


Abstract
Cyclosporin A (CsA) is a well known inhibitor of the organic anion-transporting polypeptide (OATP/Oatp) family transporters, causing a large number of transporter-mediated drug-drug interactions in clinical situations. In the present study, we examined the inhibitory effect of CsA on the hepatic uptake of sulfobromophthalein (BSP) in rats, focusing on a long-lasting inhibition. Twenty-one hours after the subcutaneous administration of CsA, the hepatic clearance of BSP was decreased. The liver uptake index study revealed that hepatic uptake of BSP was reduced in CsA-treated rats for at least 3 days. Comparison of uptake studies using isolated hepatocytes prepared from control and CsA-treated rats showed that hepatic uptake in CsA-treated rats was decreased. In primary cultured hepatocytes, after preincubation with CsA, the uptake of [3H]BSP was reduced even after removal of CsA from the incubation buffer although a preincubation time dependence was not observed. However, the expression of Oatp1a1 and Oatp1b2, which are involved in the hepatic uptake of BSP, and the amount of intrahepatic glutathione, a driving force of Oatp1a1, did not change in CsA-treated rats. Thus, we can conclude that CsA modulates the transporter function sustainably. It can cause a potent in vivo drug-drug interaction. The modulation of transporters is not caused by reduced expression or driving force of transporters. It may be affected by CsA accumulated in the liver or its metabolites. The inhibitory effect of CsA on the transporter-mediated uptake of BSP cannot be explained by a simple competitive mechanism and a novel mechanism should be considered.
Transporters as well as metabolic enzymes are being increasingly recognized as a determinant of pharmacokinetics (Mizuno and Sugiyama, 2002; Mizuno et al., 2003; Zhang et al., 2006, 2008). Because drug metabolism and biliary excretion occur after sinusoidal uptake of drugs in the liver, hepatic uptake transporters play an important role in hepatic clearances of drugs, even for those with a metabolism elimination pathway (Yamazaki et al., 1996; Shitara et al., 2006). We previously noted that a clinically reported drug-drug interaction between cerivastatin and cyclosporin A (CsA) was caused by the inhibition of hepatic uptake transporters, including organic anion-transporting polypeptide 1B1 (OATP1B1) (Shitara et al., 2003). After this report, a number of OATP1B1-mediated drug-drug interactions were reported including other HMG-CoA reductase inhibitors versus CsA, bosentan versus CsA, and repaglinide versus CsA (Simonson et al., 2004; Treiber et al., 2004; Kajosaari et al., 2005; Shitara and Sugiyama, 2006). Until now, most of the clinically reported drug-drug interactions caused by inhibition of OATP1B1 were associated with the coadministration of CsA, although interactions with rifampicin and sildenafil have also been reported (van Giersbergen et al., 2007; Treiber et al., 2007).
CsA is a well known inhibitor of OATP/Oatp family transporters, and its concentrations to produce 50% inhibition (IC50) of human OATP1B1 and OATP1B3 are 0.2 to 0.3 and 0.5 to 0.8 μM, respectively (Shitara et al., 2003; Hirano et al., 2006; Treiber et al., 2007). On the other hand, the maximum blood concentration of CsA in clinical practice is approximately 1 μM, suggesting that the unbound concentration reaches 0.1 μM because 90% of the CsA in blood binds to plasma proteins (Lemaire and Tillement, 1982; Mück et al., 1999). Taking into consideration the fact that the drug concentration at the inlet to the liver after oral administration is higher than that observed in the circulating blood, a therapeutic concentration of CsA may inhibit OATP1B1-mediated hepatic uptake (Hirano et al., 2006). However, it is hard to quantitatively explain the clinically reported severe interaction between cerivastatin and CsA, in which a 3.8-fold increase in the area under the plasma concentration-time curve (AUC) of cerivastatin was observed, considering elimination of CsA. Thus, CsA may inhibit the OATP1B1-mediated uptake by another mechanism, or mechanisms other than transporter inhibition should be considered to explain the interaction between cerivastatin and CsA. Therefore, in the present study, we investigated the mechanism of transporter inhibition by CsA using rats as an animal model.
The mechanism for inhibition of cytochrome P450 (P450) has been intensively investigated, and irreversible and quasi-irreversible inhibitors have been reported (Lin and Lu, 1998; Zhou et al., 2004). These include macrolide antibiotics, calcium channel blockers, ethynylestradiol, paroxetine, ritonavir, and others. Their inhibitory effect is enhanced by preincubation of inhibitors with P450, and the apparent IC50 value decreases (Ito et al., 1998; Lin and Lu, 1998). In addition, these inhibitors inactivate metabolic enzymes even after they are diluted, because of the irreversible inactivation (Obach et al., 2007). This means they inhibit P450 even after they are eliminated from the body and their plasma concentration becomes negligibly low. Accordingly, mechanism-based inhibitors possibly cause severe drug-drug interactions. If drugs suppress transporters irreversibly, they would potently inhibit transporters and cause remarkable drug-drug interactions. However, as of now, there have been no reports of irreversible inhibition of transporters.
