein using the spectral count data relative to the metabolic activity observed across the series of fractions. P values were determined for each correlation and corrected for multiple hypothesis testing by the false discovery rate (FDR)–based approach using the q-value library available through Bioconductor.
Kinetic Formation of M1 in HLC and DLC.
Enzyme kinetic studies of M1 formation were performed in HLC and DLC. Assay conditions were optimized with regard to protein concentration and incubation time, based on linear formation of M1 (data not shown). All incubations containing GDC-0834 (0.05–100 μM for humans and 1–100 μM for dogs) were incubated at 37°C in triplicate and initiated by the addition of cytosol, with final protein concentrations of 0.05 mg/ml (HLC) and 3.0 mg/ml (DLC). Optimized incubation times in HLC and DLC were 10 and 60 minutes, respectively. Incubations (100 µl) were terminated by protein precipitation with the addition of acetonitrile containing the internal standard (200 µl). All samples were centrifuged for 10 minutes at 2000g; the supernatants were removed, diluted 1:2 (v/v) with 0.1% FA in water, and analyzed by LC-MS/MS. Standard curves of the M1 standard prepared in each matrix were used, with a lower limit of quantitation 0.01 µM to quantify the amount of M1 formed. Estimations of the maximum rate of M1 formation (Vmax) and the Michaelis-Menten constant (Km) were performed using nonlinear regression analysis within GraphPad Prism (GraphPad Software Inc., La Jolla, CA) using the Michaelis-Menten equation (eq. 1):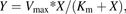 (1)where Y is enzyme velocity and X is substrate concentration.
(1)where Y is enzyme velocity and X is substrate concentration.
Formation of M1 in Fresh Blood and Plasma.
Metabolic stability studies with GDC-0834 were conducted with fresh whole blood and plasma collected from humans, rats, mice, dogs, and monkeys. Discrete incubations (100 µl) were conducted in triplicate at 37°C and initiated with the addition of GDC-0834 (final concentration, 0.8 µM). Discrete incubations corresponding to each time point were terminated at selected times with the addition of methanol (500 µl) containing the internal standard and samples prepared for LC-MS/MS analysis as described above. Standard curves of the M1 standard prepared in each matrix were used to quantify the amount of M1 formed. Samples were analyzed by LC-MS/MS for GDC-0834 and M1 formation.
Inhibition of GDC-0834 Metabolism Using Chemical Inhibitors.
Chemical inhibition studies with HLC, DLC, and human plasma were conducted using the AO inhibitors β-estradiol, DCPIP, menadione and raloxifene, the AO/CES inhibitor loperamide, the CES inhibitor BNPP, and the XO inhibitor allopurinol. The inhibitors (0–50 μM in HLC and DLC and 0–10 µM in human plasma) were coincubated with GDC-0834 (0.8 μM with 0.05 mg/ml HLC for 10 minutes, 63 μM with 3.0 mg/ml DLC for 60 minutes, or 0.8 μM in human plasma (pH 7.4) for 2 hours). At the end of the incubation period, the reactions were terminated and samples prepared for LC-MS/MS analysis as described above. Samples were analyzed by LC-MS/MS for M1 formation. Estimations of the 50% inhibitory concentration (IC50) values of the inhibitors were performed by nonlinear regression analysis by GraphPad Prism using eq. 2: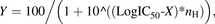 (2)where Y is response and X is the logarithm of the concentration of the inhibitor.
(2)where Y is response and X is the logarithm of the concentration of the inhibitor.
Inhibition of AO- and CES-mediated Metabolism by M1 and M2.
Inhibition studies with M1 or M2 (0–10 μM; Fig. 1) were conducted in HLC using either an AO probe substrate (phthalazine; 8 μM, 2.5 minutes with 0.05 mg/ml HLC) or a CES probe substrate (CPT-11; 5 μM, 5 minutes with 1 mg/ml HLC). Incubation conditions were determined in our laboratory for linear formation of SN-38 with respect to CPT-11 concentration, incubation time, and protein concentration (data not shown). Similar conditions were previously reported (Tabata et al., 2004). At the end of the incubation period, the reactions were terminated and samples prepared for LC-MS/MS analysis as described above. Samples were analyzed by LC-MS/MS for either the AO-mediated metabolite of phthalazine (phthalazinone) or the CES-mediated metabolite of CPT-11 (SN-38). Estimations of the IC50 values of M1 and M2 were performed by nonlinear regression analysis by GraphPad Prism using eq. 2 above.
Inhibition of AO-Mediated Metabolism by GDC-0834.
