


Abstract
We present herein a compilation and trend analysis of human i.v. pharmacokinetic data on 670 drugs representing, to our knowledge, the largest publicly available set of human clinical pharmacokinetic data. This data set provides the drug metabolism scientist with a robust and accurate resource suitable for a number of applications, including in silico modeling, in vitro-in vivo scaling, and physiologically based pharmacokinetic approaches. Clearance, volume of distribution at steady state, mean residence time, and terminal phase half-life were obtained or derived from original references exclusively from studies utilizing i.v. administration. Plasma protein binding data were collected from other sources to supplement these pharmacokinetic data. These parameters were analyzed concurrently with a range of simple physicochemical descriptors, and resultant trends and patterns within the data are presented. Our findings with this much expanded data set were consistent with earlier described notions of trends between physicochemical properties and pharmacokinetic behavior. These observations and analyses, along with the large database of human pharmacokinetic data, should enable future efforts aimed toward developing quantitative structure-pharmacokinetic relationships and improving our understanding of the relationship between fundamental chemical characteristics and drug disposition.
The prediction of human pharmacokinetics for new compounds has become an important process in drug research. Previous reports have focused on prediction methods that utilize animal pharmacokinetic data (Caldwell et al., 2004; Ward and Smith, 2004a,b; Jolivette and Ward, 2005; Evans et al., 2006; Mahmood et al., 2006; Martinez et al., 2006; Tang and Mayersohn, 2006; Fagerholm, 2007; McGinnity et al., 2007) and in vitro data (Obach et al., 1997; Lombardo et al., 2002, 2004; Nestorov et al., 2002; Riley et al., 2005; Grime and Riley, 2006). Recently, the availability of computational chemistry methodologies has increased, and these have been applied to the prediction of human pharmacokinetics and/or general absorption-distribution-metabolism-excretion-toxicology properties (Cruciani et al., 2005; Ghafourian et al., 2006; Gleeson et al., 2006; Lombardo et al., 2006; Gleeson, 2007; Gunturi and Narayanan, 2007; Norinder and Bergstroem, 2007). The construction of effective models not only requires sound computational tools but, very importantly, databases that have been carefully assembled. Human pharmacokinetic databases are challenging to compile because each data point typically derives from a separate report in which experimental approaches differ from report to report. Such variables include the numbers and types of study subjects (e.g., healthy versus diseased, gender, age, etc.), the routes of administration and doses, sample collection times, methods of analysis of the samples, and the types of pharmacokinetic parameters reported.
To develop computational models for the prediction of human clearance (CL), volume of distribution (VD), and absolute oral bioavailability, it is essential that data used in model training sets are obtained from studies in which the dose was administered intravenously. It is also important that the methods of calculations for reported pharmacokinetic parameters be done consistently. (For example, values reported generally as VD can vary from report to report and include central VD, terminal phase VD, or steady-state VD.) The fairly large set of human pharmacokinetic data reported in Appendix II of the famous textbook, The Pharmacological Basis of Therapeutics (Goodman and Gilman, 2006), has been frequently cited as a source of data for computational model construction. However, it is important to note that this data set was primarily intended for health care professionals and medical students to understand the pharmacokinetic basis for dosing regimens of frequently used drugs, rather than used for the development of structure-pharmacokinetic relationships. Hence, many of the pharmacokinetic parameters reported are derived from oral administration, and many of the VD values reported include terminal phase VD. This can confound the performance of models.
The objectives of this study were 2-fold: 1) to develop an exhaustive database of human pharmacokinetic parameters exclusively from i.v. administration that can be used by scientists involved in early drug research in the construction of models for predicting pharmacokinetic parameters for new compounds and 2) from this data set, to gain some initial insight into the relationships between chemical properties derived from structural attributes to human pharmacokinetic parameters. To the first objective, we have carefully and exhaustively mined the scientific literature for the human pharmacokinetic parameters, CL, steady-state volume of distribution (VDss), MRT, and t1/2, measured after i.v. administration. We successfully obtained human i.v. pharmacokinetic data for 670 compounds, which, to our knowledge, represents the largest database of this type. We have also included plasma protein binding data for most of these compounds. This database is reported herein and also provided in tabular format as supplemental data that can be downloaded from the web site of this journal. As such, it can be of use to scientists seeking to develop computational models and correlative approaches to the prediction of human pharmacokinetics of new compounds. For the second objective, we have examined the relationships between human i.v. pharmacokinetic parameters (VDss,VDss,u, CL, CLu, MRT, t1/2, and fu) and various computed fundamental physicochemical parameters (e.g., logP, charge, etc.) using this database. The trends observed can be instructive to those engaged in the design of new drugs. The knowledge that can be gained by utilizing this large database in model construction should be of considerable use to scientists involved in the discovery of new drugs.
Materials and Methods
Collection and Selection of Data for the Database. Pharmacokinetic data are reported in a large array of manners. Various units and symbols are used, some reports utilize compartmental analysis, whereas others use a noncompartmental approach, and the level of detail describing the conduct of the study included in the methods sections can vary considerably. This requires very careful scrutiny when mining these data for fundamental pharmacokinetic parameters. The pharmacokinetic data included in this database are strictly from i.v. administration. There are no data included from oral, i.m., or any other dosing routes. Intravenous data were from rapid bolus injection or infusions.
Of the pharmacokinetic parameters that were gathered for this database, the one that varied the most and required the most attention to how it was determined is the volume of distribution. For this database, we sought the VDss because this volume term describes the overall distributional behavior most generally. However, many authors of pharmacokinetic studies report only the terminal phase volume of distribution, and many others report volume of distribution without denoting whether it was the terminal phase VD, the central VD, or the steady-state VD. Thus, the preference for papers selected for inclusion in this database specifically reported VDss. When the usage of the VD term was vague (i.e., lacking the subscript designating the value as VDss or another VD), but a clear description of the pharmacokinetic calculations was included and indicated that the term was in fact VDss, the value was included. In other papers, microconstants from compartmental analysis were reported without including VDss. In these cases, the rate constants and the reported central volume of distribution were used to calculate VDss using one or the other of the following equations, depending upon what was reported: 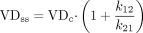 VDc refers to the volume of the central compartment and was sometimes called V1, VP, or V0 in the reports. In other compartmental models, more microrate constants were derived from the fit of the data, requiring an expansion of the equation:
VDc refers to the volume of the central compartment and was sometimes called V1, VP, or V0 in the reports. In other compartmental models, more microrate constants were derived from the fit of the data, requiring an expansion of the equation: 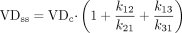 In other cases, the compartmental parameters were reported. If VDss was not included in the paper, then the equation
In other cases, the compartmental parameters were reported. If VDss was not included in the paper, then the equation 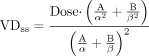 was used to calculate VDss. In a few papers, pharmacokinetic parameters were not reported, but plasma concentration versus time data were listed. In these cases, VDss was calculated using the standard equation using mean plasma concentration versus time data:
was used to calculate VDss. In a few papers, pharmacokinetic parameters were not reported, but plasma concentration versus time data were listed. In these cases, VDss was calculated using the standard equation using mean plasma concentration versus time data: 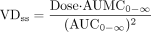 In some cases, a drug had neither the VDss reported per se, nor did it have compartmental parameters reported that could be used to calculate VDss in any description of its pharmacokinetics. In these instances, plots of plasma concentration versus time included in the report were digitized, and the resulting concentration versus time data were subjected to noncompartmental analysis. This was only done if there were no other reports on the same drug that had reported VDss values. In some cases, the only references containing pharmacokinetic values were review articles in which the cited original research data were not available. If the VD values listed in these articles were clearly stated to derive from i.v. administration, and if they were specifically described as steady-state VD values, these values were used. Product labels, in which the data listed have been reviewed and approved by the U.S. Food and Drug Administration, were sometimes the only source of VDss data. In these cases, because these had been subjected to a governmental regulatory review, they were included in this database even though the original methods and results that yielded the data were not included in the product label.