On the other hand, some drugs are reported to transcriptionally regulate transporter functions. For example, administration of indomethacin caused intestinal injury, leading to down-regulation of mRNA and protein of hepatic Oatp1b2 and Mrp2 in rats, and ethynylestradiol reduced the expression of Na+-taurocholate cotransporting polypeptide, bile salt export pump, and Mrp2 (Lee et al., 2000; Fujiyama et al., 2007). By this mechanism, drugs can cause transporter dysfunction. However, the transcriptional regulation takes a long time. More recently, therapeutic reagents such as ethacrynic acid and genipin (an ingredient in Chinese herbal medicine) reportedly have altered the membrane localization of bile canalicular transporters (Ji et al., 2004; Shoda et al., 2004). In this case, the membrane localization and the function of transporters can be regulated within a short time, which can cause a transporter dysfunction subsequently after the drug is administered.
The importance of quantitative prediction of drug-drug interactions is being increasingly recognized (Zhang et al., 2006, 2008). However, the mechanism of transporter inhibition has not been intensively studied, and, thus, most of the quantitative prediction of transporter-mediated drug-drug interactions has been performed on the basis of the reversible inhibition. In the present study, we aimed to investigate transporter inhibition by CsA, focusing especially on a long-lasting inhibitory effect.
Materials and Methods
Reagents and Animals. Sulfobromophthalein (BSP) was purchased from Sigma-Aldrich (St. Louis, MO). CsA was purchased from Wako Pure Chemicals (Osaka, Japan). [3H]BSP (5.5 Ci/mmol) was tritiated by Hartman Analytic GmbH (Brauschweig, Germany). [14C]Inulin carboxyl (1.4 mCi/g) was purchased from Moravek Biochemicals (Brea, CA). All other reagents were of analytical grade. Male Sprague-Dawley (SD) rats were purchased from Japan SLC, Inc. (Shizuoka, Japan).
Animal Study. The studies were carried out in accordance with the principles of laboratory animal care as adopted and promulgated by the National Institutes of Health (Institute of Laboratory Animal Resources, 1996) and the guidelines for animal studies provided by Chiba University. All protocols were approved by the Animal Care Committee of Chiba University. Six- to 8-week-old male SD rats were used in the experiments. Rats were housed in an air-conditioned room (25°C) under a 12-h light/dark cycle for at least 1 week before use. Food (the MF diet; Oriental Yeast Co. Ltd., Tokyo, Japan) and water were given ad libitum.
Plasma Concentration and Biliary Excretion of BSP in CsA-Treated Rats. In SD rats, 15 mg/kg CsA (dissolved in ethanol-olive oil, 1:10) or vehicle alone was administered subcutaneously. Twenty-one hours later, the femoral vein and artery of rats were cannulated with polyethylene tubes (PE-30 and -45) for the administration of BSP and blood sampling, respectively, and the bile duct was cannulated with PE-10 for the collection of bile under light ether anesthesia, followed by intravenous administration of 10 μmol/kg BSP. At designated times, blood and bile samples were collected, and the plasma was prepared by centrifugation of the blood samples (15,000g for 5 min) by using a bench-top centrifuge (Sigma 1-13; Sigma Laborzentrifugen GmbH, Ostrerode am Harz, Germany). Plasma and bile concentrations of BSP were determined from the difference in absorbance at 570 and 630 nm after alkali treatment using BSP solution (0–25 μM) as a standard by the method described by Molino and Milanese (1975). For the determination of the blood concentration of CsA, the blood samples were drawn from the jugular vein, subsequently followed by addition of EDTA (1 mg/ml). The blood and liver concentrations of CsA were determined by using a fluorescence polarization immunoassay (TDxFLx cyclosporine monoclonal whole blood assay and TDxFLx cyclosporine and metabolites whole blood assay, for CsA and CsA and its metabolites, respectively; Abbott Laboratories, Abbott Park, IL) using 0 to 1500 ng/ml and 0 to 2000 ng/ml CsA contained in the kit as a standard and quality control samples in the kit to confirm the precision of this assay.