GDC-0834 (either 0–50 or 0–100 μM) and an AO probe substrate were coincubated in HLC at 37°C (n = 3). Incubation conditions for each of the AO probe substrates were determined in our laboratory. The conditions at which the formation of the AO-mediated metabolite of each probe was linear with respect to incubation time and protein concentration were used (data not shown). AO probe substrates (and incubation conditions) were carbazeran (1 μM, 3 minutes with 1 mg/ml HLC), DACA (6.3 μM, 2.5 minutes with 0.05 mg/ml HLC), O6-benzylguanine (1 μM, 10 minutes with 1 mg/ml HLC), phthalazine (8 μM, 2.5 minutes with 0.05 mg/ml HLC), zaleplon (1 μM, 30 minute with 1 mg/ml HLC), or zoniporide (5 μM, 30 minutes with 1 mg/ml HLC). At the end of the incubation period, the reactions were terminated and samples prepared for LC-MS/MS analysis as described above. Samples were analyzed by LC-MS/MS for the AO-mediated metabolites of carbazeran (4-hydroxycarbazeran), DACA (DACA-9(10H)-acridone), O6-benzylguanine (8-oxobenzylguanine), phthalazine (phthalazinone), zaleplon (5-oxozaleplon), or zoniporide (2-oxozoniporide). Estimations of the IC50 values for GDC-0834 were performed by nonlinear regression analysis by GraphPad Prism using eq. 2 above.
LC-MS/MS Analysis.
All analytes were monitored by MS multiple-reaction monitoring using an AB Sciex Triple Quad 5500 (Redwood City, CA) coupled to an ultra high-pressure liquid chromatography pump and a CTC PAL autosampler from LEAP Technologies (Carrboro, NC). A Kinetex phenyl-hexyl column (2.6 μm, 30 × 2.1 mm) from Phenomenex (Torrance, CA) was used with mobile phases consisting of solvent A (0.1% FA in water) and solvent B (0.1% FA in acetonitrile). The flow rate was 1 ml/min, and the injection volume was 25 μl. The gradient started at 1% solvent B for 0.4 minutes, ramped up to 10% solvent B in 0.08 minutes, ramped up to 63% solvent B in 1.7 minutes, ramped up to 95% solvent B in 0.3 minutes and held for 0.3 minutes, and then stepped down to the initial conditions of 1% solvent B and held at these conditions for 0.7 minute to equilibrate the column before the next injection. The total LC-MS/MS run time was 3.48 minutes. The multiple reaction monitoring transitions in positive ion mode were: m/z 361 → 272 (carbazeran), m/z 587 → 124 (CPT-11), m/z 294 → 249 (DACA), m/z 310 → 265 (DACA-9(10H)-acridone), m/z 597.4 → 127.1 (GDC-0834), m/z 377 → 288.1 (4-hydroxycarbazeran), m/z 433.4 → 127.1 (M1), m/z 183.1 → 139.1 (M2), m/z 242 → 91 (O6-benzylguanine), m/z 258 → 91 (8-oxobenzylguanine), m/z 322 → 252 (5-oxozaleplon), 337.1 → 278.2 (2-oxozoniporide), m/z 147.0 → 90.0 (phthalazinone), m/z 393.0 → 349.0 (SN-38), m/z 306.0 → 236.0 (zaleplon), and m/z 321.1 → 262.2 (zoniporide). The positive ion mode transitions for phthalazine in single ion monitoring mode were m/z 131.1 → 131.1.
Docking of GDC-0834 in AO Active Site.
A homology model for AO was produced using the human sequence and the crystal structure of mouse AOX3 (PDB ID 3ZYV) (Coelho et al., 2012). Induced-fit docking was used to place DACA into the active site of AO. Sequence alignment used Schrödinger's ClustalW (New York, NY) and required no user input because of the high homology (79% homology) of the two primary sequences. For the portions of the mouse structure lacking sufficient electron density, the human enzyme was modeled using the energy-based method in Prime; however, residues 168–200 were not able to be replaced and were excluded in the model. Modeling was done with Schrödinger’s Prime module to generate a protein structure, followed by the induced fit docking workflow using DACA as a ligand to refine amino acid residues within 5 Å of the DACA ligand. Glide docking energies were assessed for DACA, zaleplon, RS-8359, XK-469, and GDC-0834. The structure for each of these compounds have been reported elsewhere (Alfaro and Jones, 2008; Liu et al., 2011b; Jones and Korzekwa, 2013).
Results
Mass Spectrometry Proteomics and Correlation Profiling (or Identification of AO in HLC by Correlation Profiling).
Database search results identified more than 900 proteins, including 563 proteins with spectra matching at least two unique peptides. Among the most abundant proteins in the sample series were the metabolic enzymes carbamoyl-phosphate synthase (CPSM_HUMAN; 468 total peptide-to-spectrum matches (PSMs)/77 unique peptides), fructose-bisphosphate aldolase B (ALDOB_HUMAN; 420/21), retinal dehydrogenase 1 (AL1A1_HUMAN; 347/32), aldehyde dehydrogenase (ALDH2_HUMAN; 292/31), liver carboxylesterase 1 (EST1_HUMAN; 275/33), and AO (ADO_HUMAN; 265/67) (Fig. 2A). Pearson correlations were determined for each identified protein relative to normalized metabolic activity (Fig. 2, A and B). Among the top candidates, AO was identified and displayed a pattern of peptide spectral matches that tightly correlated with amide hydrolysis activity (r2 = 0.9708) (Fig. 2, A and B).

