


Abstract
The CYP3A family is a major drug metabolism enzyme in humans. Metabolism-based inhibition of CYP3A might cause clinically significant drug-drug interactions (DDIs). To assess the risk of DDIs caused by metabolism-based inhibition (MBI) of CYP3A, we established an automated single time- and concentration-dependent inhibition assay. To create a diagram to assess DDI risk of compounds in the early discovery stage, we classified 171 marketed drugs by the possibility of the occurrence of in vivo DDI caused by MBI from the relationship between the inactivation activity determined in the MBI screening, the therapeutic blood or plasma concentration, and the in vivo DDI information. This analysis revealed that the DDI risk depends on both the MBI potential and the blood concentration of a compound, and provided the criteria of the DDI risk. In the assay, three compounds (midazolam, nifedipine, and testosterone) were compared as CYP3A probe substrates. The results show that the evaluation for MBI does not depend on the probe substrates used in the assay. In addition, we established an automated assay to distinguish quasi-irreversible and irreversible binding to CYP3A in which the quasi-irreversible inhibitors such as diltiazem, verapamil, and nicardipine were dissociated from CYP3A by the addition of potassium ferricyanide, whereas the irreversible inhibitors such as clozapine, delavirdine, and mibefradil were not. It provides useful information related to chemical structures likely to cause MBI. By using these MBI assays supported by an extensive database of marketed compounds, a systematic MBI evaluation paradigm was established and has been incorporated into our drug discovery process.
The cytochrome P450 (P450) superfamily comprises many isozymes that metabolize xenobiotic chemicals, including drugs (Guengerich, 2001). CYP1A2, 2C9, 2C19, 2D6, and 3A participate in the metabolism of approximately 80% of therapeutic drugs, such that the majority of P450-mediated drug metabolism is mediated by the CYP3A family (Wienkers and Heath, 2005). CYP3A4 is a major isoform of CYP3A. It has been reported that CYP3A5 may also contribute to the metabolism of CYP3A4 substrates because of the overlapping substrate specificity with CYP3A4 (Huang et al., 2004). Drugs metabolized by P450s may also inhibit the metabolism of coadministered drugs, which results in an increased blood concentration of the coadministered drugs (Bertz and Granneman, 1997). As a result, a patient to whom two or more drugs are administered might suffer from adverse effects induced by such a drug-drug interaction (DDI). Drugs such as terfenadine, mibefradil, cisapride, and nefazodone have been withdrawn from the market because of P450-related DDIs (Wienkers and Heath, 2005). Consequently, pharmaceutical companies now investigate the P450 inhibitory potential of drug candidates in the early stage of development (Wienkers and Heath, 2005).
P450 inhibition can be classified into three categories: reversible, quasi-irreversible, and irreversible inhibition (Murray, 1997; Lin and Lu, 1998). Reversible inhibition means that two or more drugs compete at a P450-active site and one drug inhibits the interaction of other drugs with the P450. On the other hand, a drug converted to a reactive intermediate by P450 may interact with P450 quasi-irreversibly or irreversibly and inactivate the function of P450. This type of inhibition is referred to as metabolism-based inhibition (MBI). Such MBI is thought to cause more serious P450-related DDIs than reversible inhibition, because the inhibitory effect continues long after the initial administration until new P450 is synthesized. Many metabolism-based inhibitors are currently on the market. These include macrolide antibiotics, anticancer drugs, antidepressants, anti-HIV agents, anti-hypertensive agents, and steroids and their modulators. It is interest that most of them also exhibit pharmacokinetic DDIs (Zhou et al., 2004, 2005b; Fontana et al., 2005). In some cases, the interaction is so serious that, as previously mentioned, the drugs must be withdrawn. For example, mibefradil, a calcium-channel blocker, is a potent metabolism-based CYP3A4 inhibitor. It was withdrawn from the U.S. market in 1998 because of the serious adverse interactions that occurred when mibefradil and statins, antilipemic agents such as simvastatin and lovastatin, were coadministered to patients (FDA talk paper, June 8, 1998, Roche Laboratories announces withdrawal of Posicor from the market [cited 2001 Sep 20], available from http://www.fda.gov/bbs/topics/ANSWERS/ANS00876.html; Schmassmann-Suhijar et al., 1998; Omar and Wilson, 2002; Jacobson, 2004). Because mibefradil inactivates CYP3A4, which clears statins, the plasma concentration of statins increased. This induced rhabdomyolysis, a statin-related adverse effect.
Currently, time- and concentration-dependent inhibition assays are used to determine the MBI potential of a drug or drug candidate (Silverman, 1988; Madan et al., 2002). Many pharmaceutical companies have established MBI assay systems to detect either enzymatic activity loss at a single concentration or IC50 shift caused by preincubation. Other assays are designed to obtain MBI-related kinetic parameters (Crespi and Stresser, 2000; Naritomi et al., 2004; Yamamoto et al., 2004; Atkinson et al., 2005; Lim et al., 2005; Zhao et al., 2005). Such systems use pooled human liver microsomes (HLMs), recombinant P450 isozymes, and human hepatocytes with P450 probe substrates and fluorescent substrates. However, a strategy to apply MBI screening to early drug discovery has not been well established. For CYP3A4 inhibition assays, midazolam, testosterone, terfenadine, erythromycin, and nifedipine are often used as probe substrates (Yuan et al., 2002). Although several groups have reported that the reversible CYP3A4 inhibition caused by test compounds depends on the CYP3A4 probe substrate used (Kenworthy et al., 1999; Stresser et al., 2000; Wang et al., 2000), it has remained unclear whether MBI assay results are affected by CYP3A4 probe substrates.
