


Abstract
Pharmaceutical industry investigators routinely evaluate the potential for a new drug to modify cytochrome P450 (P450) activities by determining the effect of the drug on in vitro probe reactions that represent activity of specific P450 enzymes. The in vitro findings obtained with one probe substrate are usually extrapolated to the compound's potential to affect all substrates of the same enzyme. Due to this practice, it is important to use the right probe substrate and to conduct the experiment under optimal conditions. Surveys conducted by reviewers in CDER indicated that the most common in vitro probe reactions used by industry investigators include the following: phenacetin O-deethylation for CYP1A2, coumarin 7-hydroxylation for CYP2A6, 7-ethoxy-4-trifluoromethyl coumarin O-dealkylation for CYP2B6, tolbutamide 4′-hydroxylation for CYP2C9, S-mephenytoin 4-hydroxylation for CYP2C19, bufuralol 1′-hydroxylation for CYP2D6, chlorzoxazone 6-hydroxylation for CYP2E1, and testosterone 6β-hydroxylation for CYP3A4. We reviewed the validation information in the literature on these reactions and other frequently used reactions, including caffeine N3-demethylation for CYP1A2, S-mephenytoin N-demethylation for CYP2B6, S-warfarin 7′-hydroxylation for CYP2C9, dextromethorphan O-demethylation for CYP2D6, and midazolam 1′-hydroxylation for CYP3A4. The available information indicates that we need to continue the search for better probe substrates for some enzymes. For CYP3A4-based drug interactions it may be necessary to evaluate two or more probe substrates. In many cases, the probe reaction represents a particular enzyme activity only under specific experimental conditions. Investigators must consider appropriateness of probe substrates and experimental conditions when conducting in vitro drug interaction studies and when extrapolating the results to in vivo situations.
During the drug-candidate screening and development process, investigators often conduct two types of in vitro drug metabolism studies to assess the potential for P4501-based drug interactions. One type of study characterizes the metabolic pathway of the new drug and the potential for other drugs to modify the metabolism of the new drug. The other type of study evaluates the potential for the new drug to alter the metabolism of other drugs. Due to the availability of antibodies against specific P450 enzymes, cDNA-expressed enzymes, purified enzymes, and selective chemical inhibitors, the unequivocal identification of the major P450 isoform responsible for the metabolism of a new drug can be easily established. However, predicting the potential for the new drug to alter the metabolism of other drugs usually relies on the evaluation of the effect of the new drug on the rate of a probe reaction that represents a specific P450 enzyme activity. The second type of evaluation is more challenging and is the focus of this review.
Current Practice and Potential Problems
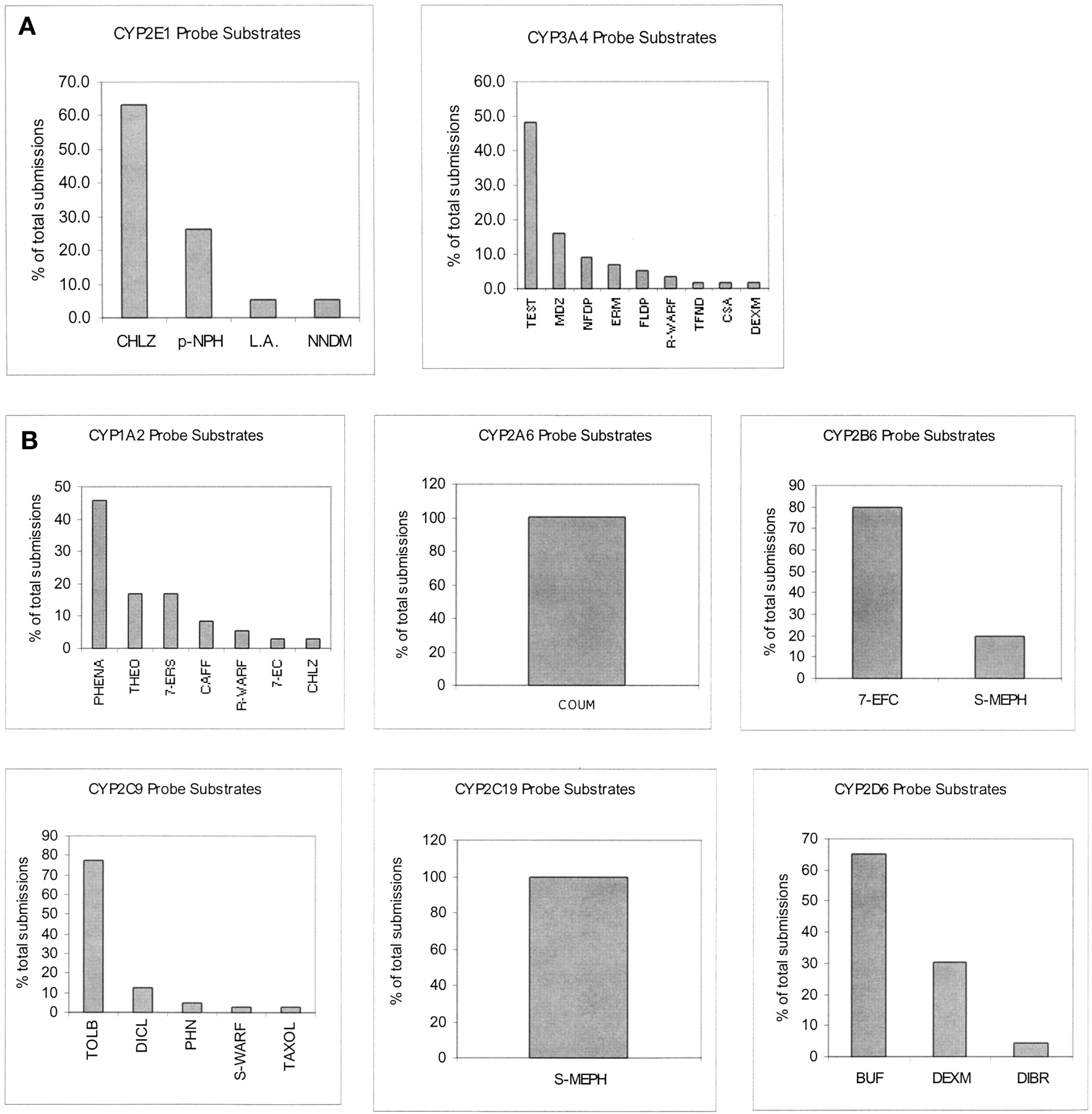
According to a survey of 194 new drugs approved in the United States from 1992 to 1997, industry investigators use different probe reactions to represent the same P450 enzyme activities for evaluating the modulatory potential of a new drug (Table1) (Yuan et al., 1999). When the same inhibitor is evaluated using different probe assays for the same P450 enzyme activity, the outcome of the drug interactions can be different. Also, investigators use different experimental conditions for the same assay. Some studies are not conducted under the optimal conditions. During the preparation of this review we surveyed an additional 44 drug applications submitted from 1997 to 1999, to determine whether recent progress in the area of in vitro drug metabolism changed the common practices. The results of the second survey are consistent with those of the previous one (Table 1, Fig. 1).