In some cases, a drug had neither the VDss reported per se, nor did it have compartmental parameters reported that could be used to calculate VDss in any description of its pharmacokinetics. In these instances, plots of plasma concentration versus time included in the report were digitized, and the resulting concentration versus time data were subjected to noncompartmental analysis. This was only done if there were no other reports on the same drug that had reported VDss values. In some cases, the only references containing pharmacokinetic values were review articles in which the cited original research data were not available. If the VD values listed in these articles were clearly stated to derive from i.v. administration, and if they were specifically described as steady-state VD values, these values were used. Product labels, in which the data listed have been reviewed and approved by the U.S. Food and Drug Administration, were sometimes the only source of VDss data. In these cases, because these had been subjected to a governmental regulatory review, they were included in this database even though the original methods and results that yielded the data were not included in the product label.

Span of pharmacokinetic values for the 670 compounds included in this analysis. A, VDss; B, CL; C, t1/2;D,fu.
From the sources that possessed adequate VDss data, could have VDss calculated from compartmental parameters, or could have VDss calculated from digitized plots, the values of plasma clearance and half-life were also obtained. Parameters (CL, VDss) that were already corrected for body weight were included as is. If they were not reported in this manner, then they were converted using the mean body weight of the subjects listed in the report. If only a range of body weights was included in the report, then the midpoint value was used for weight. In cases where no body weight information was included, a value of 70 kg was assumed. For those reports in which the parameters were corrected for body surface area, a conversion of 1.73 m2 to 70 kg was generally assumed.
The pharmacokinetic data were preferably obtained from reports in which healthy young adult subjects were studied or patient populations in which health or physiological condition is not severely compromised such that there would be reason to believe that that pharmacokinetics in the patients would be different from healthy subjects (e.g., elderly subjects, obese subjects, etc.). In some instances, data were only available from patient populations and/or populations taking concomitant medications (e.g., cancer drugs), and in these instances, the data were included. In many cases, reports of pharmacokinetic data were made with the intent to compare different populations, especially drug-drug interaction studies and studies comparing healthy individuals with those with either hepatic or renal dysfunction. In these cases, the pharmacokinetic data were only from the study groups that were used as the controls for comparison (the healthy group in hepatic or renal dysfunction studies or the groups not receiving a second drug in drug interaction studies). Other sources of i.v. pharmacokinetic data were studies in which the objective was the determination of the absolute oral bioavailability. In these instances, the pharmacokinetic parameters from the i.v. part of the study were used. Values were reported to two significant digits, and data were rounded from those reports in which the values were reported to greater than two digits. Finally, when multiple studies within a report and/or different reports were used, the data were weight-averaged by number of subjects.

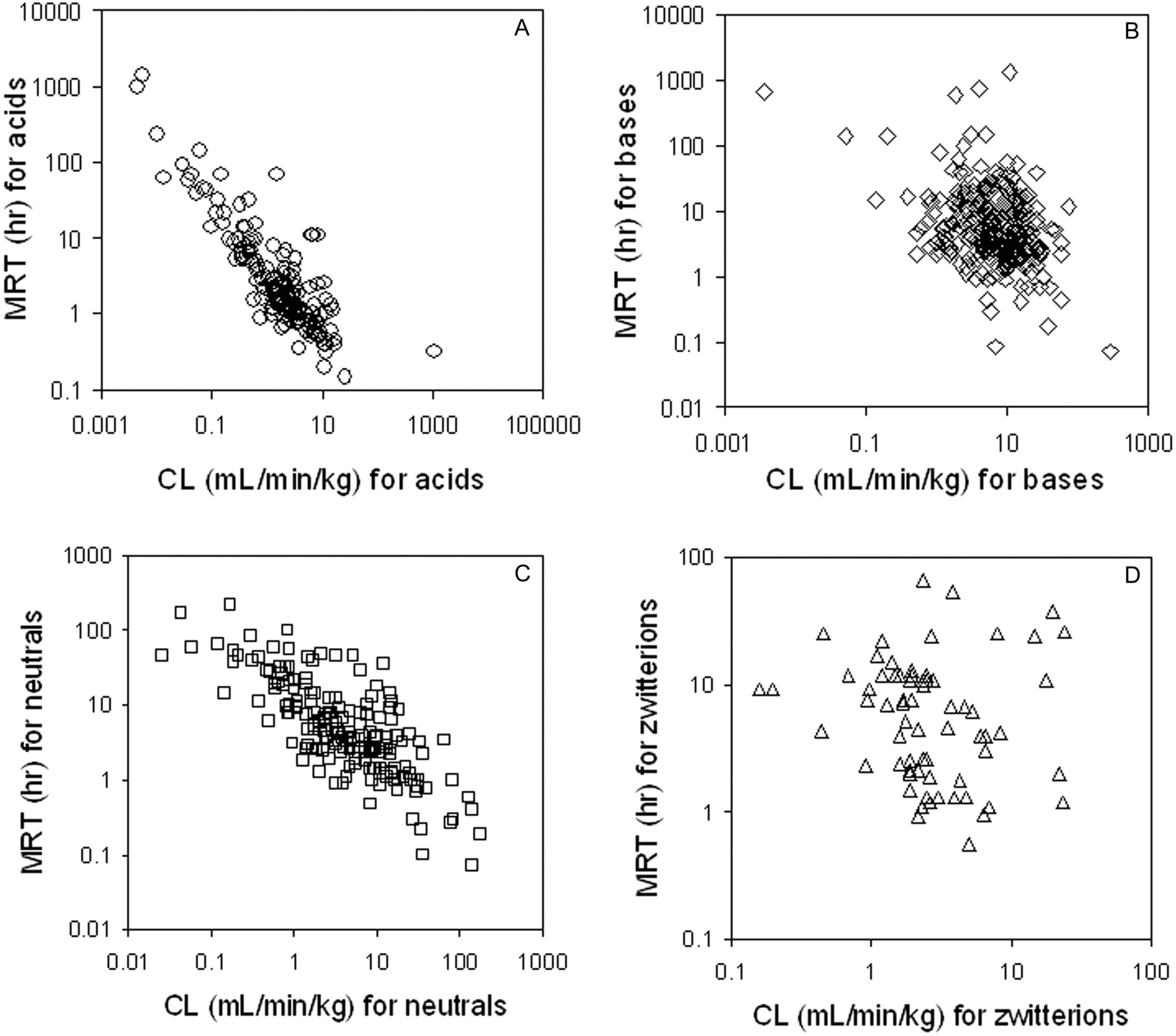
Relationship between MRT and CL for acids (A), bases (B), neutrals (C), and zwitterions (D). A color figure containing all of the compounds in one plot is available in the Supplemental Data.