Liver Uptake Index Study of [3H]BSP in Rats. Six, 21, 72, and 120 h after the subcutaneous administration of 15 mg/kg CsA, [3H]BSP and [14C]inulin dissolved in rat plasma (37°C, 18.5 and 3.7 kBq/ml, respectively) were rapidly injected into the portal vein of CsA-treated rats, immediately after ligation of hepatic artery, under light ether anesthesia. After 18 s of the bolus injection of radiolabeled compounds, the portal vein was cut and the liver was excised. The excised liver was minced in an equal volume of phosphate-buffered saline and homogenized. Then, 100 μl of the sample was transferred to a scintillation vial and dissolved in a solubilizer (Solvable; PerkinElmer Life and Analytical Sciences, Waltham, MA) at room temperature, followed by the addition of scintillation cocktail (Hionic-Fluor; PerkinElmer Life and Analytical Sciences). After that, the radioactivity taken up into the liver and in the injectate was determined in a liquid scintillation counter (LSC-5100; Aloka, Tokyo, Japan).
Western Blot Analysis. Hepatic plasma membrane fractions were prepared by the method described by Prpić et al. (1984) with modification and stored at –80°C until analysis. Then, 5 μg of protein of the samples was diluted with an equal volume of 2× sample buffer [0.1 M Tris-HCl (pH 6.8) containing 1% SDS, 12% mercaptoethanol, 16% glycerol, and 0.001% bromphenol blue] and separated on 8.5% SDS-polyacrylamide gel with a 3.75% stacking gel at 20 mA. Proteins were electrophoretically transferred to a polyvinylidene difluoride membrane (Immunobilon-P transfer membrane filter; Millipore Corporation, Billerica, MA) at 15 V for 1 h. The membrane was blocked with Tris-buffered saline containing 0.05% Tween 20 (TBS-T) and 3% bovine serum albumin (BSA) overnight at 4°C. Then, the membrane was incubated in TBS-T containing 3% BSA and 3000-fold diluted anti-rat Oatp1a1 or Oatp1b2 rabbit antisera for 1 h at room temperature. For the detection of the band, the membrane was incubated in TBS-T containing 3% BSA and 3000-fold diluted horseradish peroxidase-conjugated anti-rabbit IgG antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and the enzyme activity was assessed by using ECL Western blotting detection reagents (GE Healthcare UK, Little Chalfont, Buckinghamshire, UK) with a luminescent image analyzer (LAS1000; Fuji Film, Tokyo, Japan). The molecular weight was determined using a prestained protein marker (New England Biolabs, Ipswich, MA).
Semiquantitative Real-Time Reverse Transcription-Polymerase Chain Reaction. Total RNA was prepared from the liver of 15 mg/kg CsA- or vehicle-treated SD rats using RNA-Solv reagent (Omega Bio-Tek Inc., Doraville, GA). Reverse transcription was performed with 1 μg of the total RNA using an RNA PCR kit (AMV) (version 3.0; Takara-Bio, Shiga, Japan). Real-time polymerase chain reaction (PCR) was performed to quantify the mRNA expression of Oatp1a1 and Oatp1b2, relative to that of β-actin, using an ABI Prism 7000 system with real-time PCR Master Mix (Applied Biosystems, Foster City, CA). The primers used in the present study are listed in Table 1.
Sequences of the primers for semiquantitative real-time RT-PCR
Measurement of Glutathione Contents in the Liver of Rats. Twenty-one hours after the administration of 15 mg/kg CsA or vehicle, the livers were removed and approximately 50-mg samples were used. Samples were homogenized in a 20× volume of 0.1% EDTA disodium-25% metaphosphoric acid (7:2) and centrifuged at 15,000g and 4°C for 5 min. For 0.5 ml of the supernatant, 5 μl of 3-fluorotyrosine (2.5 mM) was added as an internal standard, followed by filtration through a 0.45-μm syringe filter (Millex-LH; Millipore Corporation). Then, 50 μl was subjected to high-performance liquid chromatography. The analyte was separated by an Inertsil ODS column (4.6-mm inner diameter × 250 mm; GL Sciences Ltd., Tokyo, Japan) with a mobile phase (0.1% trifluoroacetic acid-methanol, 18:1) at a flow rate of 1.0 ml/min. The eluate from the column was mixed with solution containing 2.31 mM o-phthalaldehyde and 17.1 mM 2-mercaptoethanol in 100 mM carbonate buffer (pH 10.5), which was delivered at a rate of 0.2 ml/min. The mixture was then passed through a stainless steel coil at 70°C to facilitate derivatization. A fluorescence detector was used and operated at an excitation wavelength of 355 nm and an emission wavelength of 425 nm. The concentration of GSH was calculated with reference to the height of a standard GSH sample (0–10 μM; Wako Pure Chemicals).