Proteomic correlation profiling revealed that aldehyde oxidase (AO/ADO) is an enzyme present in cytosolic fractions containing the hydrolytic enzyme activity involved in the metabolism of GDC-0834. (A) Table of top 15 most abundant proteins (UniProt) identified in fractions 24–31 based on total peptide spectral matches (PSMs); AO/ADO is shown in red. The hydrolytic enzyme activity (GDC-0834→M1) is shown in blue. Pearson correlations of each protein relative to GDC-0834→M1 metabolic activity are reported. Adjusted P values were corrected for multiple hypothesis testing using the q-value FDR-based approach. (B) Proteins ranked by Pearson correlation. The red diamond highlights AO/ADO.
Kinetic Formation of M1 in HLC and DLC.
The kinetic parameters describing the metabolism GDC-0834 to M1 were determined by incubating parent compound with HLC or DLC. Amide hydrolysis in humans was be more efficient than in dogs, with Vmax of 409 ± 29 compared with 20.3 ± 9.6 pmol/min per milligram protein, Km of 0.800 ± 0.027 compared with 63 ± 5 μM, and intrinsic clearance (Vmax/Km) of 0.511 compared with 0.00025 ml/min per milligram of protein in humans and dogs, respectively (Supplemental Fig. 1, A and B).
Formation of M1 in Fresh Blood and Plasma.
The formation of M1 over time from GDC-0834 (0.8 μM initial concentration) was determined in fresh blood and plasma collected from humans, rats, mice, dogs, and monkeys. A time-dependent formation of M1 was observed in humans, rats, and mice blood and plasma. In dogs and monkeys, a time-dependent formation of M1 was observed in plasma, but levels in blood were below the limit of quantification (<0.001 μM) (Fig. 3). The amounts of formation of M1 were more pronounced in mouse blood and plasma with 0.016 and 0.120 μM formed by 180 minutes, respectively. In contrast, the amounts of M1 formed were lower in the blood and plasma of humans, rats, dogs, and monkeys, with 0.003 and 0.010 μM (humans), 0.002 and 0.006 μM (rats), <0.001 and 0.003 μM (dog), and <0.001 and 0.001 μM (monkeys) present, respectively. The loss of GDC-0834 was minimal in the blood and plasma in all species, with half-life (t1/2) values >500 minutes in both matrices (data not shown).

Formation of M1 after incubation of GDC-0834 (0.8 µM) in (A) fresh whole blood and (B) plasma in human (○), rat (▪), mouse (▲), dog (♦), and monkey (●).
Inhibition of GDC-0834 Metabolism Using Chemical Inhibitors.
The inhibitory properties of AO inhibitors (β-estradiol, DCPIP, menadione, and raloxifene), an AO/CES inhibitor (loperamide), a CES inhibitor (BNPP), or an XO inhibitor (allopurinol) on GDC-0834 metabolism in HLC, DLC, and human plasma were examined (Table 1). Concentration-dependent inhibition of M1 formation in HLC was observed for the AO, AO/CES, and CES inhibitors tested, with IC50 values ranging from 0.33 to 2.14 μM for the AO inhibitors, 0.15 μM for loperamide, and 0.50 μM for BNPP. Allopurinol did not inhibit the metabolism of GDC-0834 and displayed an IC50 >50 μM. In DLC, inhibition of GDC-0834 metabolism and M1 formation was observed only with the CES inhibitor BNPP (IC50 = 15.6 μM); all other inhibitors, including loperamide, had IC50 >50 μM. In human plasma, inhibition of M1 formation was observed only with the CES inhibitor BNPP (IC50 = 6.52 μM) and the AO/CES inhibitor loperimide (IC50 = 4.73 μM); the AO inhibitors did not inhibit the formation of M1.
Mean IC50 values (±S.D.; n = 3) for the inhibition of M1 formation in human (HLC), dog (DLC) liver cytosol and human plasma by aldehyde oxidase (AO), carboxylesterase (CES), and xanthine oxidase (XO) inhibitors
Inhibition of AO- and CES-mediated Metabolism by M1 and M2.
The inhibitory properties of M1 and M2 were examined in HLC. M1 and M2 were weak inhibitors of AO- and CES-mediated metabolism of phthalazine and CPT-11, respectively. IC50 values were >10 μM (Supplemental Fig. 2, A–D).
Inhibition of AO-mediated Metabolism by GDC-0834.