For compounds possessing adequate i.v. pharmacokinetic data, the corresponding protein binding values in human plasma (or serum) were searched. In all cases, original research reports containing these values were sought, but protein binding data were included even in those cases where the original methods and results were not present (e.g., product labels, review articles). We then calculated the logK as the logarithm of the bound/free ratio using the fu data reported. However, in many instances, no published protein binding data could be found. The physicochemical parameters calculated for all compounds and described under Results were extracted from the table available for each compound through SciFinder (2007), generated via version 8.14 of the ACD Labs software (ACD Labs, Toronto, ON, Canada).
Results
Characteristics of the Pharmacokinetic and Physicochemical Values. Through extensive mining of the scientific literature and in some cases reanalysis of concentration versus time data, the human i.v. pharmacokinetic parameters, VDss, CL, MRT, and t1/2, were found or calculated for 670 compounds. In addition, human plasma protein binding values were obtained for 554 of the 670 compounds. These data are listed in Table 1, and a spreadsheet containing these values along with the literature references is included as an attachment in the Supplemental Data. The data span over considerable ranges (Fig. 1). Volume of distribution at steady state ranged from a low of 0.035 to 700 l/kg, for indocyanine green and hydroxychloroquine, respectively. The vast majority (90%) reside in a 100-fold range between 0.1 and 10 l/kg, with mean and median values of 4.2 and 0.96 l/kg, respectively. Forty-one percent of the compounds had VDss values less than 0.7 l/kg (Table 2), a value generally accepted as representative of total body water. Eight percent of compounds had VDss values greater than 10 l/kg, indicating an extensive level of tissue partitioning.
Summary of i.v. pharmacokinetic parameters and plasma protein binding values for 670 compounds in humans
Characteristics of the human i.v. pharmacokinetic parameters and plasma protein binding for 670 compounds
Plasma clearance values ranged from 0.0037 (7-hydroxystaurosporine) to 1070 (artesunate) ml/min/kg (Fig. 1), with mean and median values of 10 and 4.0 ml/min/kg, respectively. Approximately three fifths of the compounds resided in a range between 1 and 10 ml/min/kg, with 16% possessing clearance values below 1 ml/min/kg (very low clearance). Fifty-six of the compounds had CL values greater than liver blood flow (Table 2), suggesting the possibility of extrahepatic clearance mechanisms in these cases or blood/plasma ratios in excess of unity. Many of the compounds possessing high CL values are agents like anesthetics, pain medications, or cytotoxic cancer chemotherapeutics, which are drug classes that are frequently administered via the i.v. route to optimize therapy or use, and other high-CL compounds are prodrugs or drugs in which pharmacologically active metabolites are responsible for the bulk of the effect (e.g., dolasetron, esmolol, etc.).
Terminal phase half-life values ranged from 4 min (indocyanine green) to 50 days (suramin) (Fig. 1), with two thirds residing between 1 and 12 h (Table 2). (Half-lives for the bisphosphonate class of drugs may be underestimated because they sequester into bone but may not be detectable in the systemic circulation.) The average t1/2 was 18 h, and the median value was 4.1 h. Because half-life is derived from CL and VD, it cannot be directly related to physiological properties such as hepatic blood flow or volume of total body water. Half-life values can be partitioned into zones loosely based on dosing regimen frequency values (provided that there is not a considerable difference between pharmacokinetics and pharmacodynamics), and it is a common practice in pharmaceutical research to seek drugs that would be amenable to convenient q.d. dosing regimens. However, based on this, over three quarters of compounds would likely require dosing greater than once per day because they have t1/2 values below 12 h (Table 2). This same conclusion could be drawn if MRT were used to address this question. Interestingly, a plot of MRT versus CL (Fig. 2) shows a clustering of data points that implies that to obtain a MRT of at least 5 h for an acidic drug, a CL value lower than 0.5 ml/min/kg is required, a consequence of the low VDss for acidic compounds.
The ranking of protein binding values is in Fig. 1. The values ranged between no binding (several compounds) and 99.98% bound (amiodarone), with mean and median free fractions of 0.38 and 0.26, respectively. Two thirds of the compounds in the set are less than 90% bound, and about one eighth could be considered highly bound (>99%; Table 2).
The 670 compounds in this data set span a wide range of fundamental physicochemical characteristics (Fig. 3). The typical drug-like space for molecular mass (200–600 Da) is represented by 80% of the compounds, with a median value of 342 Da and a range from 3 (lithium) to 1816 (dalbavancin). The median value for polar surface area (PSA) was 87 Å2. The set encompasses 159 acids, 271 bases, 173 neutral compounds, and 67 zwitterions (Fig. 3). These were categorized with a cutoff value of 10% ionized (anionic for acids; cationic for bases, both for zwitterions) at pH 7, using the calculated pKa values from ACD Labs. Lipophilicity, as described by clogP and clogD7.0, had median values of 1.92 and 0.42, respectively. The median values for numbers of rotatable bonds, hydrogen bond acceptors, and hydrogen bond donors were 5, 6, and 2, respectively. It can be noted that, even though these data were derived from i.v. dosed compounds, the median values for logP and for the number of hydrogen bond acceptors and hydrogen bond donors were well below the limits set by the Lipinski “rule of 5” (Lipinski et al., 1997), as perhaps may be expected for compounds either marketed or in clinical development. Furthermore, the number of rotatable bonds was below the limit reported by Veber et al. (2002) for permeable compounds.