Probe reactions used to characterize enzyme activities, surveyed from 194 drugs approved from US-FDA from 1992 to 1997 period

Probe reactions used to characterize enzyme activities, surveyed from 44 drugs approved in 1997 to 1999 period.
The phenomenon of different assays providing different results for the same enzyme is most notable for CYP3A4 activity, as recent publications indicate. Wang et al. (2000) examined the mutual inhibition among the four commonly used CYP3A4 substrates testosterone, terfenadine, midazolam, and nifedipine. They found that although testosterone partially inhibits hydroxylation of terfenadine and midazolam, it does not inhibit nifedipine oxidation. Based on a study of the modulatory effect of 34 compounds on 10 commonly used CYP3A4-mediated reactions,Kenworthy et al. (1999) reported that the effect is substrate-dependent. Haloperidol, for example, activates dextromethorphan N-demethylation by 20%, but it inhibits nifedipine oxidation by 96%, even though CYP3A4 catalyzes both reactions. Stresser et al. (2000) showed that the extent of substrate dependence for the quantitative inhibition parameters (IC50) is as large as 195-fold among four tested CYP3A4 reactions.
The in vitro experimental conditions may influence the accurate assessment of drug interaction potential. Reports indicate that various solvents, for example, may have different effects on P450 probe reactions (Chauret et al., 1998; Hickman et al., 1998; Busby et al., 1999). At 0.2% (v/v), acetonitrile does not affect chlorzoxazone 6-hydroxylation (used as the CYP2E1 probe reaction), but dimethyl sulfoxide (DMSO) at 0.2% (v/v) inhibits the reaction by >80%. As a result, an incubation with DMSO is less sensitive and is subject to greater error when determining whether a new drug inhibits the same reaction. Through a careful enzyme kinetic study, Tang et al. (2000)showed that acetonitrile (3%, v/v) increases intrinsic clearance for CYP2C9-based diclofenac hydroxylation by 87%, but it decreases CYP2C9-based celecoxib hydroxylation by 25%.
In addition to using the appropriate solvent in the incubation, a probe reaction should proceed under initial rate conditions. To proceed under initial rate conditions, the experiment should use optimal experimental conditions, such as substrate concentrations, incubation time, and enzyme protein content. Deviation from optimal experimental conditions may result in an underestimation or overestimation of changes in enzyme activity, and thereby lead to incorrect conclusions regarding the drug interaction potential of the new drug.
The potential influence of probe substrates and experimental conditions on the assessment of in vitro drug interactions has a significant impact on the drug development process and regulatory decisions. The in vivo drug interaction guidance published by the Food and Drug Administration in 1999 (www.fda.gov/cder/guidance) indicates that investigators may use in vitro drug interaction data to conclude that a new drug does not inhibit a specific P450 activity (Food and Drug Administration guidance). In practice, the in vitro evidence is usually collected from one probe reaction per enzyme, and the conclusion is extrapolated to all substrates for the same enzyme. The significant regulatory impact of this approach and potential problems associated with current practice observed in our surveys prompted us to evaluate the appropriateness of in vitro methodologies that pharmaceutical industry investigators commonly use to study P450-based drug interactions. We hope that this evaluation leads industry investigators to adopt a more consistent and accurate in vitro approach. Our ultimate goals are to promote 1) development of in vitro results that provide a reliable extrapolation to in vivo drug interactions; and 2) consistent regulatory submissions that allow comparisons across different drug applications and product labels. Although it is acceptable for industry investigators to use different probe substrates for the same enzyme activity, as a step toward standardization we want to provide guidance regarding the preferred probe substrates and experimental conditions.
Evaluation Approach
We conducted two in-house surveys, as described previously, to determine which probe reactions pharmaceutical industry investigators use for each enzyme (Table 1; Fig. 1). Detailed evaluations were conducted for the most commonly used probe reaction(s) for each enzyme (Fig. 1), as well as those reactions deemed to have additional value for in vivo use. Although we do not consider the potential for in vivo use a necessary criterion in the selection of a preferred substrate, we recognize that some investigators prefer using the same probe in vitro and in vivo. The evaluation primarily focused on the specificity, selectivity, and sensitivity of a reaction for the enzyme that it represents. We reviewed the literature information from in vitro P450-based metabolism studies using purified enzymes, cDNA-expressed enzymes, selective chemical inhibitors, inhibitory antibodies as well as studies on enzyme kinetic analyses. For our evaluation, an ideal probe substrate is the one with a simple metabolic scheme, so that the formation rate of a metabolite specifically reflects the activity of one distinct P450 enzyme. Preferably, the metabolite formed does not undergo sequential metabolism. The reaction should be selective, with at least 80% of the formation of a metabolite being carried out by a single enzyme. In addition to the above-mentioned scientific criteria, the following practical criteria are relevant: the commercial availability of the assayed molecular species (i.e., parent drug and the metabolite); the availability of an assay that is sensitive, rapid, and simple; and reasonable in vitro experimental conditions. We also address cautions to exercise and difficulties encountered when extrapolating in vitro information to in vivo use for some reactions.