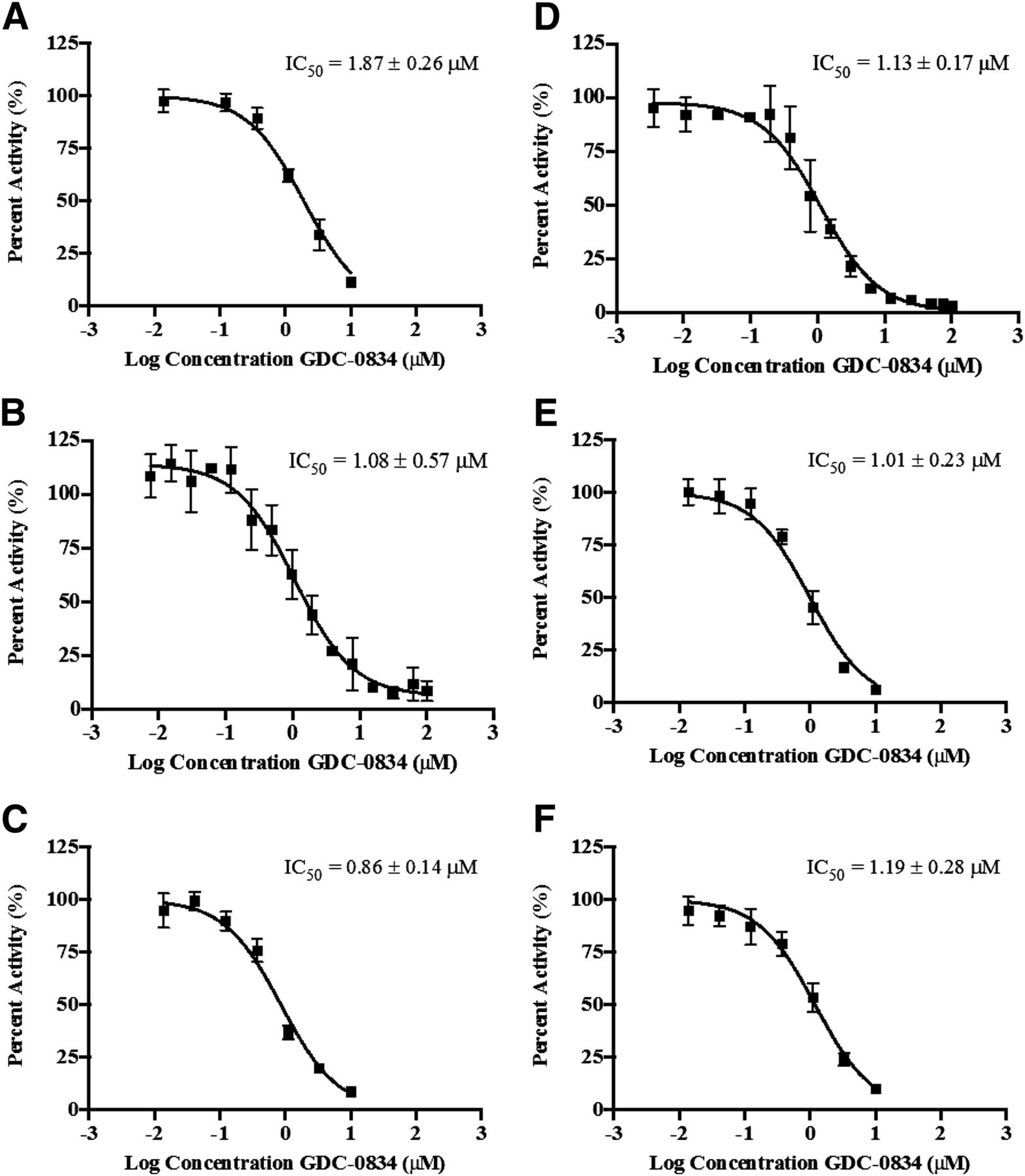
Inhibitory properties of GDC-0834 on AO probe substrates carbazeran, DACA, O6-benyzlguanine, phthalazine, zaleplon, and zoniporide were investigated in HLC. GDC-0834 inhibited the formation of the AO-mediated metabolites with IC50 values ranging from 0.86 to 1.87 μM (Fig. 4, A–F).

IC50 curves for the inhibition by GDC-0834 (0–50 or 0–100 μM) of AO-mediated metabolism of AO probe substrates in human liver cytosol (A) carbazeran (formation of 4-hydroxycarbazeran), (B) DACA (formation of DACA-9(10H)-acridone), (C) O6-benzylguanine (formation of 8-oxobenzylguanine), (D) phthalazine (formation of phthalazinone), (E) zaleplon (formation of 5-oxozaleplon), and (F) zoniporide (formation of 2-oxozoniporide). Data are the mean ± S.D. of triplicate determinations. The lines represent the best fit to the data using nonlinear regression. DACA = N-[(2-dimethylamino) ethyl]acridine-4-carboxamide.
Docking of GDC-0834 in AO Active Site.