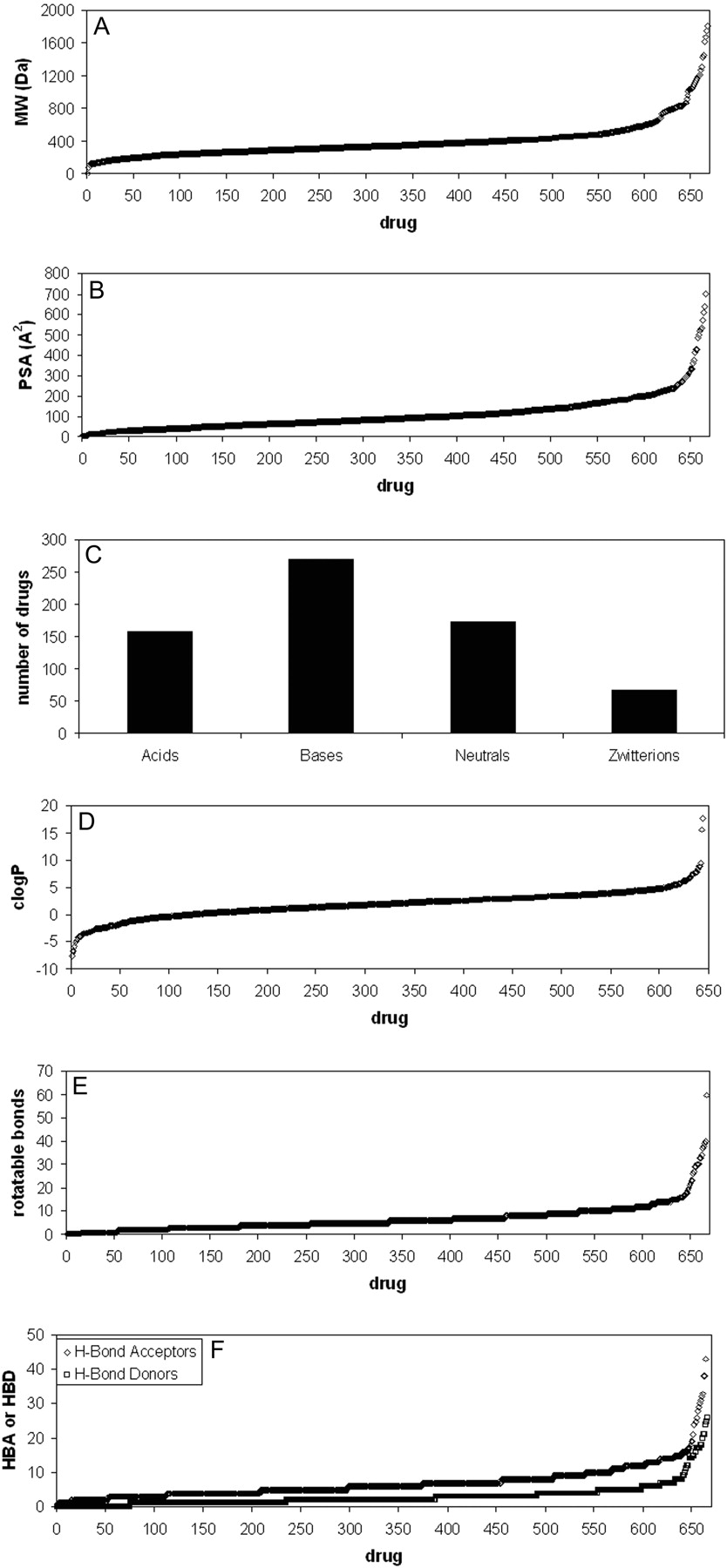
Spans of physicochemical properties of the 670 compounds included in this analysis. A, molecular weight; B, polar surface area; C, charge; D, lipophilicity; E, number of rotatable bonds per molecule; F, number of hydrogen bond acceptors and donors per molecule.

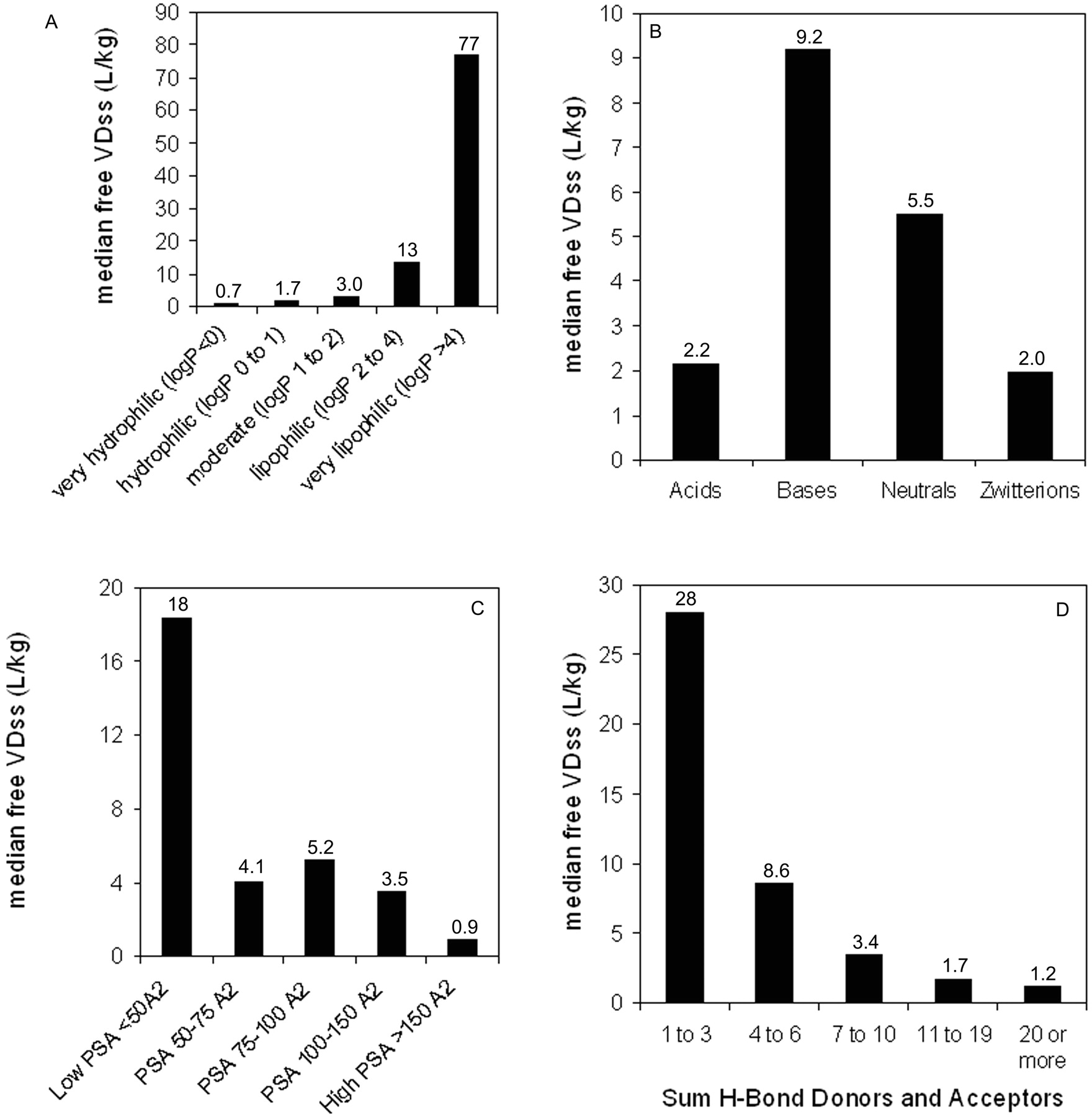
Relationship between median human VDss values and selected physicochemical parameters. A, lipophilicity; B, spans for human VDss values by charge type (horizontal line corresponds to median value); C, polar surface area; D, sum of hydrogen bond acceptors and donors.