In the present study, to establish a risk assessment system for DDIs caused by MBI, an automated screening system was established to check the CYP3A MBI potential at a single preincubation time and concentration of a test compound. Then, three different compounds (midazolam, testosterone, and nifedipine) were compared as probe substrates. Next, 171 marketed compounds were tested using this assay. Finally, an automated screening system was established to distinguish between quasi-irreversible and irreversible binding to CYP3A. Based on these results and the additional incorporation of previously reported information, the relationship between the MBI screening results, DDI risk, and MBI reversibility was analyzed to establish a systematic MBI evaluation paradigm.
Materials and Methods
Materials. Midazolam maleate salt, testosterone, nifedipine, dextrorphan tartrate, 17α-ethynylestradiol, ketoconazole, potassium ferricyanide, and glucose 6-phosphate were purchased from Sigma (St. Louis, MO). 1′-Hydroxymidazolam, 6β-hydroxytestosterone, and oxidized nifedipine were purchased from Ultrafine (Manchester, UK). Sodium phosphate buffer (0.5 M), MgCl2 · 6H2O, and dimethyl sulfoxide were purchased from Nacalai Tesque (Kyoto, Japan). β-NADP+ and glucose-6-phosphate dehydrogenase were purchased from Oriental Yeast Co. Ltd. (Tokyo, Japan). Methanol and acetonitrile were purchased from Kanto Chemical Co. Inc. (Tokyo, Japan). Formic acid was purchased from Kishida Chemical Co. Ltd. (Osaka, Japan). Pooled HLMs were purchased from XenoTech LLC (Lenexa, KS). All marketed compounds used as inhibitors were purchased from commercial sources.
Single Time- and Concentration-Dependent Inhibition Assay. The MBI assay was automated by using a Beckman BiomekFX (Beckman Coulter Inc., Fullerton, CA). The assay was designed to run duplicates of 16 reactions: 13 test compounds, 1 MBI-positive reference (50 μM ethynylestradiol), and 1 MBI-negative reference (100 nM ketoconazole), together with 1 vehicle control (reaction mixture without an inhibitor) in a 96-well flat-bottom microplate (Nalge Nunc, Rochester, NY) heated to 37°C on an aluminum plate. The preincubation solutions contained either 0.5 (for midazolam and nifedipine) or 1.0 (for testosterone) mg/ml HLMs in 0.1 M sodium phosphate buffer, and the incubation solutions contained 0.1 M sodium phosphate buffer and one of the following as a substrate: 25 μM midazolam, 200 μM testosterone, or 50 μM nifedipine. The final solvent concentration in the incubation solutions was 1% (v/v) acetonitrile. Each of the solutions were prepared in separate reagent reservoirs (Beckman Coulter Inc.). Preincubation solutions (160 μl) and 180 μl of the incubation solutions were transferred to the wells of the assay plate, and then 20 μl of the test compound solutions or control solutions (10% v/v dimethyl sulfoxide) were added to the preincubation solutions. The preincubation reactions were initiated by the addition of 20 μl of an NADPH-generating system consisting of 25 mM β-NADP+, 250 mM glucose 6-phosphate, 20 units/ml glucose-6-phosphate dehydrogenase, 100 mM MgCl2 · 6H2O, and 0.1 M sodium phosphate buffer. For the 0-min preincubation, 20 μl of each preincubation mixture was immediately transferred into 180 μl of the incubation solution and then incubated for 10 min. For the 30-min preincubation, each preincubation mixture was diluted 10-fold with the incubation solution after 30 min. At the end of the incubation reactions, 50-μl aliquots of each incubation solution were added to 200 μl of methanol containing 200 nM dextrorphan as an internal standard in separate wells of a 96-well round-bottomed microplate (Agilent Technologies, Naerum, Denmark) cooled to 4°C on an aluminum plate. Samples were centrifuged at 2000g for 10 min, and the supernatants were transferred to a Millipore MultiScreen (Millipore, Molsheim, France) and filtered by centrifugation at 2000g for 10 min. The samples in the 96-well round-bottomed microplate were kept at –20°C until LC/MS analysis.
Multiple Time- and Concentration-Dependent Inhibition Assay. The automated MBI assay described above was used with modification of the preincubation times and concentrations of the test compounds. The preincubation solutions were reacted for 0, 15, and 30 min with five concentrations of the test compounds.
Reversibility of MBI. The reversibility of MBI was investigated by oxidation with potassium ferricyanide. A Beckman BiomekFX was used to run seven compounds and one control reaction (without an inhibitor) in a 96-well flat-bottomed microplate (Nalge Nunc) heated to 37°C on an aluminum plate. The compositions of the preincubation solutions and the incubation solutions were the same as those described above (Single Time- and Concentration-Dependent Inhibition Assay). After a 0-min or 30-min preincubation, 50 μl of each preincubation solution was added to 50 μl of the solutions containing 0.1 M sodium phosphate buffer with or without 2 mM potassium ferricyanide in the assay plate, and then incubated for 10 min. After the 10-min reaction, each reaction mixture was diluted 5-fold with the incubation solution. At the end of the incubation reactions, samples were treated as described previously.
LC/MS Analysis. All samples were analyzed by an LC/MS system consisting of a Waters Alliance 2790 and a Micromass ZQ (Waters, Manchester, UK). Chromatographic separation was performed with a Chromolith Flash RP-18e column (4.6 mm × 25 mm; Merck KGaA, Darmstadt, Germany) coupled with a Chromolith Guard Cartridge RP-18e (4.6 mm × 25 mm; Merck KGaA). The mobile phase consisted of solvent A, 0.1% (v/v) formic acid in water, and solvent B, 0.1% (v/v) formic acid in acetonitrile, with an A/B gradient of 95:5 (0–0.5 min) and 65:35 (2.0–4.5 min) for 1′-hydroxymidazolam, 95:5 (0–0.5 min) and 70:30 (2.0–5.0 min) for 6β-hydroxytestosterone, and 95:5 (0–0.5 min) and 50:50 (0.6–4.0 min) for oxidized nifedipine with a flow rate of 0.6 ml/min. The m/z value of 1′-hydroxymidazolam is 341.9, that of 6β-hydroxytestosterone is 304.9, that of oxidized nifedipine is 344.8, and that of dextrorphan is 257.9.