Uptake of [3H]BSP into Isolated Rat Hepatocytes. Twenty-one hours after the administration of 15 mg/kg CsA or vehicle, isolated rat hepatocytes were prepared from the rats by the collagenase perfusion method (Yamazaki et al., 1993). Isolated hepatocytes (viability >90%) were suspended in Krebs-Henseleit buffer (KHB), adjusted to 4.0 × 106 viable cells/ml and stored on ice. Before the uptake study, hepatocytes were incubated at 37°C for 3 min, and the uptake reaction was initiated by the addition of equal volume of KHB prewarmed at 37°C containing [3H]BSP (3.7 kBq/ml) and unlabeled BSP. At 0.5 and 2 min, the reaction was terminated by separating hepatocytes from the substrate solution. For this purpose, an aliquot of 100 μl of incubation mixture was collected and placed in a centrifuge tube containing 50 μl of 2 N NaOH under a layer of 100 μl of oil (density = 0.105, a mixture of silicone and mineral oil; Sigma-Aldrich), and subsequently the sample tube was centrifuged for 10 s at 15,000g using a bench-top centrifuge (Sigma 1-13). During this process, the hepatocytes pass through the oil layer into the alkaline solution. After an overnight incubation at room temperature to dissolve hepatocytes in alkali, the centrifuge tube was cut, and each compartment was transferred to a scintillation vial. The compartment containing dissolved hepatocytes was neutralized with 50 μl of 2 N HCl. Samples were mixed with scintillation cocktail (Cleasol I; Nakalai Tesque, Kyoto, Japan), and the radioactivity in each compartment was determined in a liquid scintillation counter (LSC-5100). The protein amount in the hepatocyte suspension was determined by the method of Lowry et al. (1951) with BSA as a standard.
Uptake of [3H]BSP into Primary Cultures of Rat Hepatocytes. Isolated parenchymal hepatocytes (viability >90%) were prepared by the collagenase perfusion method and suspended in Williams' medium E supplemented with 10% fetal bovine serum, 1 mg/ml streptomycin sulfate, 1000 U/ml penicillin G sodium, and 2.5 μg/ml amphotericin B. Isolated hepatocytes were seeded on 12-well culture plates coated with collagen type I at a density of 5.0 × 105 cells/well and cultured under 5% CO2 in air at 37°C for 4 h. Then, cells were washed twice with ice-cold KHB. Just before the uptake study, the ice-cold KHB was replaced with KHB at 37°C and prewarmed for 10 min. The uptake was initiated by replacing KHB with that containing [3H]BSP (0.83 kBq/ml) and unlabeled BSP to give a final concentration of 0.1 μM. The reaction was terminated by removing the substrate solution by suction and washed four times with ice-cold KHB after 5 min because we confirmed the linearity of the uptake for at least 5 min. To examine the effect of CsA treatment, rat hepatocytes were exposed to 0 to 10 μM CsA for designated times during the culture and washed twice with ice-cold KHB to remove culture media and CsA, and the uptake studies were conducted without CsA in the incubation buffer. For the inhibition study, hepatocytes were exposed to 0 to 10 μM CsA for 0, 20, or 60 min during the culture and washed twice with ice-cold KHB, subsequently followed by the uptake study in the presence of the same concentrations of CsA. Cells were dissolved in 0.5 ml of 0.1 N NaOH overnight, followed by neutralization with 0.5 ml of 0.1 N HCl. Then, 900-μl aliquots and 100 μl of the incubation buffer were transferred to scintillation vials, and the radioactivity associated with cells and that in the incubation buffer were counted (LSC-5100). For the protein assay 50 μl of the cell lysate was used.
Data Analysis. The plasma AUC of BSP after intravenous administration was calculated by the trapezoidal method. The total body clearance (CLtot) and biliary excretion clearance (CLbile) of BSP were calculated by the following equations: 
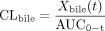 where AUC0–t and Xbile(t) are the AUC value from time 0 to t (minutes) and cumulative biliary excreted amount of BSP at t (minutes), respectively.
where AUC0–t and Xbile(t) are the AUC value from time 0 to t (minutes) and cumulative biliary excreted amount of BSP at t (minutes), respectively.
The time courses of the uptake of [3H]BSP into hepatocytes were expressed as the uptake volume (microliters per milligram of protein) for the radioactivity taken up into cells (disintegrations per minute per milligram of protein) divided by that in incubation buffer (disintegrations per minute per microliter). The uptake clearance (CLuptake) of [3H]BSP was calculated from the slope of the uptake volume versus time plot. The kinetic parameters for the uptake of [3H]BSP in isolated rat hepatocytes were estimated using the following equation: 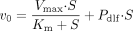 where v0 is the initial uptake rate (picomoles per minute per milligram of protein) of BSP, S is the initial concentration of BSP (micromolar). Km, Vmax, and Pdif are the Michaelis constant (micromolar), maximum uptake rate (picomoles per minute per milligram of protein), and nonsaturable uptake clearance (microliters per minute per milligram of protein) for the uptake of BSP, respectively.
where v0 is the initial uptake rate (picomoles per minute per milligram of protein) of BSP, S is the initial concentration of BSP (micromolar). Km, Vmax, and Pdif are the Michaelis constant (micromolar), maximum uptake rate (picomoles per minute per milligram of protein), and nonsaturable uptake clearance (microliters per minute per milligram of protein) for the uptake of BSP, respectively.