Trends in the Data Set: VDss versus Physicochemical Properties. The data set was mined for any trends between the physicochemical values obtained and the human VDss values. In no case was there a relationship such that any single physicochemical property could be considered uniquely predictive of VDss; nevertheless, there were trends in the data suggesting a significant contribution of some physicochemical properties to VDss. Trends could not be easily observed when values were partitioned by means because of the scatter and overlap in the data (as exemplified in Fig. 4B); however, trends could be observed for median values. Median VDss values showed trends with clogP, PSA, number of H-bond acceptors and donors (Fig. 4), and charge type (Table 3). These four properties have varying extents of inter-relatedness. The properties of high polar surface area, high numbers of H-bond acceptors/donors, and low lipophilicity tend to reside in similar sets of molecules, and median VDss values trend higher for low PSA, low numbers of H-bond acceptors/donors, and high lipophilicity (Fig. 4). Negative charge (acids) have lower median and mean VDss values than bases, with neutral molecules and zwitterions in between (Table 3). The median values of VDss for the acids remain around 0.2 l/kg, irrespective of clogP, whereas for bases, neutrals, and zwitterions, there is an upward trend observed for VDss with increasing lipophilicity (Fig. 5).
Comparison of median values for total and free VDss and CL values relative to charge type Gadoversetamide, gentamicin, and lithium carbonate are not included in the charge classes.
Data were also examined after correcting VDss for free fraction (i.e., free VDss = VDss/fu), which is more indicative of the extent of tissue partitioning. As with total VDss, trends for free VDss were observed primarily through examination of median values because there is considerable scatter among the data. The same four physicochemical parameters, lipophilicity, PSA, charge type, and sum of hydrogen bond donors and acceptors, appear to be the properties that contribute to free VDss (Fig. 6). An interesting trend emerged when the relationship between VDss and lipophilicity was stratified by charge type; a considerable improvement was observed when the trend was examined for free VDss than for total VDss (Fig. 5). For example, there was no trend in the relationship between VDss and lipophilicity for acids, but when examined for free VDss, there was a trend that increasing lipophilicity leads to greater free VDss values. This tends to indicate that plasma protein binding dominates in the distribution of negatively charged compounds.
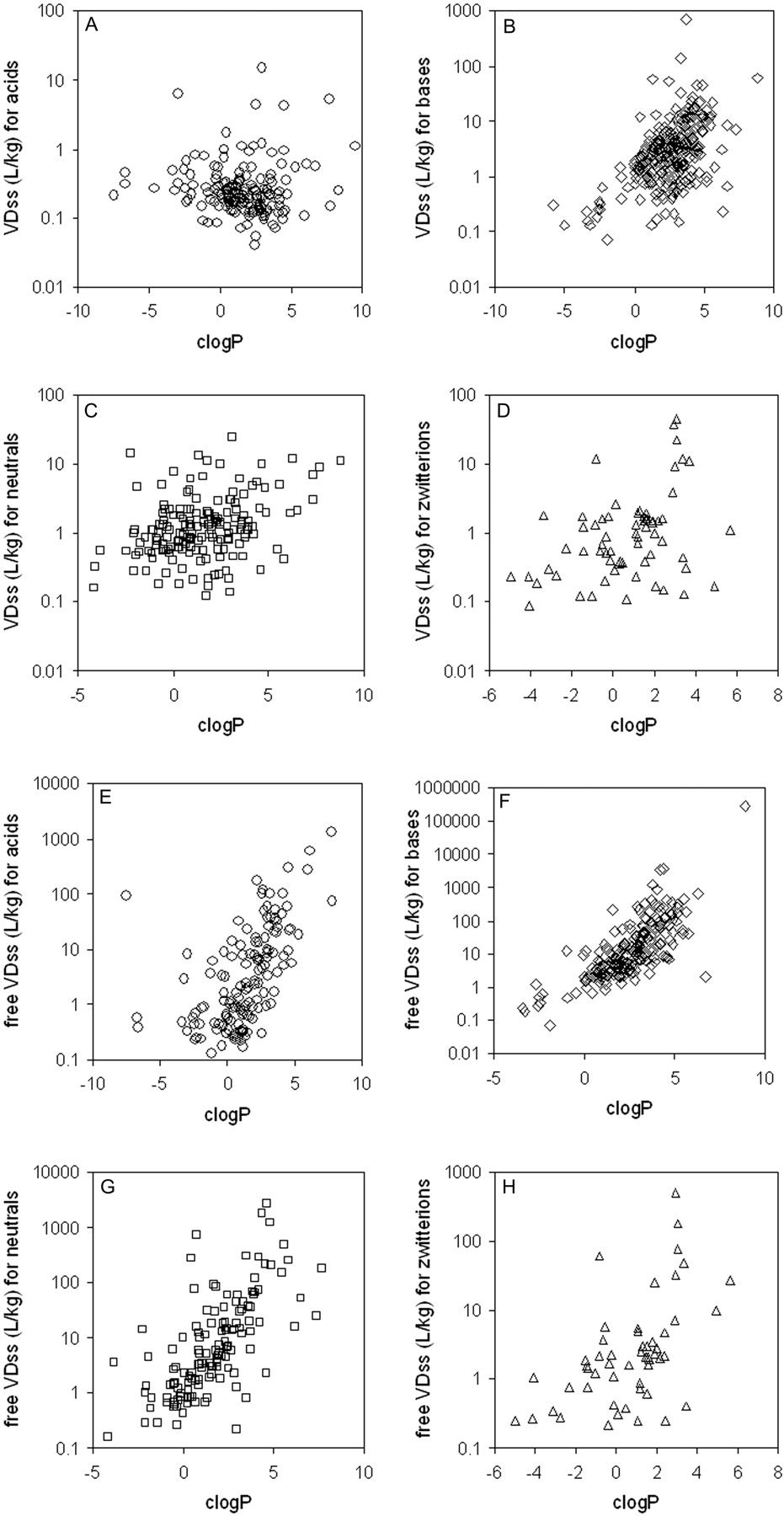
Relationship between VDss and lipophilicity, separated by charge type. A, VDss versus clogP for acids; B, VDss versus clogP for bases; C, VDss versus clogP for neutrals; D, VDss versus clogP for zwitterions; E, free VDss versus clogP for acids; F, free VDss versus clogP for bases; G, free VDss versus clogP for neutrals; H, free VDss versus clogP for zwitterions. DHA-paclitaxel (clog P 15.7) and eritoran (clogP 17.8) were excluded for easier visualization of the data. (Note: Color figures containing all of the compounds in one plot are available in the supplemental data.)