Data Analysis. The concentrations of metabolites in the samples were calculated by using MassLynx V4.0 (Waters). The percentage of the metabolic activity (% of control(0 min) and % of control(30 min)) was obtained for each sample after a 0-min or 30-min preincubation with an inhibitor, compared with each control sample after a 0-min or 30-min preincubation without an inhibitor as follows. 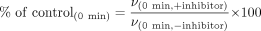
 v(0 min, ±inhibitor) is the metabolic activity after 0-min preincubation with (+) or without (–) an inhibitor, and v(30 min, ±inhibitor) is the metabolic activity after 30-min preincubation with (+) or without (–) an inhibitor. Using these values, the percentage of the enzymatic activity remaining (% remaining) after the 30-min preincubation relative to the 0-min preincubation was calculated as follows.
v(0 min, ±inhibitor) is the metabolic activity after 0-min preincubation with (+) or without (–) an inhibitor, and v(30 min, ±inhibitor) is the metabolic activity after 30-min preincubation with (+) or without (–) an inhibitor. Using these values, the percentage of the enzymatic activity remaining (% remaining) after the 30-min preincubation relative to the 0-min preincubation was calculated as follows. 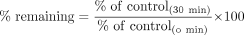
The relationship between the therapeutic plasma concentrations and % remaining for CYP3A was plotted using Spotfire DecisionSite 8.0 (Spotfire KK, Tokyo, Japan). The compounds were placed into one of three groups and marked according to the increase in AUC of coadministered drugs: 1) reported AUC ratio of coadministered drugs ≥2, 2) reported AUC ratio of coadministered drugs <2, and 3) no DDI information.
KI and kinact were determined from the results of the multiple time- and concentration-dependent MBI assay. The natural logarithm of % remaining was plotted against the preincubation times for each concentration of test compound investigated. The slope from the linear regression analysis gave the observed inactivation rate constant (kobs) for each concentration; kobs and the inhibitor concentration (I) were fitted into the following expression by using GraFit Version 5.0.10 (Erithacus Software Limited, Surrey, UK).  For the reversibility of MBI, when the P450 enzymatic activity was reduced by a 30-min preincubation with a compound and the activity could be restored more than 20% with the addition of potassium ferricyanide, the compound was judged to be a quasi-irreversible inhibitor.
For the reversibility of MBI, when the P450 enzymatic activity was reduced by a 30-min preincubation with a compound and the activity could be restored more than 20% with the addition of potassium ferricyanide, the compound was judged to be a quasi-irreversible inhibitor.
Results
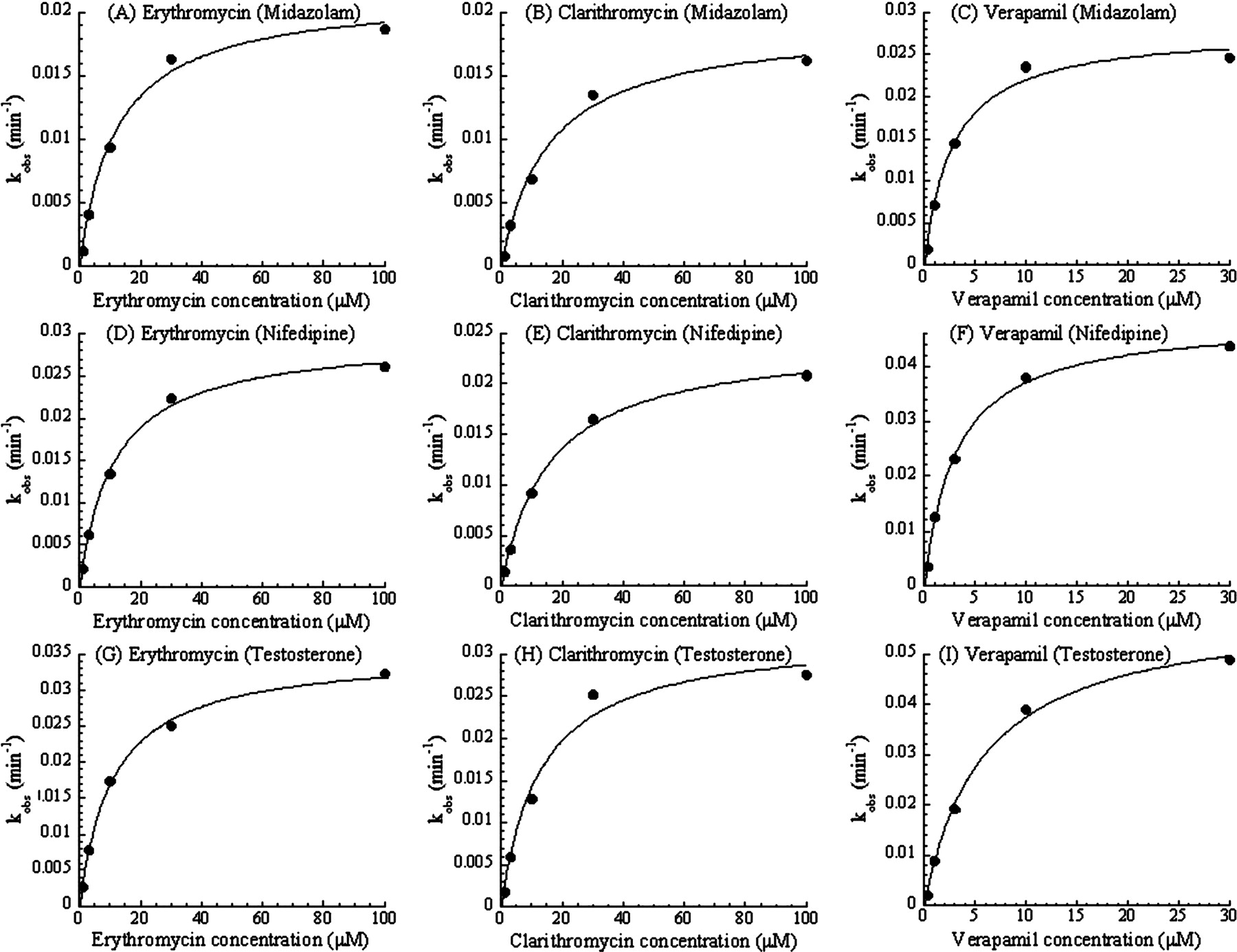