Results
CYP1A2.
Human liver microsomes (HLMs) contain relatively high constitutive levels of CYP1A2 (10–15% of the total P450 content of human liver), but not CYP1A1, which is more readily detected in extra-hepatic tissues under induced conditions. Environmental factors affect CYP1A1 and CYP1A2 expression levels, complicating the in vitro-to-in vivo extrapolation. CYP1A2 metabolizes many clinically important drugs such as amitriptyline, imipramine, theophylline, clozapine, tacrine, and zileuton. According to our survey, 45% of the submissions use phenacetin O-deethylation to form acetaminophen to represent CYP1A2 activity (Fig. 1). However, industry investigators also use several substrates other than phenacetin to evaluate CYP1A2 activity. We chose to review caffeine N3-demethylation, in addition to phenacetin O-deethylation, because it is a widely used in vivo substrate.
Phenacetin O-Deethylation.
Phenacetin is an analgesic and antipyretic drug no longer marketed for human use in the United States. The frequent use of this substrate in vitro may be due to the availability of the parent compound and metabolite, and the fast and simple high-performance liquid chromatography-ultraviolet detection assay with high sensitivity for the reaction.
In HLMs, the O-deethylation of phenacetin displays biphasic kinetics (Boobis et al., 1981; Tassaneeyakul et al., 1993; Kobayashi et al., 1998). Studies with cDNA-expressed CYP1A2 (Venkatakrishnan et al., 1998), chemical inhibitors (Boobis et al., 1981; Sesardic et al., 1990;von Moltke et al., 1996), and monoclonal antibodies (Sesardic et al., 1988; Tassaneeyakul et al., 1993) show that the high-affinity component of phenacetin O-deethylation is CYP1A2. TheKm value of this pathway is reported at 10 to 50 μM, at least 10-fold lower than that of the low-affinity component. At a substrate concentration of 100 μM, the contribution of CYP1A2 is estimated to be 86%, but the contribution is reduced to 50% at a substrate concentration of 500 μM (von Moltke et al., 1996;Venkatakrishnan et al., 1998). At concentrations ≥500 μM, several enzymes, especially CYP2C9, contribute significantly to theO-deethylation of phenacetin in HLMs.
Study with organic solvents indicates that at solvent concentrations ≤1% (v/v), phenacetin O-deethylation is not significantly affected by DMSO and methanol (Chauret et al., 1998; Busby et al., 1999). In summary, at substrate concentrations that reflect lowKm enzyme activity (i.e., at concentration lower than 100 μM), phenacetinO-deethylation is the preferred probe reaction for detecting CYP1A2-based drug interaction potential in vitro.
Caffeine N3-Demethylation.
As with phenacetin O-deethylation, the rate of caffeineN3-demethylation to form paraxanthine is biphasic in HLMs. CYP1A2 is responsible for the high-affinity component with aKm value of 200 to 500 μM, and unidentified P450s are responsible for the low-affinity pathway, with aKm value of 20 to 30 mM (Grant et al., 1987; Tassaneeyakul et al., 1993, 1994). Studies using cDNA-expressed enzymes, monoclonal antibody against CYP1A2, chemical inhibitors, and enzyme kinetics validate the involvement of CYP1A2 in the high-affinity pathway (Grant et al., 1987; Butler et al., 1989; Tassaneeyakul et al., 1992). At 1 mM caffeine, CYP1A2 contributes to only 70% of the paraxanthine formation (Tassaneeyakul et al., 1992). At substrate concentrations ≤0.1 mM, the paraxanthine formation rate reflects CYP1A2 activity. However, due to the detection limit on conventional high-performance liquid chromatography system, caffeineN3-demethylation often is carried out at high substrate concentrations, usually at 0.5 to 5 mM unless radiolabeled drug or liquid chromatography/mass spectrometry is used, and at high microsomal protein concentrations, up to 2 mg/ml (Grant et al., 1987). In addition, caffeine N3-demethylation is sensitive to solvent effects. Methanol at 1% (v/v) inhibits the reaction by >80%, whereas acetone and acetonitrile at the same concentration stimulate the reaction by >200% (Hickman et al., 1998).
Taken together, caffeine is not a preferred in vitro substrate for CYP1A2 activity compared with phenacetin. Literature indicates an ongoing effort to develop a more sensitive detection assay to facilitate the study of this metabolic pathway. If one chooses to use this substrate to represent CYP1A2 activity in vitro, one should use substrate concentrations below 0.1 mM and be cautious on the choice of organic solvent.
CYP2A6.
CYP2A6 is an important enzyme for precarcinogen activation and oxidation of certain drugs. It exhibits significant ethnic-related genotypic or phenotypic deficiency (Shimada et al., 1996). CYP2A6 substrates include coumarin, aflatoxin B1, nicotine,N-nitrosodiethylamine, N-nitrosodimethylamine, and N-nitrosonornicotine. Our surveys indicate 7-hydroxylation of coumarin is the only reaction that industry investigators use to assess CYP2A6 activity (Fig. 1; Table 1).
Coumarin 7-Hydroxylation.
Studies with CYP2A6 inhibitory monoclonal antibody show that at substrate concentrations ≤10 μM, more than 90% of the 7-hydroxylation of coumarin in HLMs is carried out by CYP2A6, demonstrating the unequivocal role of CYP2A6 in coumarin 7-hydroxylation (Li et al., 1997; Sai et al., 1999; Yang et al., 1999). Among nine cDNA-expressed enzymes, only CYP2A6 catalyzes this reaction (Ono et al., 1996). Consistently, kinetic studies in HLMs show monophasic formation of 7-hydroxy-coumarin at commonly used substrate concentrations of 0.1 to 10 μM, with aKm value of 0.5 to 2 μM (Shimada et al., 1996; Draper et al., 1997).