Trends in the Data Set: CL versus Physicochemical Properties. Similar to the observations with VDss, readily discernible trends could be observed between some physicochemical properties and CL; however, no relationship was tight enough to suggest that any single property could be quantitatively predictive of CL. Decreases in median CL were observed with increases in PSA or sum of hydrogen bond acceptors and donors (Fig. 7). Only a weak trend could be observed between median CL and lipophilicity. Bases had generally greater CL values than acids, neutrals, or zwitterions (Fig. 7). The relationship between lipophilicity and median CL was considerably strengthened when CL values were converted to free CL values (Figs. 8 and 9). Interestingly, unlike the median CL values, the difference in median free CL values for acids, bases, and neutrals was not observable (Table 3). When CL values were examined for any relationship to lipophilicity after stratification by charge type, no apparent trends could be observed, but when corrected for protein binding (i.e., free CL = CL/fu), the relationship showed a weak trend, with free CL increasing with increasing lipophilicity (Fig. 9).
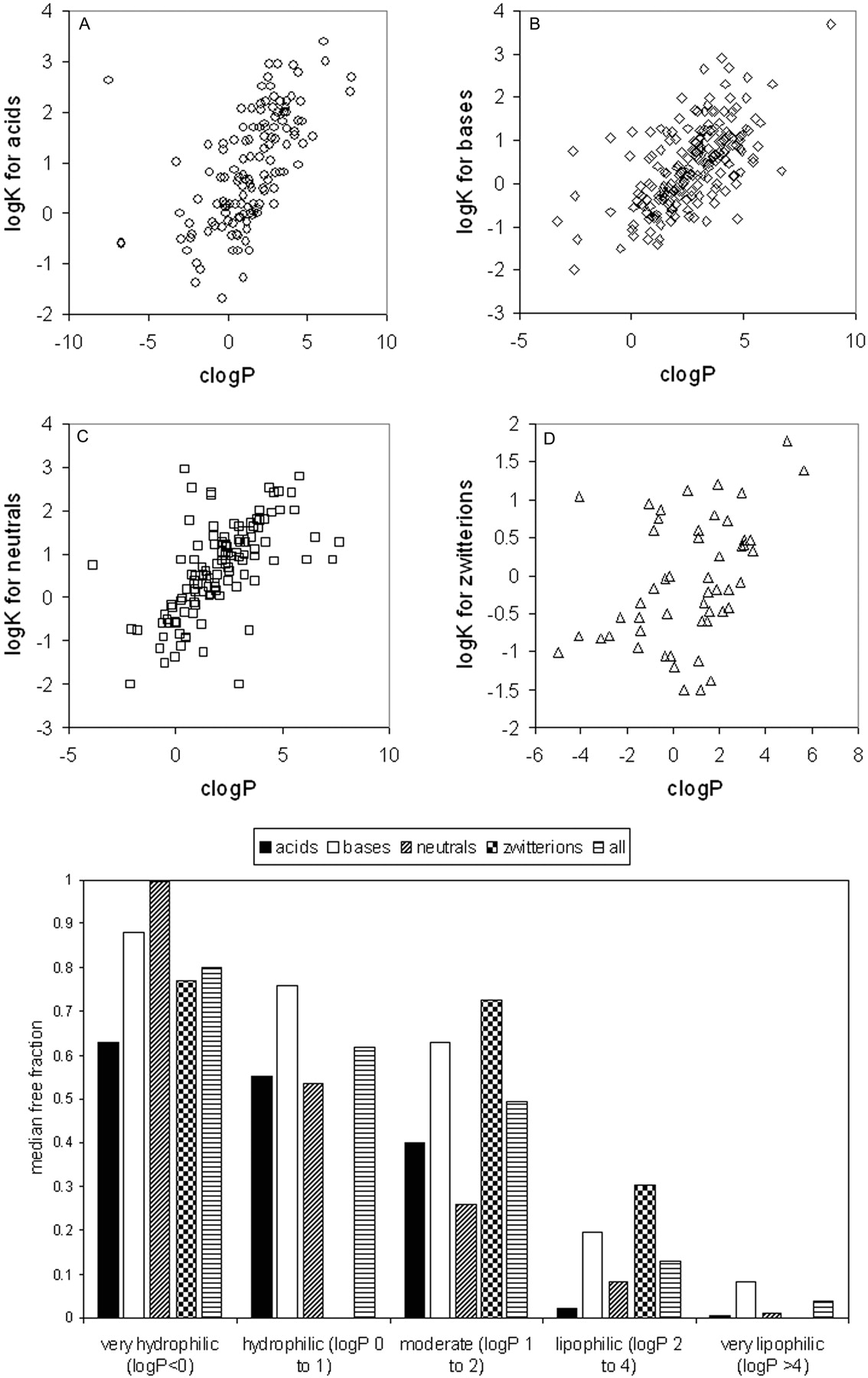
Trends in the Data Set: Protein Binding versus Physicochemical Properties. Of the physicochemical properties examined, relationships were only observed between protein binding and lipophilicity and charge type (Fig. 10). With greater lipophilicity, protein binding tends to increase, and this trend becomes apparent for all charge types when protein binding is expressed as the logarithm of the apparent affinity constant, logK (or log[bound/free]) as shown in Fig. 10.
Discussion
The utility of human pharmacokinetic databases in the development of a greater understanding of the relationship between chemical structure and pharmacokinetic behavior is unquestionable. Over the years, trends have been noted in the relationship between PK and structure (and physicochemical properties), but these have been for limited compound sets, in many cases within a single class of drugs (Smith, 1997; van de Waterbeemd et al., 2001). In this report, we have gathered a database of human pharmacokinetic parameters of a size never before assembled. Each value was obtained after careful analysis of original scientific literature (or in a few cases from drug product labels approved by government regulatory authorities). In many cases, different investigators chose to analyze data and report pharmacokinetic parameters in different manners (e.g., noncompartmental versus compartmental analysis, VDss versus VDβ, i.v. versus oral dosing, etc.). Thus, we reviewed each original report to ensure that the parameters reported were consistent from compound to compound and, if not, to reanalyze the data to obtain the desired parameters. Unlike other commonly cited pharmacokinetic databases (Goodman and Gilman, 2006), the database values presented in this report are exclusively from studies in which drugs were administered intravenously. Thus, these values are not confounded by effects of slow and incomplete absorption or extensive first pass extraction. The database is offered as supplemental data to this report so that other scientists can easily download it and use it to test various hypotheses and develop quantitative structure pharmacokinetic relationships models. However, the following limitations of this database should be appreciated: 1) study designs vary from drug to drug with regard to dose level, number of subjects, blood sampling times, analytical procedures, etc.; and 2) values reported are means and do not account for intersubject variability. Nevertheless, the database should be able to be used to provide insight into the relationship between structure and pharmacokinetics and useful in building computational models. Efforts on the latter are underway, and a discussion of initial efforts on the former is described below.