Substrate Dependence of the CYP3A MBI Screening. To check the substrate dependence in the designed MBI assay, three typical probe compounds (midazolam, nifedipine, and testosterone) were used as the CYP3A substrates. Table 1 shows the interday variation in the assay for each probe substrate in the presence of 50 μM ethynylestradiol as an MBI-positive reference or 100 nM ketoconazole as an MBI-negative reference. Figure 1 shows plots comparing the % remaining of 24 marketed compounds as determined by the screening with different substrates. Figure 1 shows that a good relationship exists between the % remaining for midazolam and nifedipine, and for midazolam and testosterone, respectively. In addition, KI and kinact values of three MBI compounds for each probe substrate were obtained from the results of the multiple time- and concentration-dependent inhibition assay shown in Fig. 2, and are listed in Table 2. Kinetic parameters of the test compounds were similar among the assays with the different probe substrates.
Interday variation of % remaining of ethynylestradiol (50 μM) as a MBI-positive reference and ketoconazole (100 nM) as a MBI-negative reference in the automated CYP3A MBI screening using each substrate
The interday data were obtained from the average of the % remaining data from several different studies in duplicate on three different days.
KI and kinact of erythromycin, clarithromycin, and verapamil obtained from the multiple time- and concentration-dependent inhibition assay with each CYP3A substrate
These parameters were determined as described under Materials and Methods (Data Analysis) based on the result shown in Fig. 2.

Comparison of % remaining of marketed compounds for each CYP3A substrate. Each point was obtained from the MBI screening using midazolam, nifedipine, or testosterone as a probe substrate. The comparison between midazolam and nifedipine is shown in A, and that between midazolam and testosterone is shown in B. Each point is a mean of duplicate analyses.
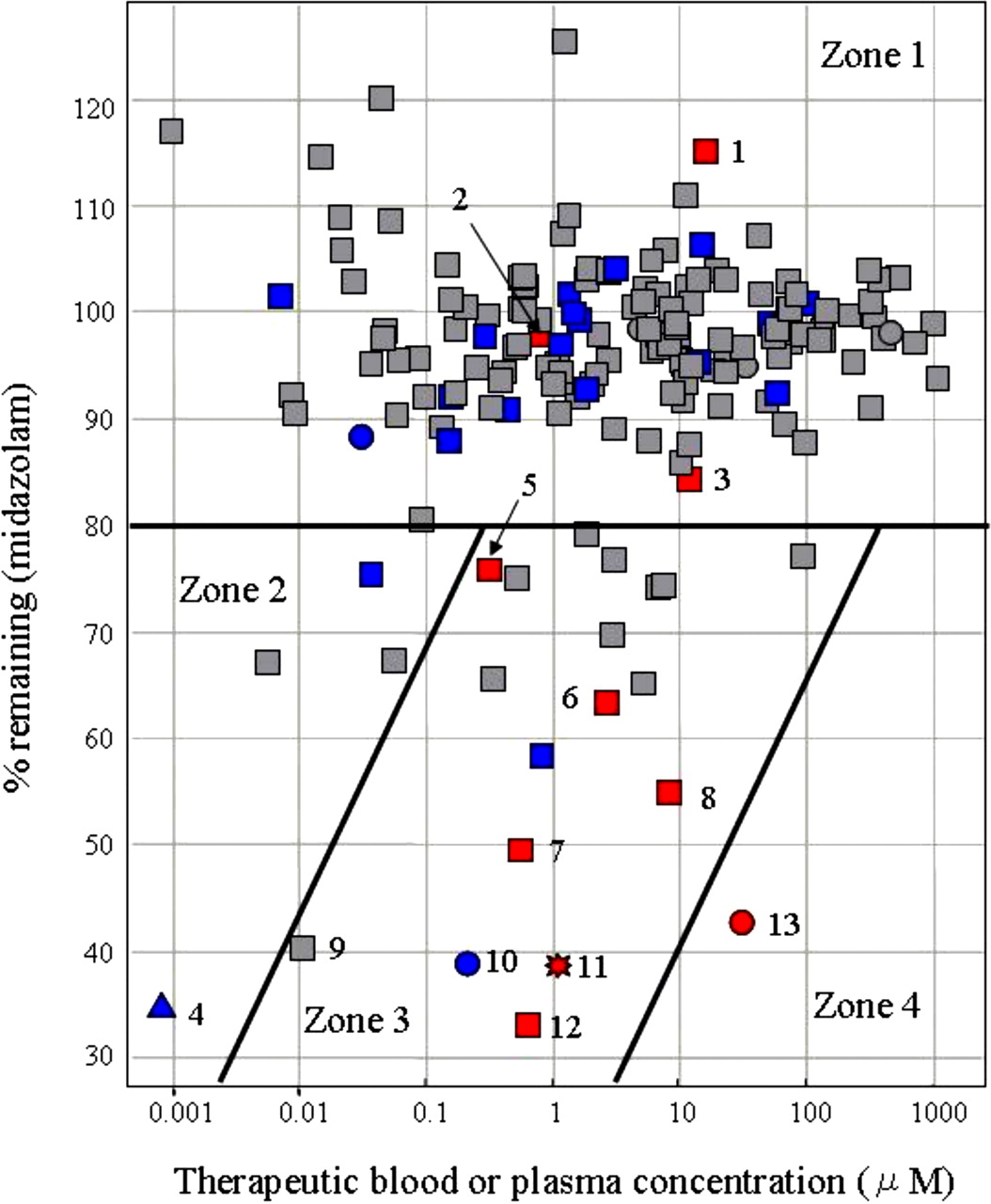
Risk Assessment of DDIs Caused by MBI of CYP3A. To assess in vivo DDI risk from the MBI screening results, 171 marketed compounds, including compounds that have been previously reported to cause MBI (Zhou et al., 2004, 2005b; Fontana et al., 2005), were evaluated in the established screening system. Midazolam was used as the substrate. The concentration of each test compound was initially set at 100 μM. For drugs with strong reversible inhibitory effects, the compound was diluted to a suitable concentration, shown in Fig. 3. The relationship between the screening data and the therapeutic blood or plasma concentrations of these compounds (Schulz and Schmoldt, 2003) was then analyzed by combining the in vivo DDI information (Ito et al., 2004; Obach et al., 2006) and is shown in Fig. 3.