The CLuptake values of [3H]BSP in the primary culture of rat hepatocytes obtained in the presence of CsA were fitted to the following equation to calculate IC50 for the uptake of [3H]BSP: 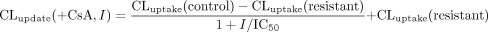 where I is the concentration of CsA added to the incubation buffer and CLuptake(+CsA, I) is the uptake clearance in the presence of CsA at the concentration of I. CLuptake(control) is the uptake clearance in the absence of CsA and CLuptake(resistant) is the uptake clearance, which is not affected by CsA.
where I is the concentration of CsA added to the incubation buffer and CLuptake(+CsA, I) is the uptake clearance in the presence of CsA at the concentration of I. CLuptake(control) is the uptake clearance in the absence of CsA and CLuptake(resistant) is the uptake clearance, which is not affected by CsA.
The data obtained in the liver uptake index (LUI) studies were expressed as percent LUI, which represents the ratio of the hepatic extraction of [3H]BSP to that of [14C]inulin. The percent LUI value was obtained by the following equation: 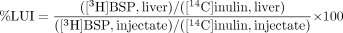 All fitting analyses were performed using a computerized non-least-squares method WinNonlin Professional (version 5.2.1; Pharsight, Mountain View, CA) to obtain parameters with a computer-calculated S.E. value, which indicates the precision of the estimated parameter but is not an estimate of variability. Statistical comparisons among multiple groups were carried out using Student's t test or Dunnett's test.
All fitting analyses were performed using a computerized non-least-squares method WinNonlin Professional (version 5.2.1; Pharsight, Mountain View, CA) to obtain parameters with a computer-calculated S.E. value, which indicates the precision of the estimated parameter but is not an estimate of variability. Statistical comparisons among multiple groups were carried out using Student's t test or Dunnett's test.
Results
Pharmacokinetics of BSP in CsA-Treated and Control Rats. The plasma concentration and biliary excretion of BSP in rats with or without CsA treatment are shown in Fig. 1. BSP was predominantly excreted into bile in both groups of rats. Its plasma concentration was increased, and biliary excretion was decreased in CsA-treated rats. The plasma clearances of BSP in CsA-treated and control rats were 15.2 ± 2.5 and 41.5 ± 2.3 (mean ± S.E., p < 0.01), respectively. Their corresponding biliary excretion clearances were 15.0 ± 2.8 and 44.1 ± 4.0 (mean ± S.E., p < 0.01), respectively.
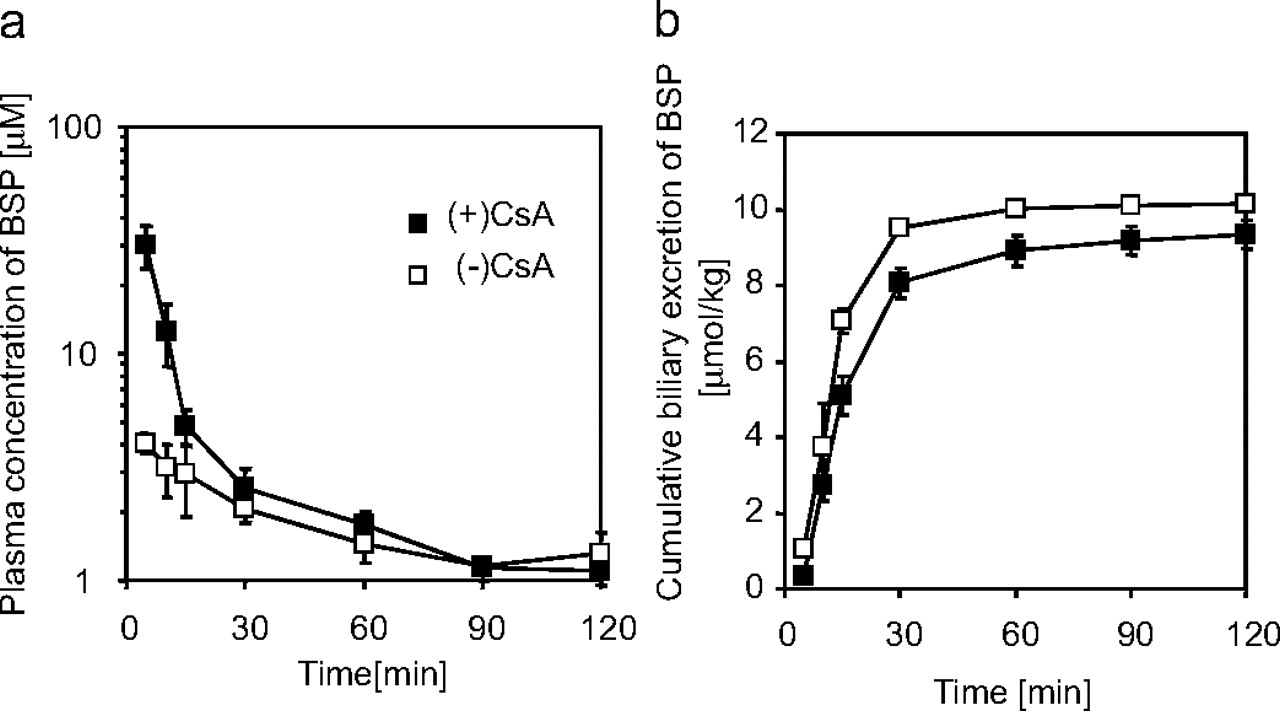
Plasma concentration or biliary excretion of BSP in CsA-treated or control rats. Plasma concentration (a) or cumulative biliary excretion (b) of BSP was examined after its intravenous administration (10 μmol/kg) in rats at 21 h after subcutaneous administration of 15 mg/kg CsA (▪) or vehicle (□). Each point represents the mean ± S.E. (n = 3).