The set of compounds reported in this trend analysis encompasses a wide variety of structures and therapeutic areas and physicochemical properties as shown under Results, and Table 2 shows the pharmacokinetics parameters grouped according to defined thresholds. For VDss, a large proportion of the values (approximately 42%) falls within total body water (0.7 l/kg). Only a small subset of these drugs was confined to blood (or plasma) volume, with a value close to the generally accepted distribution volume of albumin, or 0.1 l/kg (McGinnity et al., 2007). [The latter can be approximated to the above value from the extravascular/intravascular ratio of 1.4, as in the Oie-Tozer equation (Oie and Tozer, 1979) assuming a blood volume of 0.07 l/kg.] This would indicate that although most of these compounds do not extensively partition into tissues, they can distribute throughout bodily fluids and, therefore, may be considered unbound in tissues and potentially capable to reach the intended target. If a volume of 0.7 l/kg is taken as the threshold for total body water, about 60% of the compounds have values exceeding that threshold, with almost one half of these compounds (29% of total) having volumes in excess of 2 l/kg. These compounds may be expected to partition moderately to extensively into tissues and contribute significantly to the residence time (or t1/2) of the drug. However, no direct correlation can be drawn between VDss values and the ability of a compound to reach the intended pharmacological target or to diffuse into a particular organ because these values are overall “averages” across all tissues and fluids.
Relationship between median human free VDss values and selected physicochemical parameters. A, lipophilicity; B, charge; C, polar surface area; D, sum of hydrogen bond acceptors and donors.

Relationship between median human CL values and selected physicochemical parameters. A, lipophilicity; B, spans for human CL values by charge type (horizontal line corresponds to median value); C, polar surface area; D, sum of hydrogen bond acceptors and donors.

Relationship between median human free CL values and selected physicochemical parameters. A, lipophilicity; B, charge; C, polar surface area; D, sum of hydrogen bond acceptors and donors.
Overall, nearly 85% of the compounds reside in the moderate to low range (<15 ml/min/kg or hepatic extraction ratio approximately taken as ≤0.7) of clearance, which, in conjunction with a moderate to high volume of distribution, might contribute to yielding a fairly large residence or terminal half-life because a good proportion of compounds resides in ranges of volume of distribution that exceed total body water. Nevertheless, only a minority of compounds, about 20%, have a t1/2 profile consistent with a once-a-day dosage regimen, and about one half show a half-life equal to or lower than 4 h. A very significant proportion, about one third, show an intermediate half-life that would still be generally considered unsuitable for a once-a-day dosage (barring nondirect pharmacokinetic/pharmacodynamic relationships or tolerance to a high Cmax/Ceff ratio). However, depending on the therapeutic target, a rapid onset and short half-life may be desirable, and a once-a-day dosing regimen may not always be needed especially for acute therapy. Conversely, absorption kinetics may be such that t1/2 via the oral route exceeds that following iv administration, permitting once-a-day dosage regimens.
Physicochemical properties and their impact on drug metabolism and pharmacokinetics is a broad and complex topic. Unfortunately, there is no single unifying descriptor that is able to explain drug pharmacokinetic behavior. The rule of 5 concept developed by Lipinski et al. (1997) utilizes thresholds of logP, molecular weight, hydrogen bond acceptors, and hydrogen bond donors to classify oral drug absorption. However, with respect to other PK parameters, typically two key descriptors, lipophilicity (expressed as logP or logD) and charge/ionization type, have been employed with general trends observed in certain drug classes (Smith, 1997; van de Waterbeemd et al., 2001). Lipophilicity effectively models a number of partitioning and distribution processes, including cell membrane permeation, membrane and protein binding, and affinity for drug-metabolizing enzymes, whereas charge type reflects ion-pair interactions with plasma proteins, membrane lipids, and specific drug-metabolizing enzymes.
Plasma protein binding typically shows a sigmoidal relationship with lipophilicity as demonstrated by van de Waterbeemd et al. (2001) on a data set of ∼150 acidic, basic, and neutral compounds. When expressed as the logarithm of the apparent affinity constant (logK), this sigmoidal trend becomes linear and is exemplified in the data set of 554 compounds presented here (Fig. 10). Increasing lipophilicity typically yields increased protein binding because the interaction with albumin and α1-acid glycoprotein is driven by hydrophobic forces. Acidic compounds show correspondingly higher binding relative to bases and neutrals due to an ion-pair interaction with a basic residue within albumin (Ghuman et al., 2005). Basic compounds tend to show high affinity for α1-acid glycoprotein due to an electrostatic interaction with acidic residues (Kremer et al., 1988). Overall, plasma protein binding influences the disposition profile in terms of both CL and VDss because only free drug is available for elimination and distribution into peripheral tissues. By converting CL and VDss into their free forms, the confounding impact of plasma protein binding, and the physicochemistry that drives it, was removed. This led to much improved trends between physicochemical properties and free CL and VDss.

Relationship between CL and lipophilicity, separated by charge type. A, CL versus clogP for acids; B, CL versus clogP for bases; C, CL versus clogP for neutrals; D, CL versus clogP for zwitterions; E, free CL versus clogP for acids; F, free CL versus clogP for bases; G, free CL versus clogP for neutrals; H, free CL versus clogP for zwitterions. DHA-paclitaxel (clog P 15.7) and eritoran (clogP 17.8) were excluded for easier visualization of the data. Color figures containing all of the compounds in one plot are available in the Supplemental Data.

Relationship between protein binding and physicochemical properties. A, acids; B, bases; C, neutrals; D, zwitterions; lower panel, median free fraction versus lipophilicity. A color figure containing all of the compounds in one plot is available in the Supplemental Data.
In a physiological sense, steady-state volume of distribution can be described by the Gillette or Oie-Tozer equations (Oie and Tozer, 1979), where the weighted mean ratio of plasma binding to tissue binding is the crucial term. Factors driving the unbound fraction in plasma are as described above, whereas the unbound fraction in tissue is dependent on cell membrane permeability and nonspecific hydrophobic interaction with cellular lipids and protein. In addition, bases typically exhibit an ion-pair interaction, with the charged polar head group of membrane phospholipids contributing to an increased tissue affinity and therefore VDss. Furthermore, basic compounds have the potential for specific interaction with subcellular organelles such as lysosomes and mitochondria through a pH partition mechanism, as illustrated by some of the psychotropic drugs (Daniel and Wojcikowski, 1997). The lipophilic and charge dependence of tissue affinity has been nicely demonstrated in correlations of free VDss and logD on small compound data sets (Smith, 1997; van de Waterbeemd et al., 2001), corroborating our findings with 670 compounds. Additionally, we observed a trend with polar surface area and numbers of hydrogen bond donors and hydrogen bond acceptors; increasing polarity leads to a reduction in the median VDss. These polarity descriptors, to some extent inversely related to lipophilicity, demonstrate that VDss cannot be explained by logP and charge type alone and highlight the utility of multivariate in silico approaches to VDss prediction (Gleeson et al., 2006; Lombardo et al., 2006). The magnitude of this data set may now allow exploration of specific functional moieties or substituents that play an integral role in tissue distribution and VDss.