Although the experimental conditions for coumarin 7-hydroxylation are straightforward, the reaction rate is subject to various solvent effects. Acetone and acetonitrile at 1% (v/v) each inhibit the reaction rate by >40%, but DMSO and methanol at 1% (v/v) have little effect (Draper et al., 1997; Chauret et al., 1998; Hickman et al., 1998). With the appropriate organic solvent, coumarin 7-hydroxylation is a preferred probe reaction to detect CYP2A6-based drug interaction potential in vitro.
CYP2B6.
Cytochrome P450 2B6 is the only member of the CYP2B family expressed in humans. However, it has not been studied extensively due to unavailability of a probe substrate and reported low levels of the enzyme in human tissues (Shimada et al., 1994). Recent studies indicate that the quantity of CYP2B6 in the liver is underestimated due to a lack of sensitive techniques and antibodies. In our surveys, we observe that few industry investigators characterize the activity of this enzyme in in vitro studies.
There are very few xenobiotics recognized as substrates of CYP2B6. The results of our first survey of drugs approved from 1992 to 1997 indicate that 7-ethoxy-4-triflouromethylcoumarin (7-EFC)O-deethylation is the reaction that some industry investigators use to represent CYP2B6 activity. Recent literature studies show, however, that 7-EFC is metabolized to 7-hydroxy-4-trifluoromethylcoumarin by CYP1A2 and CYP2E1 as well as CYP2B6 (Ekins et al., 1997), making this substrate nonselective for CYP2B6. Thus, we do not consider 7-EFC a preferred candidate for CYP2B6 probe substrate.
S-Mephenytoin N-Demethylation.
The more recently conducted second survey of 44 submissions to United States Food and Drug Administration indicates that some investigators use S-mephenytoin N-demethylation to nirvanol to represent CYP2B6 activity (Fig. 1). The available evidence in the literature supports the selectivity of this reaction for CYP2B6. Ko et al. (1998) report biphasic kinetics for nirvanol formation, with high- and low-affinity Km values of 174 and 1900 μM, respectively. However, Heyn et al. (1996) assume monophasic kinetics and report a mean Km value of ∼800 μM. The discrepancy seems to be due to the differences in the substrate concentration ranges used by these investigators.
Due to conflicting literature data, limited information is available to validate the specificity of S-mephenytoin for CYP2B6. At 200 μM S-mephenytoin, 500 μM orphenadrine, an inhibitor of CYP2B6, inhibits nirvanol formation by 84% (Heyn et al., 1996). In addition, anti-CYP2B6 antibody inhibits N-demethylation by up to 79% at a substrate concentration of 100 μM (Stresser and Kupfer, 1999). In contrast, incubation of S-mephenytoin with the CYP2C9 inhibitor sulfaphenazole indicates that CYP2C9 is responsible for the high-affinity component of the reaction (Ko et al., 1998). These investigators find that although both recombinant CYP2B6 and CYP2C9 form nirvanol, CYP2B6 forms it at a 4-fold higher rate. Using microsomal intrinsic clearances of 0.98 and 2.1 μl/min/mg for the high- and low-affinity enzymes, respectively, Ko et al. (1998)conclude that CYP2B6 is the main enzyme responsible for formation of nirvanol at substrate concentrations higher than 1000 μM and that CYP2C9 has a major contribution at lower and more clinically relevantS-mephenytoin concentrations. These results indicate a major contribution of CYP2B6 in the formation of nirvanol under high micromolar concentrations.
In summary, the available data suggest that as a probe reaction to represent CYP2B6 activity, S-mephenytoinN-demethylation needs to proceed at high substrate concentrations that reflect low-affinity enzyme activity. In the absence of another suitable substrate, this fairly selective reaction is recommended to detect CYP2B6-based drug interaction potential in vitro.
CYP2C9.
CYP2C9 is a member of the CYP2C subfamily, the second largest P450 subfamily after CYP3A. It exhibits great genetic variation among individuals and is involved in the metabolism of many clinically important drugs that have a narrow therapeutic range, including carbamazepine, phenytoin, and warfarin. Current knowledge is just beginning to allow a clear separation of CYP2C8 from CYP2C9. According to our survey, tolbutamide 4′-hydroxylation is the preferred probe reaction used in 80% of the submissions to characterize both enzymes collectively (Fig. 1). Among other drugs that are used as probe substrates for CYP2C9, we reviewed the warfarin assay because it is one of the in vivo probes most extensively studied by industry investigators (Marroum et al., 2000).
Tolbutamide 4′-Hydroxylation.
Tolbutamide 4′-hydroxylation is the initial and rate-limiting step of tolbutamide elimination. Studies with cDNA expressed enzymes show that at substrate concentrations ≤500 μM, 90% of tolbutamide is hydroxylated by CYP2C9 and 10% by CYP2C8 (Minors et al., 1988; Relling et al., 1990; Ono et al., 1996). An immunoblotting assay with antibody developed against CYP2C9 provides further evidence to support the predominant role of CYP2C9 in tolbutamide 4′-hydroxylation (Edwards et al., 1998). In HLMs with substrate concentrations up to 2.0 mM, tolbutamide hydroxylation exhibits simple Michaelis-Menten kinetics with apparent Km values ranging from 60 to 400 μM, with most values between 100 and 200 μM (Miners et al., 1988; Bourrie et al., 1996). However, recent reports show that CYP2C19 may also catalyze the reaction with aKm value similar to CYP2C9 (Lasker et al., 1998; Wester et al., 2000). The contribution of CYP2C19 to overall tolbutamide 4′-hydroxylation may be minimal, considering the limited protein expression of this enzyme in normal human liver. However, CYP2C19's catalytic role in CYP2C9-deficient liver may be important.
Tolbutamide has a slow turnover rate. The initial rate conditions exist even with incubation times up to 3 h and HLM protein concentrations of 1.6 mg/ml (Miners and Birkett, 1996). Based on our survey, some investigators use a 90-min incubation time and a protein concentration of 2 mg/ml. Although not reported in these studies, the use of high protein concentrations and long incubation times can deplete inhibitors contained in the incubation mixture.