LUI Study in CsA-Treated Rats. Hepatic uptake of [3H]BSP was evaluated in CsA-treated and CsA-untreated control rats by the LUI method. Percent LUI is plotted against the time after the subcutaneous administration of CsA (Fig. 2a). It was reduced after the injection of CsA and reached 54.0 ± 8.0% of control at 6 h after its injection (mean ± S.E., p < 0.05 versus control). Blood and liver concentrations of CsA after its administration were measured for 120 h (Fig. 2, b and c). The blood concentration of CsA reached 0.604 μM at 21 h after its injection, with the percent LUI value accounting for 70.6 ± 6.8% of control. Three days after the injection, although the blood concentration of CsA was reduced to 0.196 μM, the percent LUI still remained low (68.7 ± 11.6% of the control), whereas 5 days after the injection of CsA, it recovered to 91.3% of the control with a blood concentration of CsA of 0.0898 μM. CsA was highly accumulated in the liver, and intrahepatic concentration at 6 h after the administration of CsA was 1.89 ± 0.44 nmol/g of liver (n = 3, mean ± S.E.). It reached 4.22 ± 0.43 nmol/g of liver at 21 h and decreased to 0.942 ± 0.264 nmol/g of liver at 120 h. The liver/blood concentration ratio [(nanomoles per gram of liver)/(nanomoles per milliliter of blood)] was 5.37 to 10.5.
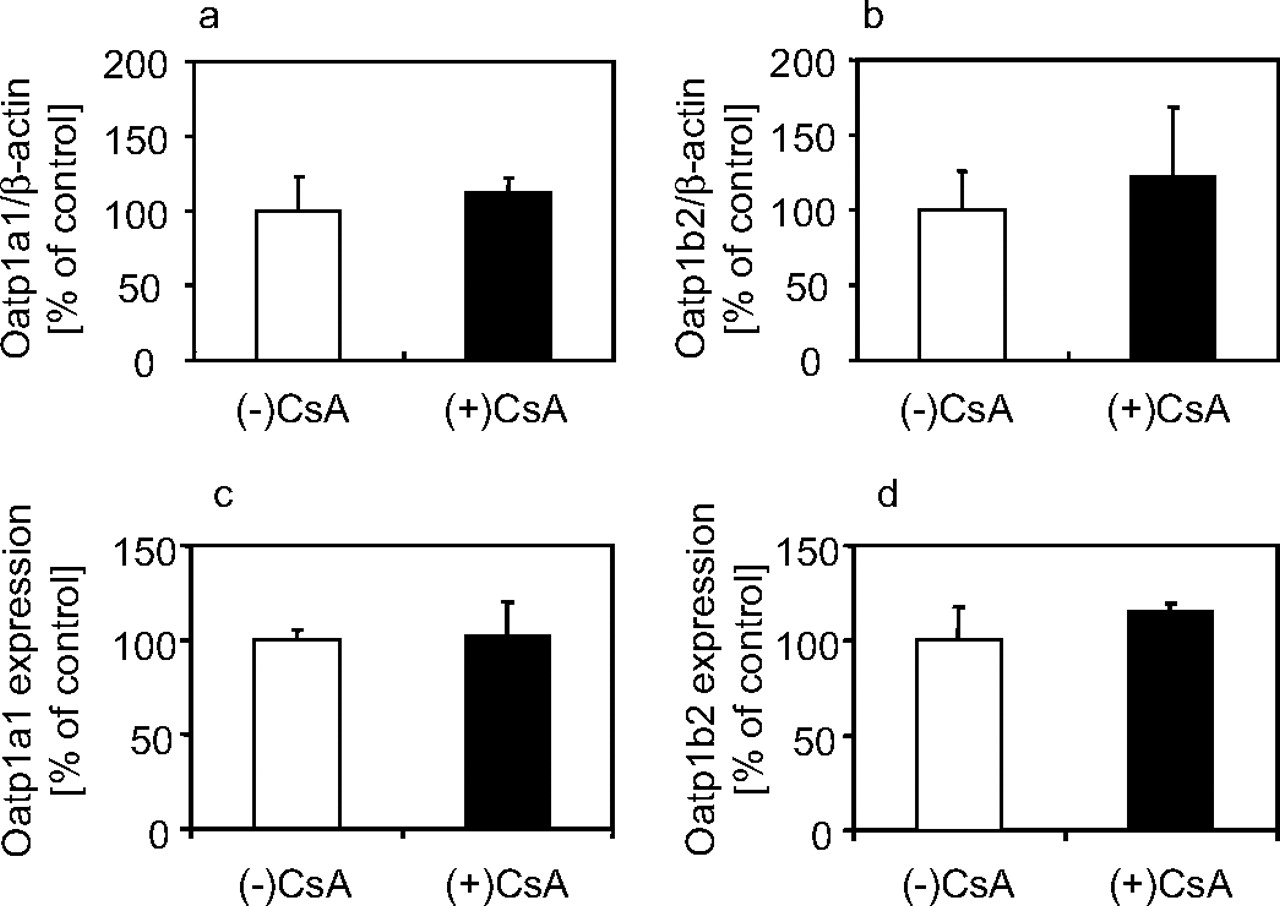
Transporter Expression and Intrahepatic GSH in CsA-Treated Rats. Because the hepatic uptake of [3H]BSP was decreased in CsA-treated rats, the expression levels of hepatic uptake transporters were examined. Real-time reverse transcription-PCR (RT-PCR) revealed that the expression levels of mRNA for Oatp1a1 and Oatp1b2 were not changed in CsA-treated and control rats (Fig. 3, a and b). Western blot analyses showed that their protein expression in the hepatic plasma membrane was not altered. In addition, the amount of intrahepatic GSH, a driving force of Oatp1a1 (Li et al., 1998), was measured. Intrahepatic concentrations of GSH were 2.58 ± 0.23 and 2.63 ± 0.64 μmol/g of liver (mean ± S.E., n = 3) for CsA-treated and control rats, respectively.