Mammals have evolved a number of clearance mechanisms designed to render lipophilic xenobiotics more water soluble to allow efficient renal or biliary elimination. Introducing hydrophilicity into xenobiotics is typically a concerted effort of functionalization and conjugative metabolic reactions, mediated by cytochromes P450 and UDP-glucuronosyl transferases or sulfotransferases, respectively. The Km for cytochrome P450 reactions is inversely related to lipophilicity because the binding of substrate to enzyme relies on hydrophobic interactions (Lewis and Dickins, 2003). Positive correlations of free metabolic clearance and logD support this finding, being irrespective of charge type (Smith, 1997). We observed a general trend of increasing free CL with logP, supporting earlier work with this much expanded data set. The variation within this trend contains latent information not described solely by lipophilicity. Renal CL, for example, is typically inversely correlated with partition or distribution coefficients because increasing lipophilicity allows efficient tubular passive reabsorption in the nephron of the kidney. Trends with polarity descriptors (hydrogen bond donors/acceptors and PSA) were also observed such that median CL decreases as polarity is amplified, in line with established reasoning on metabolic CL structure-activity relationships. Latent information within the global trend of logP and CL also concerns functionality and substituent effects on CL. One such example is the N-dealkylation of the benzodiazepines, clonazepam, oxazepam, diazepam, and temazepam, where differences in CL are related to the propensity of N-demethylation and not bulk lipophilicity (Smith, 1997). These examples of functionalization effects should be well represented in this 670-compound data set and provide an opportunity for further exploration.
Future efforts are directed at the utilization of this data set in gaining a better understanding of the relationship between chemical structure and pharmacokinetic behavior. Although the analysis in this report focused on gross physicochemical properties such as lipophilicity and charge, further questions can be asked regarding the impact of specific chemical substituents on human pharmacokinetic parameters and, potentially, lead to structure-property relationships. In particular, the exploration of the predictability of clearance and half-life using in silico, in vitro-in vivo, or in vivo correlative methods could be attempted using this large and accurate data set of human data. Volume of distribution, as an example, may be more related to overall physicochemical properties, which would explain the previous successes in model building for this parameter(Gleeson et al., 2006; Lombardo et al., 2006), whereas clearance may have a greater dependence on specific chemical substituents.
Efforts are now underway, by our groups, to use these data to further our understanding of the complex relationships between structure and PK parameters, using several approaches along the lines described above. This large set of carefully compiled human PK data and the basic structure-PK relationships observed thus far should be of use in the design of new medicines.
Acknowledgments
We thank S. Harriman, G. Liang, A. Amaral, M. Gunduz, and J. Zhan (Novartis Institutes for Biomedical Research Cambridge) for searching and providing some of the PK data reported.
Footnotes
-
The complete list of compounds with pharmacokinetic data, Chemical Abstracts Services numbers, full references, and comments by the authors of this manuscript are available as an Excel file. A color version of Figs. 2, 5, 9, and 10 is also available in a Word file as Supplemental data.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.020479.
-
ABBREVIATIONS: CL, clearance; VD, volume of distribution; VDss, steady-state volume of distribution; PSA, polar surface area; PK, pharmokinetic; DHA, docosahexaenoate; MRT, mean residence time; VDss,u, free volume of distribution at steady-state; CLu, free plasma clearance; fu, free fraction in plasma; BB 83698, N-[(1S)-1-[[4-(1,3-benzodioxol-5-ylmethyl)-1-piperazinyl]carbonyl]-2,2-dimethylpropyl]-α-[(formylhydroxyamino)methyl]-(αR)-cyclopentanepropanamide; BMS-214662, 2,3,4,5-tetrahydro-1-(1H-imidazol-5-ylmethyl)-3-(phenylmethyl)-4-(2-thienylsulfonyl)-(3R)-1H-1,4-benzodiazepine-7-carbonitrile; CB 10-277, p-(3,3-dimethyl-1-triazeno)benzoic acid; DP-b 99, N,N′-[1,2-ethanediylbis(oxy-2,1-phenylene)]bis[N-(carboxymethyl)-1,1′-bis[2-(octyloxy)ethyl] ester (9CI)-glycine; IVL745, N-[[3-methoxy-4-[[[(2-methylphenyl)amino]carbonyl]amino]phenyl]acetyl]-glycyl-N-[(3,4-dimethoxyphenyl)methyl]-(9CI)-β-alanine; KRN-5500, 4-deoxy-4-[[2-[[2E,4E)-1-oxo-2,4-tetradecadien-1-yl]amino]acetyl]amino]-N-1H-purin-6-yl-l-glycero-β-l-manno-heptopyranosylamine; KW-2170, 5-[(3-aminopropyl)amino]-7,10-dihydroxy-2-[[(2-hydroxyethyl)amino]-methyl]-6H-pyrazolo[4,5,1-de]acridin-6-one; MEN-10755, 7-[[4-O-(3-amino-2,3,6-trideoxy-α-l-lyxo-hexopyranosyl)-2,6-dideoxy-α-l-lyxo-hexopyranosyl]oxy]-7,8,9,10-tetrahydro-6,9,11-trihydroxy-9-(hydroxyacetyl)-5,12-naphthacenedione; NK 611, [[2-deoxy-2-(dimethylamino)-4,6-O-(1R)-ethylidene-β-d-glycopyranosyl]oxy]-5,8,8a,9-tetrahydro-5-(4-hydroxy-3,5-dimethoxyphenyl)-furo[3′,4′:6,7]naphtho[2,3-d]-1,3-dioxol-6(5aH)-one; PNU-145156E, 7,7′-[carbonylbis[imino(1-methyl-1H-pyrrole-4,2-diyl)carbonylimino(1-methyl-1H-pyrrole-4,2-diyl)carbonylimino]]bis-1,3-naphthalenedisulfonic acid; RPR 109881A, β-[[(1,1-dimethylethoxy)carbonyl]amino]-α-hydroxy-(1S,2S,4S,7R,8aR,9aS,10aR,12aS,12bR)-7,12a-bis(acetyloxy)-1-(benzoyloxy)-1,3,4,7,8,9,9a,10,10a,12,12a,12b,-dodecahydro-2-hydroxy-5,13,13-trimethyl-8-oxo-2,6-methano-2H-cyclodeca[3,4]cyclopropa[4,5]benz[1,2-b]oxet-4-yl benzenepropanoic acid ester(αR,βS); Sch34343, 3-[[2-[(aminocarbonyl)oxy]ethyl]thio]-6-[(1R)-1-hydroxyethyl]-7-oxo-(5R,6S)-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid; UK-240,455, N-(6,7-dichloro-1,2,3,4-tetrahydro-2,3-dioxo-5-quinoxalinyl)-N-(2-hydroxyethyl)-methanesulfonamide.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. - Received January 16, 2008.
- Accepted April 18, 2008.

- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}