Tolbutamide hydroxylation reaction is very sensitive to the organic solvent effect. At a concentration of 1% (v/v), isopropanol, DMSO, and methanol each inhibit tolbutamide hydroxylation by ≥40% (Hickman et al., 1998). But acetonitrile does not affect the reaction significantly at the same concentration (Chauret et al., 1998; Hickman et al., 1998;Tang et al., 2000). Interestingly, Tang et al. (2000) observe that the solvent effect varies with different probe reactions for CYP2C9; and at concentration >1%, acetonitrile significantly activates tolbutamide hydroxylation in a solvent concentration-dependent manner (Tang et al., 2000).
The collective evidence indicates that tolbutamide 4′-hydroxylation is an appropriate in vitro probe reaction for CYP2C9 activity. But investigators should pay attention to the incubation conditions, the use of organic solvent, and the expression level of CYP2C9 in HLMs when using this reaction to determine CYP2C9-based drug interaction potential in vitro.
S-Warfarin 7′-Hydroxylation.
Several P450s metabolize warfarin, but with different regio- and stereoselectivity. At therapeutic doses, >85% ofS-warfarin is biotransformed to 6′- and 7′-hydroxyS-warfarin in a 1:3 ratio (Toon et al., 1986). With purified and cDNA expressed P450s, CYP2C9 has the highest activity towardS-7′-OH-warfarin formation, followed by CYP1A2 and CYP3A4 (Rettie et al., 1992). In HLMs, the formation ofS-7′-OH-warfarin is inhibited strongly by sulfaphenazole and correlates with tolbutamide 4′-hydroxylation (Hall et al., 1994). At substrate concentrations up to 200 μM, typical Michaelis-Menten kinetics is observed, with a Km of 1 to 5 μM (Lang and Bocker, 1995; Hemeryck et al., 1999). The formation of 6′-OH-metabolite is also carried out by CYP2C9. It has the sameKm value as that of 7′-OH metabolite formation, but with one-third of theVmax value (Rettie et al., 1992; Kunze et al., 1996). However, at substrate concentrations ≥50 μM, at least one other pathway (possibly CYP3A4) also contributes to 6′-OH-formation.
Although not a substrate for CYP2C9, R-warfarin competitively inhibits the formation of 7′-OH-S-warfarin with a Ki value of 6 to 8 μM (Kunze et al., 1991). This inhibition introduces potential complexity in assessing CYP2C9-based drug interactions and thus, commercially available racemic warfarin should not be used as a substrate. Currently, no information on the solvent effect onS-Warfarin 7′-hydroxylation has been reported.
We conclude that if one can overcome the practical challenges,S-warfarin 7′-hydroxylation may also be a good reaction for probing CYP2C9-based drug interaction at substrate concentrations reflecting the high-affinity (low Km) enzyme activity toward this reaction.
CYP2C19.
CYP2C19 is a genetically polymorphic enzyme responsible for the metabolism of mephenytoin, omeprazole, diazepam, and many psychotherapeutic agents. Poor metabolizers represent ∼2.5 to 5% of Caucasian populations, 19% of African populations, and up to 30% of Asian populations (Pollock et al., 1991; Flockhart, 1995). Mephenytoin, an anticonvulsant agent, has long been used as an in vitro and in vivo probe substrate for CYP2C19. Its unequivocal role in drug development is reflected in our survey, where it is the only drug used to assess CYP2C19 activity (Fig. 1; Table 1) Recent studies suggest that omeprazole (5-hydroxylation) may also be used as a probe for CYP2C19 activity (Flockhart, 1995). However, in vitro studies show that CYP3A4 carries out the same reaction for omeprazole. At 10 μM, the contribution of each enzyme depends on the ratio of their expression levels in HLMs (Yamazaki et al., 1997). Considering the much higher expression level of CYP3A4 compared with CYP2C19, we do not consider omeprazole a preferred in vitro probe for CYP2C19 activity.
S-Mephenytoin 4′-Hydroxylation.
Mephenytoin exists as a racemic mixture of R- andS-enantiomers and its metabolism is stereospecific. In HLMs of extensive metabolizers, S-mephenytoin is metabolized to the 4′-hydroxyl metabolite and nirvanol (Jurima et al., 1985). Studies with inhibitory antibody against CYP2C19 and purified and cDNA-expressed enzymes validate the exclusive role of CYP2C19 in 4′-hydroxylation of S-mephenytoin (Shimada et al., 1986;Wrighton et al., 1993; Inoue et al., 1997). Kinetic studies ofS-mephenytoin 4′-hydroxylation consistently show monophasic Michaelis-Menten kinetics, with a Kmof 31 to 340 μM (Jurima et al., 1985; Hall et al., 1987; Chiba et al., 1993) in HLMs. The variability inKm values reported in different studies primarily reflects different experimental conditions; but it may also reflect genetic variations, especially in HLM prepared from Asian or African individuals where the percentage of poor metabolizers is high. In the poor metabolizers, S-mephenytoin 4′-hydroxylation is not mediated by CYP2C19 but by other enzymes in place of CYP2C19. Thus, microsomes that are deficient in CYP2C19 should not be used to examine CYP2C19-based drug interactions.
S-Mephenytoin 4′-hydroxylation is sensitive to solvent effect. Dimethylformamide, DMSO, and isopropranol at 1% (v/v) are reported to inhibit S-mephenytoin 4′-hydroxylation by >70%, but acetonitrile and methanol at the same concentration do not affect the reaction significantly (Chauret et al., 1998; Hickman et al., 1998).
In summary, under optimal experimental conditions,S-mephenytoin 4′-hydroxylation is the preferred probe reaction for CYP2C19 activity. Investigators should pay attention to the use of organic solvent, and the expression level of CYP2C19 in HLMs when using this reaction to determine CYP2C19-based drug interaction in vitro.
CYP2D6.