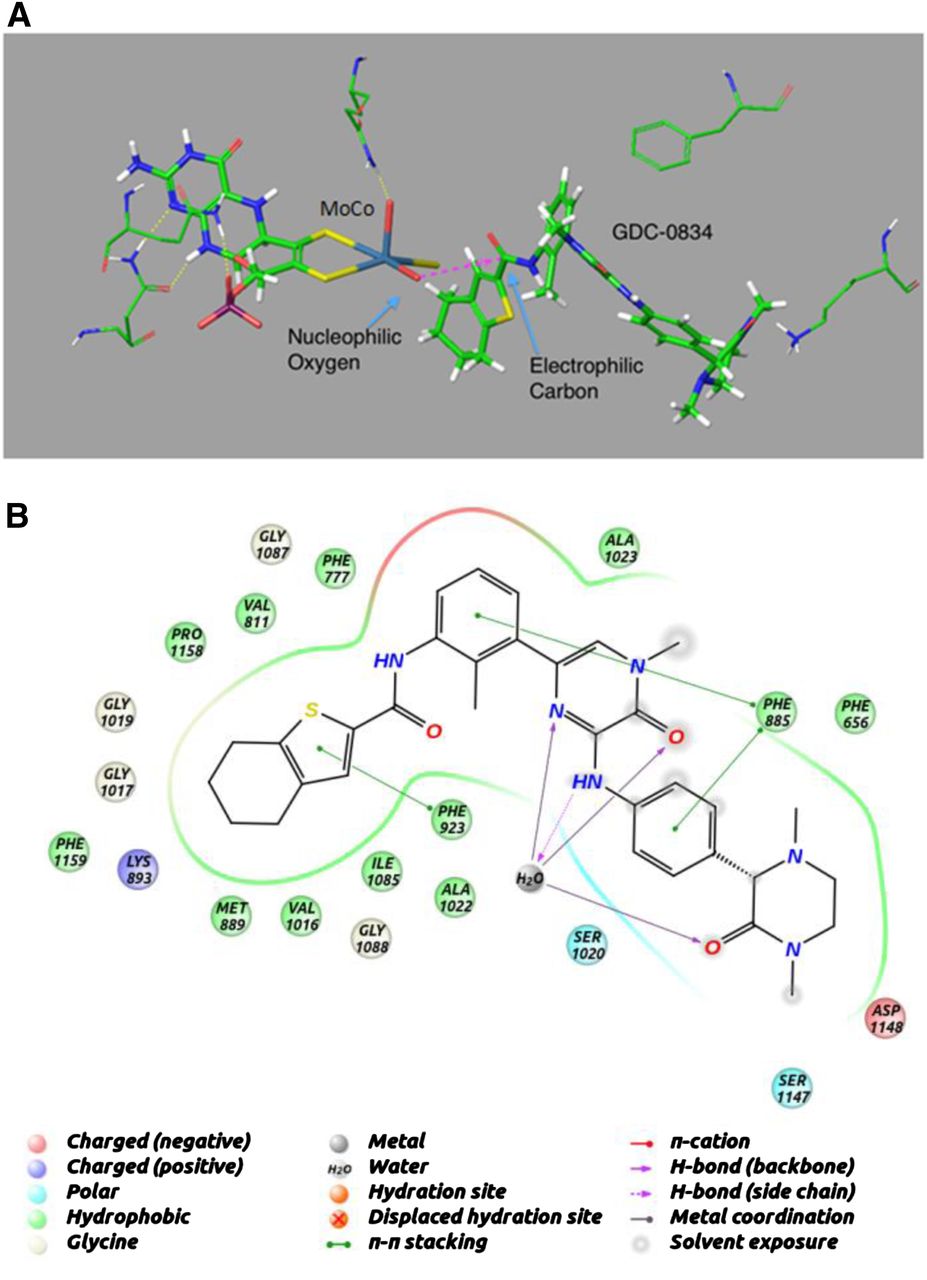
Structural modeling was used to dock GDC-0834 into the active site of AO (Fig. 5A). GDC-0834 filled the active site, and part of the molecule remained solvated. GDC-0834 bound near the molybdenum cofactor (MoCo) group in an orientation that would suggest nucleophilic attack by the hydroxyl-molybdenum (Mo) on the carbonyl of the amide bond. The −5.7 kcal/mol binding energy of GDC-0834 was similar to the other known AO substrates DACA (−6.6 kcal/mol), zaleplon (−5.0 kcal/mol), RS-8359 (−5.8 kcal/mol), and XK-469 (−5.0 kcal/mol). Figure 5B illustrates the putative interactions of GDC-0834 within the active site of AO.