To begin the analysis shown in Fig. 3, the compounds were placed into one of three groups according to the criteria listed under Materials and Methods. Compounds reported to increase the AUC ratio of coadministered drugs more than 2-fold were defined as causing significant in vivo DDIs, based on previous reports (Tucker et al., 2001; Bachmann and Lewis, 2005). Compounds for which no DDI information has been reported were regarded as causing weak or little DDI. Compounds reported to cause both MBI and DDI, such as diltiazem, verapamil, clarithromycin, erythromycin, mibefradil, and nefazodone, fell under the 80%-remaining line and in the therapeutic blood or plasma concentration range of 0.1 to 10 μM. In contrast, most compounds reported to cause neither MBI nor DDI fell over the 80%-remaining line. Thus, the 80%-remaining line was set as the threshold for the MBI screening. In the region of high therapeutic blood or plasma concentration, over 10 μM, and the low percentage remaining, under 80%, only delavirdine, an antiviral drug, fell. Based on the analysis, the DDI risk caused by MBI could be divided into four zones: little DDI risk caused by MBI (zone 1); low possibility of DDI because of the low blood or plasma concentration, even if the compounds have the potential of MBI (zone 2); high possibility of DDI caused by MBI (zone 3); and very low potential as a marketed drug because of high blood or plasma concentration and potent MBI (zone 4).
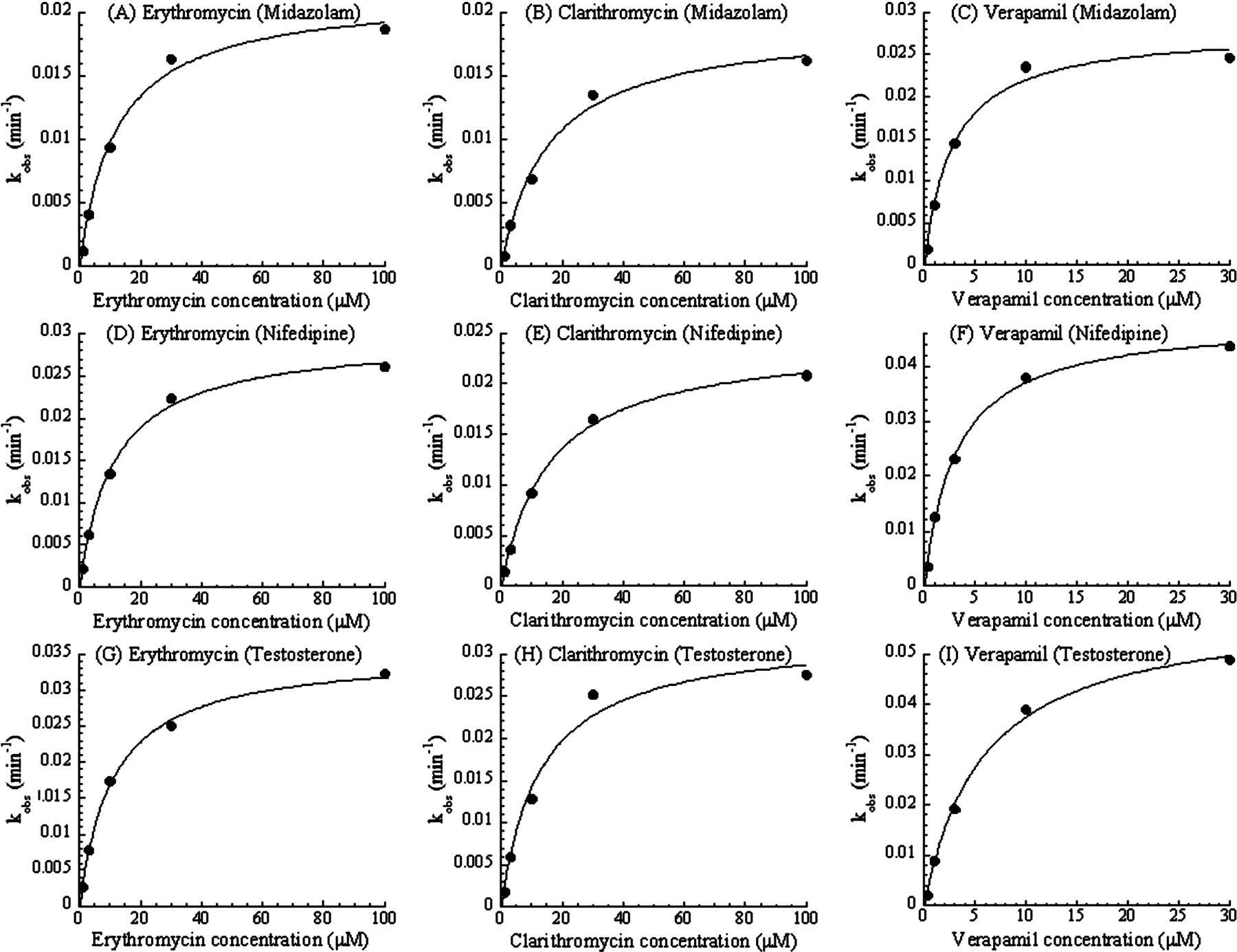
Multiple time- and concentration-dependent inhibition of the metabolism of midazolam (A, B, and C), nifedipine (D, E, and F), or testosterone (G, H, and I) caused by erythromycin (A, D, and G), clarithromycin (B, E, and H), or verapamil (C, F, and I). Each observed inactivation rate constant, kobs, was plotted against each inhibitor concentration. Each point is a mean of duplicate analyses.