CYP2D6 is a polymorphically expressed P450 enzyme. About 5 to 10% of Caucasians are poor metabolizers of CYP2D6 substrates. As with polymorphically expressed CYP2C19, using prototype substrates to assess CYP2D6-based drug interaction is meaningful only in the extensive metabolizer's liver microsomes. Although CYP2D6 only constitutes about 2% of total P450 enzymes in the liver (Shimada et al., 1994), it is responsible for metabolizing drugs in a variety of therapeutic classes, including antidepressants, antipsychotics, and β-blockers. Our survey indicates that dextromethorphan (O-demethylation) and bufuralol (1′-hydroxylation) are the two in vitro CYP2D6 probe substrates preferred by industry investigators. More than 60% of submissions used bufuralol as the probe substrate to assess CYP2D6 activity, and 30% used dextromethorphan.
Bufuralol 1′-Hydroxylation.
Bufuralol is a chiral adrenoceptor antagonist that undergoes extensive oxidative metabolism in humans (Francis et al., 1982; Dayer et al., 1983, 1986), where the aliphatic 1′-hydroxylation accounts for 95% of bufuralol clearance (Mankowski, 1999).
Enzyme kinetic studies demonstrate biphasic formation of 1′-OH-bufuralol at bufuralol concentrations up to 100 μM, where the high-affinity component has a Km of 4 to 10 μM and the low-affinity component has aKm of 83 to 600 μM (Gut et al., 1986; Kronbach et al., 1987). The similarVmax for the two components results in higher intrinsic clearance and thereby greater contribution to the overall hydroxylation by the high-affinity component. Quinidine, a selective and potent CYP2D6 inhibitor, diminishes 90 and 70% of the reaction at bufuralol concentrations of 1 and 50 μM, respectively (Mankowski, 1999). Studies with cDNA-expressed enzymes show that CYP2D6 exhibits the highest ability to form 1′-OH-bufuralol, followed by CYP2C19. CYP2D6 catalyzed the reaction with 1/10 of theKm of CYP2C19, but at a 36-fold higher rate (Mankowski, 1999). These results suggest that CYP2D6 is the enzyme responsible for the high-affinity component and CYP2C19 is responsible for the low-affinity component of 1′-OH bufuralol formation.
Using inhibitory monoclonal antibody against CYP2D6, Gelboin et al. (1997) show that, at 50 μM substrate concentration, 1′-OH-bufuralol formation is only partially carried out by CYP2D6. CYP2C19, and to a lesser extent CYP2C8/9 and CYP1A2, also contribute to the metabolism of bufuralol. Whether the formation of 1′-OH bufuralol represents CYP2D6 activity depends on its relative expression levels, compared with these other enzymes in HLMs. This study indicates bufuralol 1′-hydroxylation loses its selectivity for CYP2D6 at substrate concentrations greater than 50 μM.
It is important to consider the effect of solvents on this reaction. Studying the reaction rate with cDNA-expressed enzymes, Busby et al. (1999) show >50% inhibition of 1′-OH bufuralol formation with ethanol, DMSO, and methanol at solvent concentrations of 3% (v/v). But at 1% (v/v), acetonitrile does not inhibit the reaction significantly.
Despite the biphasic kinetics of racemic bufuralol at concentrations ranging from 1 to 1000 μM, racemic bufuralol is a good in vitro CYP2D6 probe substrate. When using it to determine CYP2D6-based drug interaction potential in vitro, investigators should pay attention to the selection of organic solvent, the expression level of CYP2D6 in HLMs; and should use low substrate concentrations that primarily reflect the high-affinity enzyme activity toward this reaction.
Dextromethorphan O-Demethylation.
Dextromethorphan, an antitussive drug, undergoes two parallel oxidative metabolic pathways, O-demethylation by CYP2D6 to form dextrorphan (DXP) and N-demethylation by CYP3A to form 3-methoxymorphinan (Jacqz-Aigrain et al., 1993). Both of these products undergo sequential metabolism in vitro, unless optimal incubation conditions are used (Hickman et al., 1998).
Biphasic enzyme kinetics are observed for DXP formation at dextromethorphan concentrations up to 2000 μM, withKm values for the high-affinity component being 2.2 to 8.5 μM (Kronbach, 1991; Jacqz-Aigrain et al., 1993). Km for the low-affinity component is at least 10-fold higher (70–1880 μM). Under optimal incubation conditions, i.e., protein concentration of 0.4 mg/ml, incubation time less than 60 min and substrate concentration less than 50 μM, the Eadie-Hofstee plot for DXP formation shows a monophasic characteristic (Kronbach, 1991; Hickman et al., 1998).
Studies with cDNA-expressed enzymes show that both CYP2C9 and CYP2D6 catalyze DXP formation. At 10 μM substrate concentrations, the CYP2D6-mediated reaction proceeds at a rate 5-fold greater than that by CYP2C9 (Ono et al., 1996). But at 500 μM, the CYP2D6 reaction rate is only one-sixth of the CYP2C9-based reaction rate. At a dextromethorphan concentration of 5 μM, quinidine completely abolishes the reaction, confirming the selective role of CYP2D6 at low substrate concentrations (Bourrie et al., 1996). Using monoclonal antibody against CYP2D6,Gelboin et al. (1997) show 50 to 93% inhibition of the formation of DXP.
Organic solvents seem to have less effect on dextromethorphan than on bufuralol, providing some advantage for using this substrate as an in vitro probe for assessing CYP2D6 activity (Hickman et al., 1998; Busby et al., 1999). But as with bufuralol, the experiment with dextromethorphan should proceed with low substrate concentrations to reflect the high-affinity enzyme (i.e., CYP2D6) activity in vitro.
Although other CYP2D6 probe substrates (such as metroprolol, spartein, and debrisoquin) can be as selective as bufurolol and dextromethorphan, the latter two are particularly attractive because of the availability of the assayed species and the fluorescent nature of these species that permits the development of a highly sensitive detection assay. Thus, at appropriate experimental conditions, bufuralol 1′hydroxylation and dextromethorphan O-demethylation are both preferred reactions to probe CYP2D6-based drug interaction potential.
CYP2E1.