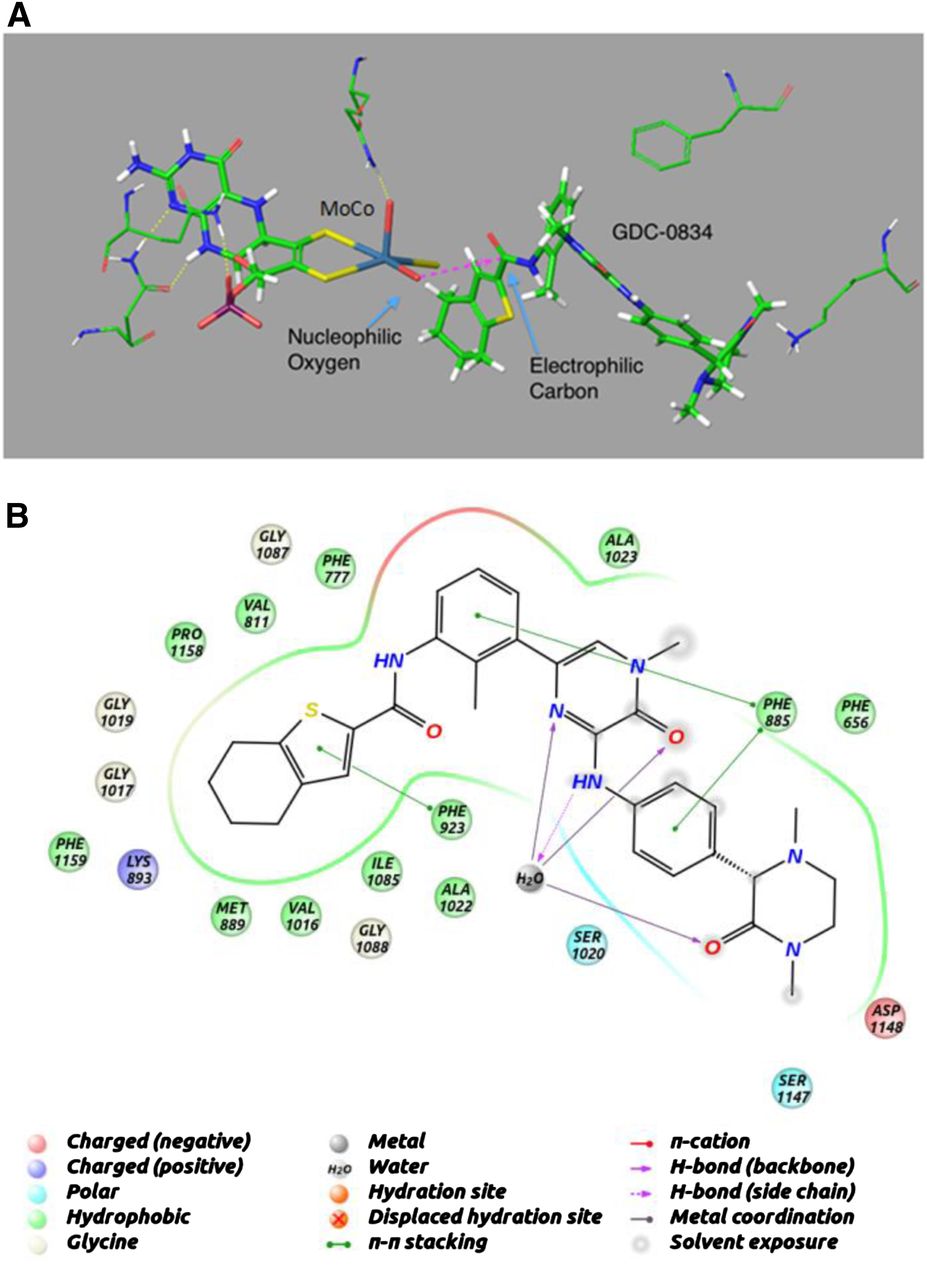
Homology model for aldehyde oxidase (AO) using the human sequence and the mouse crystal structure (PDB ID 3ZYV). (A) Induced-fit docking was used to dock GDC-0834 into the active site of AO near the MoCo group in an orientation that would suggest nucleophilic attack by the hydroxyl on the carbonyl of the amide bond. (B) Putative interactions of GDC-0834 within the active site of AO.
Discussion
Despite the uncertainties surrounding the human clearance prediction (Liu et al., 2011b), GDC-0834 was rapidly advanced to a single-dose human clinical trial to investigate its pharmacokinetic parameters due to other advantageous properties. Unfortunately, after oral administration of GDC-0834 to humans, little to no parent compound was detected as a result of extensive metabolism. Amide hydrolysis of GDC-0834, and subsequent M1 formation, was the major metabolic pathway responsible for its high clearance in the clinic (Fig. 1). The enzyme(s) responsible for the amide hydrolysis was (were) unknown before to clinical pharmacokinetic studies. Using HLC, GDC-0834 was observed to be a substrate for enzyme(s) in this subcellular fraction with a relatively low Km (0.8 µM) and relatively high rate of amide hydrolysis (approximately 400 pmol/min per milligram).