Uptake of [3H]BSP in Isolated Hepatocytes Prepared from CsA-Treated and Control Rats.Figure 4 shows the Eadie-Hofstee plot for the uptake of [3H]BSP in isolated hepatocytes from CsA-treated and control rats. Saturable uptake was observed in hepatocytes from both CsA-treated and control rats. It was reduced in hepatocytes from CsA-treated rats. Kinetic analyses revealed that Km, Vmax, and Pdif were 3.42 ± 0.57 μM, 594 ± 63 pmol/min/mg protein, and 2.74 ± 0.59 μl/min/mg protein (computer-estimated data), respectively, for control rats. On the other hand, the corresponding values for CsA-treated rats were 16.3 ± 1.8 μM, 1030 ± 90 pmol/min/mg protein, and 0.927 ± 0.420 μl/min/mg protein, respectively, suggesting that the hepatic uptake of [3H]BSP was reduced in CsA-treated rats, mainly due to the alteration in Km rather than Vmax.

Time profile of hepatic uptake of [3H]BSP in rats after CsA administration. a, hepatic uptake of [3H]BSP was examined by using the LUI method and is expressed as percent LUI, as shown under Materials and Methods, in rats at designated times after subcutaneous administration of 15 mg/kg CsA. Percent LUI was plotted to the time after administration of CsA. Each point represents mean ± S.E. (n = 4–6). b, blood concentration of CsA after the subcutaneous administration of 15 mg/kg CsA is shown. •, concentrations of CsA; ○, concentrations of CsA and its metabolites. Each point represents the mean ± S.E. (n = 5–7). c, liver concentration of CsA after the subcutaneous administration of 15 mg/kg CsA is shown. •, concentrations of CsA; ○, concentrations of CsA and its metabolites. Each point represents the mean ± S.E. (n = 3).