CYP2E1 metabolizes chlorzoxazone, acetaminophen, and the volatile anesthetics, including enflurane, sevoflurane, methoxyflurane, and isoflurane. Among these drugs, chlorzoxazone is the preferred in vitro probe substrate used in 60% of the surveyed investigator submissions (Fig. 1).
Chlorzoxazone 6-Hydroxylation (6-OH).
Chlorzoxazone is an analgesic muscle relaxant that can be used in in vitro and in vivo drug metabolism studies. After ingestion, chlorzoxazone is rapidly absorbed and extensively metabolized. In HLMs, 6-OH-chlorzoxazone is the sole metabolite formed, which makes the assay highly specific.
However, the selectivity of chlorzoxazone for CYP2E1 is controversial. A study by Peter et al. (1990) indicates that rabbit anti-P450 monoclonal antibody against human CYP2E1 inhibits 81 to 87% of chlorzoxazone 6-hydroxylation, a monophasic reaction with aKm value of 40 μM. However, Shou et al. (2000) demonstrate that inhibitory monoclonal antibody inhibits 20 to 80% of the formation of 6-OH-chlorzoxazone at a substrate concentration of 200 μM. In the majority of 18 liver donors the inhibition was around 50% (Shou et al., 2000). Gorski et al. (1997)show that rabbit anti-human CYP3A inhibits the reaction by 47%, suggesting a significant contribution of CYP3A in the reaction. Using cDNA-expressed enzymes, Ono et al. (1996) demonstrate that the same reaction is also catalyzed by CYP1A2, with aKm value being one-third of that by CYP2E1 (Ono et al., 1996). At a chlorzoxazone concentration of 10 μM, CYP2E1 catalyzes the reaction at the same rate as CYP1A2; but at 500 μM, CYP2E1 catalyzes the reaction at a rate 10 times higher than CYP1A2. The above-mentioned evidence indicates that the formation of 6-OH-chlorzoxazone is more specific for CYP2E1 activity at high substrate concentrations than at low substrate concentrations.
In HLMs, there is a prominent solvent effect on 6-OH-chlorzoxazone. Methanol at 0.2% (v/v) decreases 6-OH-chlorzoxazone by 60% (Chauret et al., 1998). Hickman et al. (1998) report that 1% (v/v) acetone, dimethylformamide, DMSO, and isopropanol inhibit the reaction by >70%. Acetonitrile, on the other hand, does not show an effect until the concentration reaches 5% (v/v).
Literature evidence indicates that chlorzoxazone 6-hydroxylation is the current preferred probe reaction for CYP2E1, but it is important to use high substrate concentrations that reflect low-affinity enzyme (i.e., CYP2E1) activity toward this reaction, and to consider solvent effects in the experiment. However, a substrate that offers better enzyme selectivity and lower solvent effect would be more desirable.
CYP3A.
CYP3A is the most abundant P450 enzyme in humans, accounting for an average 30 to 40% of total P450 protein in the liver. It has three isoforms in various tissues: CYP3A4 and CYP3A5 predominantly in liver and gut, and CYP3A7 in fetal liver. Current data indicate that CYP3A4 is the most important CYP3A member with regard to involvement in clinically significant drug interactions. Many probe reactions represent the activity of this enzyme, as reflected in our survey. The substrate used most often by industry investigators is testosterone, followed by midazolam, nifedipine, and erythromycin (Fig. 1).
Testosterone 6β-Hydroxylation.
Steroid hydroxylation has long been recognized as a CYP3A-mediated reaction. In HLMs, numerous studies demonstrate that selective CYP3A4 inhibitors diminish the 6β-hydroxylation of testosterone (Wrighton et al., 1989; Newton et al., 1995; Bourrie et al., 1996). Specific antibodies against CYP3A4 inhibit more than 90% 6β-OH-testosterone formation at substrate concentrations of 200 to 250 μM (Gelboin et al., 1995; Mei et al., 1999; Shou et al., 2000). Studies with purified human proteins (Yamazaki and Shimada, 1997) and cDNA-expressed enzymes (Waxman et al., 1991; Ono et al., 1996) provide more evidence showing that all CYP3A members, other than CYP3A7, catalyze testosterone-6β-hydroxylation; CYP3A4 exhibits the highest activity. CYP2C9 and CYP2C19 also catalyze the reaction, but at 1/10 the rate of CYP3A4. In HLMs, CYP3A4-mediated 6β-OH-testosterone formation has aKm of 50 to 100 μM. No significant solvent effect is seen with methanol and acetonitrile at solvent concentrations ≤1% in either HLMs or cDNA-expressed enzymes (Chauret et al., 1998; Busby et al., 1999). We conclude that, at substrate concentrations at or lower than 250 μM, testosterone-6β-hydroxylation rate primarily reflects the CYP3A4 activity, and thus can be used to probe drug interaction potential of a new drug toward this enzyme in vitro.
Midazolam 1′-Hydroxylation.
Midazolam is a short-acting benzodiazepine routinely used to induce sedation and anesthesia. It is available for both intravenous and oral administration, which provides a unique opportunity to study gastrointestinal-based or liver-based drug interactions in vivo.
Biotransformation of midazolam in humans yields two primary hydroxylated metabolites: 1′-OH and 4-OH, both of which are further metabolized at a much slower rate (Kronbach et al., 1989). The 1′-OH metabolite accounts for more than 90% of the total hydroxylation reaction (Ghosal et al., 1996). Mounting evidence, including studies with inhibitory antibodies, specific chemical inhibitors, and cDNA-expressed enzymes, demonstrate that CYP3A4 mediates the formation of the two metabolites (Kronbach et al., 1989; Wrighton and Ring, 1994). However, midazolam is also readily metabolized by CYP3A5 with similar Km value as by CYP3A4, although CYP3A5 shows different catalytic activities from CYP3A4 (Gibbs et al., 1999).