The most obvious enzymes considered as candidates for metabolizing GDC-0834 were esterases and amidases, which are capable of hydrolyzing amide bonds. These enzymes include carboxylesterases, cholinesterases, organophosphatases, and amidases/peptidases, where cholinesterases and aminopeptidases are most efficient in hydrolyzing the amide bonds of marketed drugs (Uetrecht and Trager, 2007) and the most active hydrolases in human small intestine and liver (Taketani et al., 2007). In vitro inhibition studies in human liver microsomes, using general inhibitors of esterases and amidases, failed to implicate these as the hydrolytic enzymes responsible for the metabolism of GDC-0834 (Liu et al., 2011b).
Preliminary in vitro metabolism experiments using various liver fractions revealed that soluble enzyme(s) present in HLC mediated the amide hydrolysis of GDC-0834. Therefore, HLC was chosen over human liver microsomes and hepatocytes since it contained the soluble enzyme(s) to facilitate fractionation and MS-proteomics analysis. In an attempt to identify the enzyme(s) responsible for the amide hydrolysis of GDC-0834, HLC was fractionated and subjected to proteomic correlation profiling analogous to methods used for matching kinases with their respective substrates (McAllister and Gygi, 2013). Proteomic analysis of the fractions was then correlated to the rate of hydrolysis of GDC-0834 (Fig. 2). The abundances of AO and CES and their tight correlations with hydrolytic activity (Pearson correlations of 0.97 and 0.87, respectively) led us to pursue in vitro chemical inhibition and enzyme activity studies in HLC to further investigate the role of AO and CES in the amide hydrolysis of GDC-0834.
Data from the chemical inhibition studies confirmed AO-mediated contribution to the amide hydrolysis of GDC-0834. A panel of widely used AO inhibitors with different chemical structures and properties, including raloxifene (Obach, 2004), DCPIP (Barr and Jones, 2011), menadione (Johns, 1967; Sahi et al., 2008; Barr and Jones, 2011), and β-estradiol (Johns et al., 1969; Barr and Jones, 2011), inhibited GDC-0834 metabolism (formation of M1) with low single-digit micromolar IC50 values (Table 1). There was also evidence for CES-mediated amide hydrolysis in HLC (and DLC and human blood and plasma; see discussion to follow). The AO/CES inhibitor loperamide (Satoh et al., 1994; Rivory et al., 1996; Williams et al., 2011) and CES inhibitor BNPP (Satoh et al., 1994) both inhibited M1 formation in HLC, whereas the XO inhibitor allopurinol (Baker and Wood, 1967) failed to inhibit GDC-0834 metabolism in HLC.
Dogs have been shown to lack AO activity (Beedham et al., 1987), and yet amide hydrolysis of GDC-0834 was observed in vivo and in vitro (Liu et al., 2011b), albeit less than in humans. In vitro enzyme identification studies using chemical inhibitors in DLC suggest that CES mediated the hydrolysis of GDC-0834 in dogs; however, the hydrolysis rate was much lower in DLC than in HLC. To detect and quantify the hydrolysis reaction in DLC, a 60-fold larger amount of cytosolic protein, 6-fold longer incubation time, and 79-fold greater concentration of GDC-0834 were used compared with that of HLC. These data highlight the extensive capability and efficiency of GDC-0834 hydrolysis in humans.
GDC-0834 was metabolically stable in whole blood and plasma, which suggested limited contribution of these matrices to the total clearance in vivo (Liu et al., 2011b). In addition, these studies allowed for the assessments of 1) hydrolysis of GDC-0834 by a hydrolase (presumably CES) since this enzyme (but not AO) is reported to be present in plasma and blood (McCracken et al., 1993; Beedham 2002; Sharma et al., 2011); and 2) the specificities of the various AO inhibitors. In blood and plasma, the loss of GDC-0834 was minimal coupled with little M1 formation (<2% by 180 minutes) in all species, except in mouse plasma (15% by 180 minutes) (Fig. 3). Chemical inhibition studies in human plasma confirmed the specificities of the AO inhibitors used to implicate AO in HLC. In human plasma, which contains CES but not AO, the AO inhibitors did not inhibit the formation of M1, whereas BNPP and loperamide did. These data support the findings of AO involvement in HLC using AO chemical inhibitors, and implicates AO-mediated hydrolysis in HLC, a matrix that contains both AO and CES.
In addition to the potent inhibition of GDC-0834 metabolism by AO inhibitors, GDC-0834 also inhibits the metabolism of several known AO substrates carbazeran, DACA, O6-benzylguanine, phthalazine, zaleplon, and zoniporide (Beedham et al., 1990; Beedham et al., 1995; Kawashima et al., 1999; Schofield et al., 2000; Obach et al., 2004; Dalvie et al., 2012; Hutzler et al., 2012) (Fig. 4, A–F). The measured IC50 values for the inhibition of six known AO substrates range between 0.86 and 1.87 μM and are close to the measured Km of 0.8 μM for the cytosolic hydrolysis of GDC-0834. These results provide additional evidence that GDC-0834 likely interacts at the active site of AO to serve as a substrate and potent AO inhibitor. The metabolites M1 and M2 resulting from hydrolysis were not AO or CES inhibitors; therefore, the inhibition of AO was due to GDC-0834.