Reversibility of MBI. To distinguish between quasi-irreversible and irreversible binding to CYP3A by MBI compounds, an automated system to assay the reversibility of MBI was established. Compounds determined to be metabolism-based inhibitors from the MBI screening data were tested in this new assay, and the results are shown in Table 3. The enzymatic activity of CYP3A inactivated after a 30-min preincubation with diltiazem, verapamil, nicardipine, amlodipine, erythromycin, clarithromycin, or troleandomycin was restored by over 20% after oxidation with potassium ferricyanide. The metabolic intermediates of compounds have been reported to make complexes with the heme of CYP3A4 (Ma et al., 2000; Zhou et al., 2005a). In contrast, the enzymatic activity of CYP3A reduced after a 30-min preincubation with clozapine, delavirdine, mibefradil, or ethynylestradiol did not recover after oxidation with potassium ferricyanide (Zhou et al., 2005a). Reactive metabolites of these compounds are reported to bind covalently either to cellular or microsomal proteins, or to heme. In addition, prazosin, bromocriptine, bepridil, bupivacaine, and buprenorphine were shown to be irreversible inactivators.
Summary of reversibility results of binding to CYP3A by metabolism-based inhibitors obtained from the MBI reversibility assay, and previously reported information
Each datum is a mean of duplicate analyses. Each % of control datum after a 0- or 30-min preincubation followed by the incubation with (+K3Fe(CN)6) or without (–K3Fe(CN)6) potassium ferricyanide is shown below. Judgment of the reversibility was determined as described under Materials and Methods.
MBI Evaluation Paradigm. Using three automated MBI assays and the diagram shown in Fig. 3, a systematic MBI evaluation paradigm was established (Fig. 4).
Discussion
To assess the risk of DDIs caused by MBI of CYP3A, we established an automated screening system to evaluate CYP3A enzymatic activity remaining (% remaining) after a 30-min preincubation with a single concentration of a test compound and obtained extensive data of marketed compounds. Because in vivo DDI risk depends on both the MBI potential and the blood concentration of a compound, the risk of DDIs caused by MBI was classified considering these two factors (Fig. 3). Fluvoxamine, fluconazole, and cimetidine, which are known to cause significant in vivo DDIs, were located in zone 1, the zone of lowest DDI risk caused by MBI. Although they are CYP3A inhibitors, they were located here because they inhibit CYP3A directly, not in a metabolism-based manner. Ethynylestradiol, which is located in zone 2, is a potent metabolism-based inhibitor, but it does not cause in vivo DDI because of its usually low blood concentrations of ≤1 nM (Kuhnz et al., 1996; Palovaara et al., 2000). Compounds reported to cause both MBI and significant in vivo DDIs, such as diltiazem, verapamil, clarithromycin, erythromycin, mibefradil, and nefazodone, were mainly located in zone 3. Mibefradil and nefazodone were withdrawn from the market in 1998 and 2003, respectively, because of this CYP3A4-related DDI (Wienkers and Heath, 2005). Nicardipine, a potent metabolism-based inhibitor located in zone 3, causes mild in vivo DDI by increasing the AUC of coadministered drugs to approximately 1.5-fold.