In HLMs at incubation times up to 5 min, midazolam metabolism follows simple Michaelis-Menten kinetics. But at high substrate concentrations, substrate-inhibition kinetics become apparent for 1′-OH-midazolam formation, but not for 4-OH-midazolam formation (Kronbach et al., 1989). A similar phenomenon is reported with cDNA-expressed enzymes (Ghosal et al., 1996). Although formed by the same enzyme, theKm value for 1′-hydroxylation of midazolam is 3 to 5 μM, and for 4-hydroxylation is 40 to 60 μM.
Midazolam is insoluble in water, so an organic solvent is often used in the in vitro experiment to dissolve this substrate. A previous study indicates that acetone may enhance midazolam metabolism in HLMs (Kronbach et al., 1989), but other solvent effects have not been reported. Because of the complexity of CYP3A-based drug-drug interactions (as delineated below) and the common use of this reaction to assess CYP3A activity, a thorough study of the solvent effect on midazolam is needed. Under optimal experimental conditions, midazolam 1′-hydroxylation seems to be a good in vitro probe reaction for CYP3A activity at substrate concentrations less than 10 μM. However, using this reaction does not allow one to determine whether the interaction is based on CYP3A4 or CYP3A5, unless the study is conducted in HLMs without detectable CYP3A5.
Because most of the reported studies do not differentiate CYP3A4 activity from CYP3A5, and use CYP3A4 to reflect both or either enzyme activity, we follow the same nomenclature in the following text.
Use of Two or More Probe Reactions for CYP3A4-Based Drug Interactions.
Although the knowledge of CYP3A4-catalyzed reactions is growing fast, in vivo CYP3A4-based drug metabolism and interactions remain among the most difficult scenarios to predict in vitro. The difficulty arises from the complex substrate-enzyme interaction at the molecular level (Ueng et al., 1997), the involvement of efflux transport systems in the substrate's disposition in vivo (Takano et al., 1998; Yumoto et al., 1999), and the contribution of gastrointestinal metabolism (Gorski et al., 1998).
Based on chemical inhibition characterizations and substrate correlation analyses, testosterone and midazolam seem to belong to two distinct groups of CYP3A4 substrates (Kenworthy et al., 1999; Stresser et al., 2000). Although the metabolic activity of testosterone is highly correlated with those of CYP3A4/5 substrates with large molecular weight, such as erythromycin or cyclosporin A, it is only weakly related to those of benzodiazepines such as midazolam. Thus, the effect of a CYP3A4 modulator depends on the substrate used in the experiment (Kenworthy et al., 1999). Fluconazole, for example, displays 65% inhibition in midazolam assay but 37% in testosterone assay. Nimodipine, on the other hand, displays 60% inhibition in midazolam assay but 96% in testosterone assay. Besides these two groups of substrates, additional groups such as those represented by nifedipine or benzoxyl-resorufin may also exist. Carbamazepine, which shows a negligible effect on testosterone and midazolam, inhibits nifedipine by 100% (Stresser et al., 2000). Although the mechanism of this substrate-dependent phenomenon is not known, two or more in vitro probe substrates from different groups may be needed to accurately predict CYP3A4-based drug interactions in vivo.
Conclusions
In this survey study, we review the most commonly used in vitro probe substrates from pharmaceutical industry submissions. These probe substrates have been widely studied and their characteristics are described in the literature. Table 2summarizes our recommended reactions for all of the evaluated P450 enzymes.
Summary of probe substrate metabolic reactions used in in vitro drug metabolism studies
Because the selectivity of represented P450 depends on the specific experimental conditions, the use of appropriate experimental conditions in in vitro studies is crucial. In particular, it is important to understand the enzyme kinetics of the reaction and the involvement of high-affinity and low-affinity enzymes (when multiple enzymes metabolize the same reaction) to determine the appropriate substrate concentrations to use. To study high-affinity enzyme, one should use substrate concentrations that reflect the lowKm enzyme activity (e.g., bufuralol 1′-hydroxylation and dextromethorphan O-demethylation for CYP2D6); and for a low-affinity enzyme, one should use high substrate concentrations (e.g., S-mephenytoinN-demethylation for CYP2B6 and chlorzoxazone 6-hydroxylation for CYP2E1). Because of the observed significant solvent effects on reaction rates (especially at solvent concentration >1%), if possible, investigator should avoid using organic solvent or use it at low strength. For CYP3A4, two or more probe reactions may be needed to yield an overall evaluation of potential drug interaction. For other enzymes, such as CYP2E1, further investigations for a better probe reaction may be needed.
Opinions regarding the most appropriate probe substrates for individual P450 enzymes are evolving. In addition to the probe substrates presented in this article, industry and academia investigators use other appropriate probe substrates. The proceedings of a consensus meeting convened in 2000 include a representative list of preferred and acceptable in vitro probe substrates (Tucker et al., 2001). Under appropriate experimental conditions, these substrates provide useful information.
The in vitro probe reaction is a useful tool to screen for potential in vivo drug interactions. Due to genetic variation, the influence of environmental or hormonal factors, as well as intrinsic limitations of in vitro systems, the quantitative prediction of in vivo drug interactions for an individual patient remains a challenge. However, with the rapid growth of our knowledge and technology in drug metabolism and disposition, quantitative prediction may be achievable in the future. The conduct of high-quality in vitro studies is the first step toward this goal.
Acknowledgments
We acknowledge Dr. Larry Lesko for strong support of this research. We also greatly appreciate the generous support from Dr. Anthony Lu, who kindly offered unlimited encouragement and expertise on drug metabolism during various phases of this project.
Footnotes
-
This work was supported by the Intramural Regulatory Science and Review Enhancement grant awarded by the Center for Drug Evaluation and Research, United States Food and Drug Administration in 1998. However, the views expressed in this manuscript are personal and may not represent the agency's position.
- Abbreviations used are::
- P450
- cytochrome P450
- DMSO
- dimethyl sulfoxide
- HML
- human liver microsome
- 7-EFC
- 7-ethoxy-4-trifluoromethyl coumarin
- DXP
- dextrorphan
- 6-OH
- 6-hydroxylation
- Received July 31, 2002.
- Accepted September 17, 2002.
- U.S. Government




{kind=link}