The inhibition of M1 formation by AO inhibitors and the inhibition of AO activity by GDC-0834 support AO involvement in the amide hydrolysis reaction and interaction between GDC-0834 and the active site of AO. The in vitro data surrounding AO prompted us to investigate the AO active site using docking experiments. GDC-0834, DACA, zaleplon, RS-8359, and XK-469 were docked in the active site of AO and the induced fit homology model was able to bind each of the substrates. For DACA, zaleplon, RS-8359, and XK-469, a metabolically active orientation was observed for the top two or three binding scores. The docking score for GDC-0834 was within the same range as the known AO substrates. In the docking experiment, the amide bond of GDC-0834 appears to be oriented near the MoCo group in close proximity to the reactive hydroxyl moiety of the MoCo group. The enzyme is able to accommodate a large substrate such as GDC-0834 because the binding pocket is close to the surface of the enzyme. A large part of the substrate remains exposed to solvent as proposed in Fig. 5.
With the use of in silico modeling, it is possible to speculate that the amide bond of GDC-0834 could be coordinated in such a way to be attacked by the hydroxymolybdenum for the initial step to form a tetrahedral intermediate (Fig. 6). This step requires a nucleophilic hydroxyl moiety. Whereas the oxidized hydroxymolybdenum species is capable of nucleophilic attack, as is evident by the nucleophilic aromatic substitution normally catalyzed by this reaction, we propose that MoIV oxidative state is more suited for this reaction because of its higher electron density. This would require build-up of the MoIV oxidative state in much the same way that AO-mediated reductions of compounds, such as nitrites, need a substrate to initially be oxidized (Weidert et al., 2014). If this is the case, the substrate that is oxidized is presently unknown but could be an endogenous aldehyde or azaheterocyclic substrate in the cytosol. The tetrahedral intermediate formed by either the MoIV or MoVI oxidative state collapses to form M1 and possibly a bound ester to molybdenum, which then could be further hydrolyzed to release the corresponding acid metabolite. This reaction does not require any electron transfer; therefore, the catalytic cycle for amide hydrolysis would be complete after breakdown of the tetrahedral intermediate to release product.

Proposed reaction for the amide hydrolysis mediated by aldehyde oxidase.
In conclusion, these data show that AO is involved in the amide hydrolysis of GDC-0834 and suggest that, in addition to AO-mediated oxidative and reductive metabolism of xenobiotics (Kitamura et al., 2006), hydrolysis may represent an additional metabolic activity mediated by this enzyme. Therefore, it is prudent to recognize the role of AO in metabolism, including amide hydrolysis reactions, to avoid poor pharmacokinetics in drug discovery and development stages.
Acknowledgments
The authors gratefully acknowledge the assistance of Amish Karanjit, Daisy Bustos, Robert Cass, Kathryn Kassa, Hoa Le, Jane Lovelidge, Sharmin Jaffer, Chenghong Zhang, Qinghua Song, Chris Nelson, Richard Vandlen, and James Driscoll. The authors also thank Wendy Young, Kevin Ford, and Fabio Broccatelli for helpful comments and discussions.
Authorship Contributions
Participated in research design: Sodhi, Barr, Jones, Halladay.
Conducted experiments: Sodhi, Wong, Barr, Jones, Kirkpatrick, Halladay.
Contributed new reagents or analytic tools: Sodhi, Barr, Jones, Kirkpatrick, Halladay.
Performed data analysis: Sodhi, Wong, Liu, Barr, Jones, Halladay.
Wrote or contributed to the writing of the manuscript: Sodhi, Khojasteh, Hop, Jones, Halladay.
Footnotes
- Received October 28, 2014.
- Accepted April 6, 2015.
↵1 Current affiliation: Anacor Pharmaceuticals, Inc., Palo Alto, California.
The in silico modelling work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant GM100874] (J.P.J., J.T.B.)
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AO
- aldehyde oxidase
- BNPP
- bis-(p-nitrophenyl) phosphate
- BTK
- Bruton’s tyrosine kinase
- CES
- carboxylesterase
- CPT-11
- irinotecan
- DACA
- N-[(2-dimethylamino) ethyl]acridine-4-carboxamide
- DCPIP
- dichlorophenolindophenol
- DLC
- dog liver cytosol
- FA
- formic acid
- GDC-0834
- (R)-N-(3-(6-(4-(1,4-dimethyl-3-oxopiperazin-2-yl)phenylamino)-4-methyl-5-oxo- 4,5-dihydropyrazin-2-yl)-2-methylphenyl)-4,5,6,7-tetrahydrobenzo[b] thiophene-2-carboxamide)
- HLC
- human liver cytosol
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MoCo
- molybdenum cofactor
- MS
- mass spectrometry
- SN-38
- 7-ethyl-10-hydroxycamptothecin
- XO
- xanthine oxidase
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}