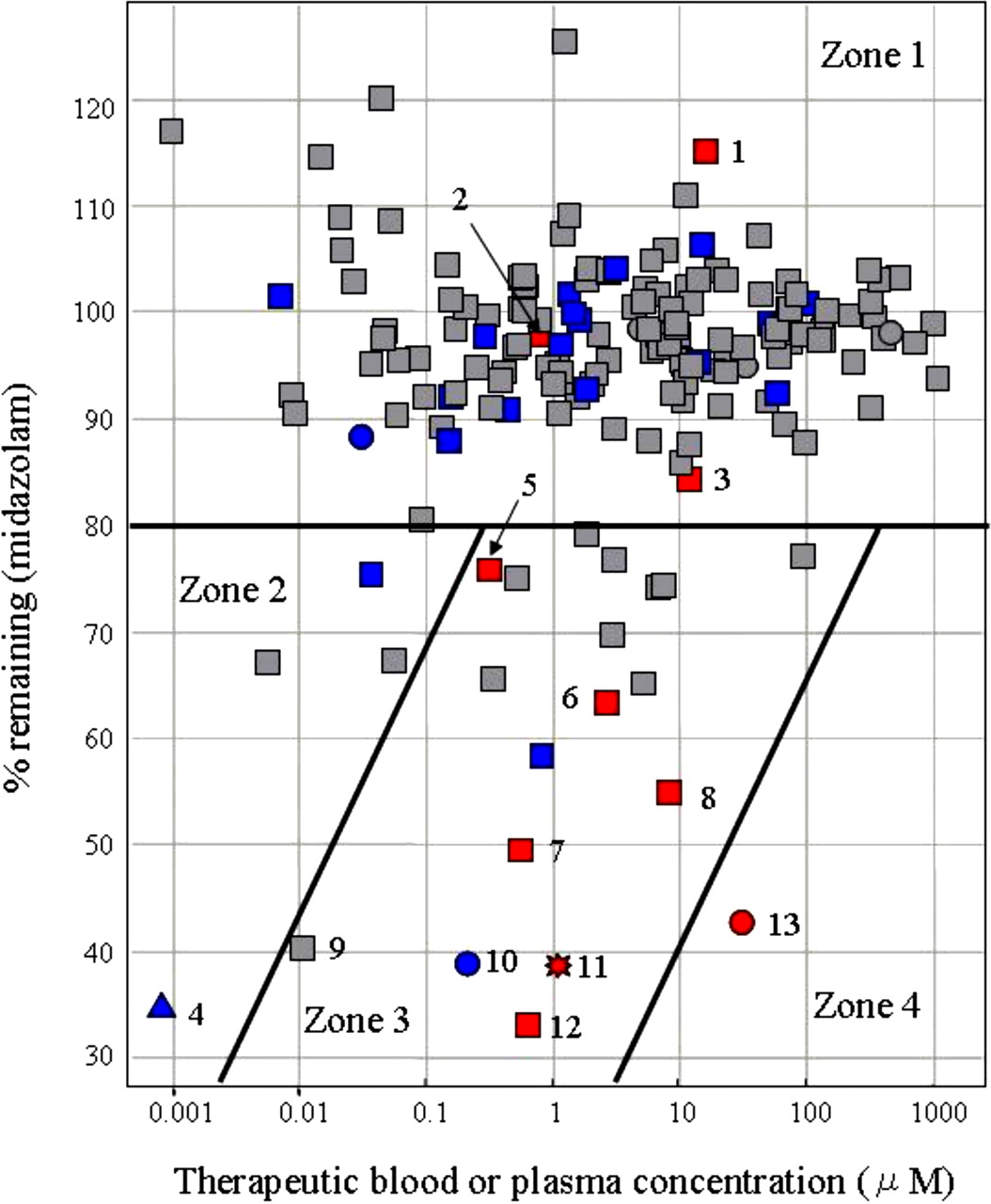
A diagram showing the relationship between % remaining and the therapeutic blood or plasma concentration of 171 marketed compounds in which in vivo DDI information was incorporated. The % remaining data were generated from the MBI assay with midazolam as a substrate. Previously reported therapeutic blood or plasma concentrations and in vivo DDI information for these compounds were used. The concentrations of the compounds tested are as follows: 100 μM (square), 50 μM (triangle), 10 μM (circle), and 1 μM (star). These points were also marked according to the amount of increase in the AUC of coadministered drugs: reported AUC ratio of coadministered drugs ≥2 (red), reported AUC ratio of coadministered drugs <2 (blue), and no DDI information (gray). The risk assessment for DDI caused by MBI can be classified into four zones: no DDI risk caused by MBI (zone 1), low possibility of DDI because of the low blood or plasma concentration even if the compounds have the potential of MBI (zone 2), high possibility of DDI caused by MBI (zone 3), and very low potential as a marketed drug because of high blood or plasma concentration and potent MBI (zone 4). The number added to the symbol corresponds to the compound name as follows: 1, fluconazole; 2, fluvoxamine; 3, cimetidine; 4, ethynylestradiol; 5, diltiazem; 6, clarithromycin; 7, verapamil; 8, erythromycin; 9, buprenorphine; 10, nicardipine; 11, mibefradil; 12, nefazodone; and 13, delavirdine.
Buprenorphine, a semisynthetic opioid derivative, is also located in zone 3. It has been reported that the fatal DDI between buprenorphine and benzodiazepines occurs because of their synergistic pharmacologic effect unrelated to their pharmacokinetic interaction. Buprenorphine is known to be a weak reversible CYP3A4 inhibitor. Its Ki value is large enough compared with the therapeutic concentration to allow a margin of safety (Ibrahim et al., 2000; Elkader and Sproule, 2005). However, results from this study show that buprenorphine is a potent metabolism-based inhibitor of CYP3A; consequently, the severe DDIs seen with benzodiazepines might result, in part, from pharmacokinetic interactions.
The only currently marketed drug in zone 4 is delavirdine, an antiviral drug. Because delavirdine is used in anti-HIV therapy, its level of use can be regarded as exceptional. To maintain the plasma concentration of amprenavir, another antiviral drug metabolized by CYP3A4, at a sufficient level for anti-HIV therapy, amprenavir is often used in combination with delavirdine (Tran et al., 2002). Thus, this analysis shows that compounds located in zones 3 and 4 have strong in vivo DDI potential through their actions on CYP3A.
The relationship diagram (Fig. 3) shows that in order to avoid DDIs in clinical trials or after drug approval, it is important either to reduce the MBI potential of new compounds or decrease their therapeutic blood concentrations by increasing their pharmacologic activity. The diagram provides criteria to assess the DDI risk that might be useful for suggesting new synthesis directions to medicinal chemists. Moreover, in the early drug discovery process, we can estimate the therapeutic concentration of compounds based on the in vitro pharmacologic data or based on information about drugs that has been used already in the same therapeutic area of the clinical stage. Using the estimated therapeutic concentration, it is possible to assess the risk of DDI from the relationship diagram.
The effect of the CYP3A probe substrates used in MBI assays on the assay results was also tested. Several CYP3A4 substrates, for example, midazolam, testosterone, terfenadine, erythromycin, and nifedipine, are often used as probe substrates in enzyme inhibition assays (Yuan et al., 2002). It has been reported that the observed inhibitory effect of test compounds on CYP3A4 metabolic activity depends on the CYP3A4 probe substrate, because it is thought that CYP3A4 has multiple substrate-binding sites that complicate analysis of the interaction between CYP3A4 and its substrates (Kenworthy et al., 1999; Stresser et al., 2000; Wang et al., 2000). Consequently, to avoid under- or overestimation of in vivo DDIs resulting from CYP3A4 inhibition, it is recommended to use multiple probe substrates for in vitro enzyme inhibition assays. However, it has not been reported whether the MBI observed with a given compound depends on CYP3A4 probe substrates or not; therefore, three probe substrates, midazolam, nifedipine, and testosterone, were compared in the MBI assays. The % remaining data for the 24 marketed compounds obtained from the MBI screening with each probe substrate were well correlated, showing that % remaining values do not depend on the probe substrates used for the MBI screening (Fig. 1). In addition, for each probe substrate, the KI and kinact values of three compounds known to cause MBI, erythromycin, clarithromycin, and verapamil, were obtained by using the multiple time- and concentration-dependent inhibition assay (Table 2). These parameters for the three probe substrates were also similar. Thus, the MBI potential of the compounds detected using these MBI assays appears to be independent of the probe substrates used in the assays. Based on these results, midazolam was selected as the probe substrate for the following assays because abundant in vivo DDI information is available on midazolam. The result is thought to be reasonable, because reactive intermediates generated from MBI compounds can bind the apoprotein and heme of P450 irreversibly or quasi-irreversibly, and inactivate metabolic activities; thus, other compounds might not be able to access these inactivated enzymes.
It is also said that the MBI compounds, which in turn bind covalently to cellular or microsomal proteins, might induce hepatotoxicity in addition to DDI (Walgren et al., 2005; Zhou et al., 2005a). A large number of compounds including methylenedioxybenzenes, alkylamines, and hydrazines form metabolite intermediate complexes (Murray, 1997; Lin and Lu, 1998). Compounds containing terminal acetylenes are converted to reactive metabolites that irreversibly alkylate the heme of P450, and electrophilic reactive metabolites or radical species converted by the metabolism of several compounds bind to the apoprotein of P450 covalently (Murray, 1997; Lin and Lu, 1998). Therefore, information about MBI reversibility might be useful for determining chemical structures likely to cause MBI. To distinguish between quasi-irreversible and irreversible binding to CYP3A by MBI compounds, an automated system to assess MBI reversibility was established. Quasi-irreversible metabolite intermediate complexes can be dissociated by oxidation with potassium ferricyanide; the enzymatic activity of P450 then recovers. In contrast, the enzymatic activity inhibited by irreversible binding is not restored (Lin and Lu, 1998). Compounds determined to be metabolism-based inhibitors in the MBI screening were tested by this method (Table 3). CYP3A enzymatic activity inactivated after a 30-min preincubation with quasi-irreversible inhibitors was restored more than 20% by the addition of potassium ferricyanide in the assay mixture (Ma et al., 2000; Fontana et al., 2005). On the contrary, reduced enzymatic activity resulting from a 30-min preincubation with compounds whose reactive metabolites are reported to bind irreversibly either to cellular or microsomal proteins, or to the heme of P450, could not be restored by potassium ferricyanide treatment (Fontana et al., 2005; Zhou et al., 2005a). It has been reported that an irreversible inhibitor, clozapine, is associated with idiosyncratic toxicity, rather than MBI (Walgren et al., 2005). More studies are needed to clarify the relationship between MBI and hepatotoxicity.

Systematic MBI evaluation paradigm using three automated MBI assays. In the single time- and concentration-dependent MBI assay, a compound under the 80%-remaining threshold is classified as having moderate to high DDI risk depending on its therapeutic concentration. Binding to P450 is then evaluated by using the MBI reversibility assay to determine chemical structures likely to cause MBI. If further detailed prediction of in vivo DDI is needed, values for KI and kinact are obtained by using the multiple time- and concentration-dependent inhibition assay.
In conclusion, an automated single time- and concentration-dependent inhibition assay for high-throughput screening was established. Marketed compounds tested by this assay were then categorized based on both the screening results and clinical DDI information, revealing the relationship between the MBI potential, the therapeutic blood or plasma concentrations, and the DDI risk. The results of this study also show that different CYP3A probe substrates used in MBI assays do not affect the evaluation for MBI. In addition, an automated MBI reversibility assay was established to distinguish between quasi-irreversible and irreversible binding to P450. Based on these observations, a systematic MBI evaluation paradigm using these MBI assays, shown in Fig. 4, was established and incorporated into the drug discovery process at this company. It enables the evaluation of the MBI potential of new compounds effectively and early in their development. This system allows in vitro assessment of DDI risk without extensive and expensive in vivo testing. We hope that this system, and others likely to follow, will enable the speedy development of safer drugs.
Acknowledgments
We thank Takae Tanaka for excellent technical support.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.015016.
-
ABBREVIATIONS: P450, cytochrome P450; MBI, metabolism-based inhibition; DDI, drug-drug interaction; HLM, human liver microsome; LC/MS, liquid chromatography/mass spectrometry.
- Received January 30, 2007.
- Accepted March 27, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}