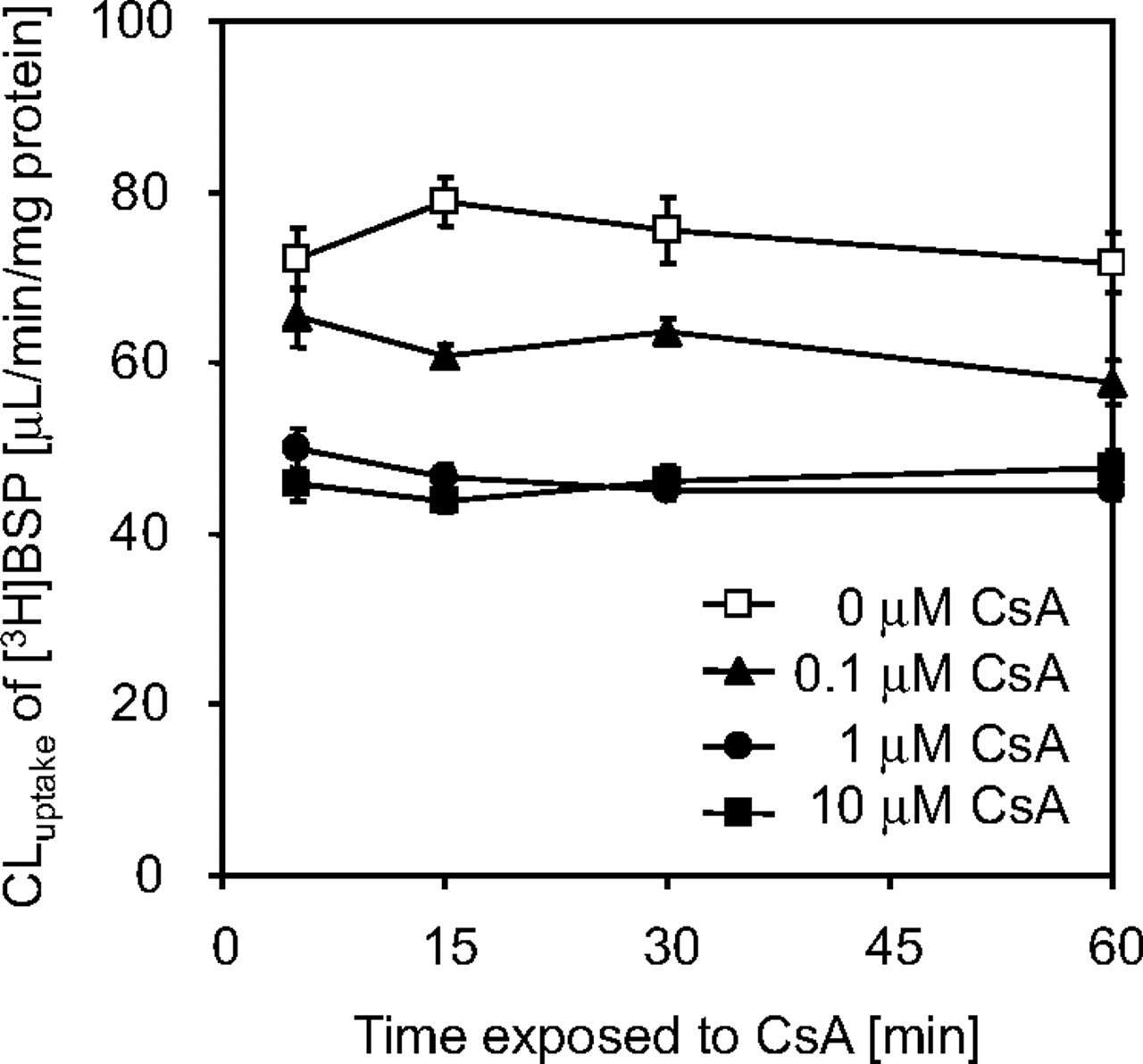
Uptake of [3H]BSP in Primary Culture of Rat Hepatocytes Exposed to CsA. In primary culture of rat hepatocytes, which was exposed to CsA during the culture, uptake of [3H]BSP was examined after removal of CsA from the incubation buffer (Fig. 5). As shown in this figure, exposure of rat hepatocytes to CsA during the culture resulted in the reduction in the uptake of [3H]BSP in a concentration-dependent manner. The saturable portion of [3H]BSP uptake was completely lost by treatment with 1 μM CsA. Exposure time dependence was not observed in the effect of CsA, except that 0.1 μM CsA tended to slightly reduce the uptake for the first 15 min.

Expression levels of Oatp1a1 and Oatp1b2 in livers of CsA-treated or control rats. Expression levels of mRNA for Oatp1a1 (a) or Oatp1b2 (b) were measured by a real-time RT-PCR method at 21 h after subcutaneous administration of 15 mg/kg CsA or vehicle. Their expression levels were normalized by mRNA for β-actin. Each bar represents the mean ± S.E. (n = 3). Protein expression levels of Oatp1a1 (c) or Oatp1b2 (d) were measured by Western blot analysis. Each bar represents the mean ± S.E. (n = 3).
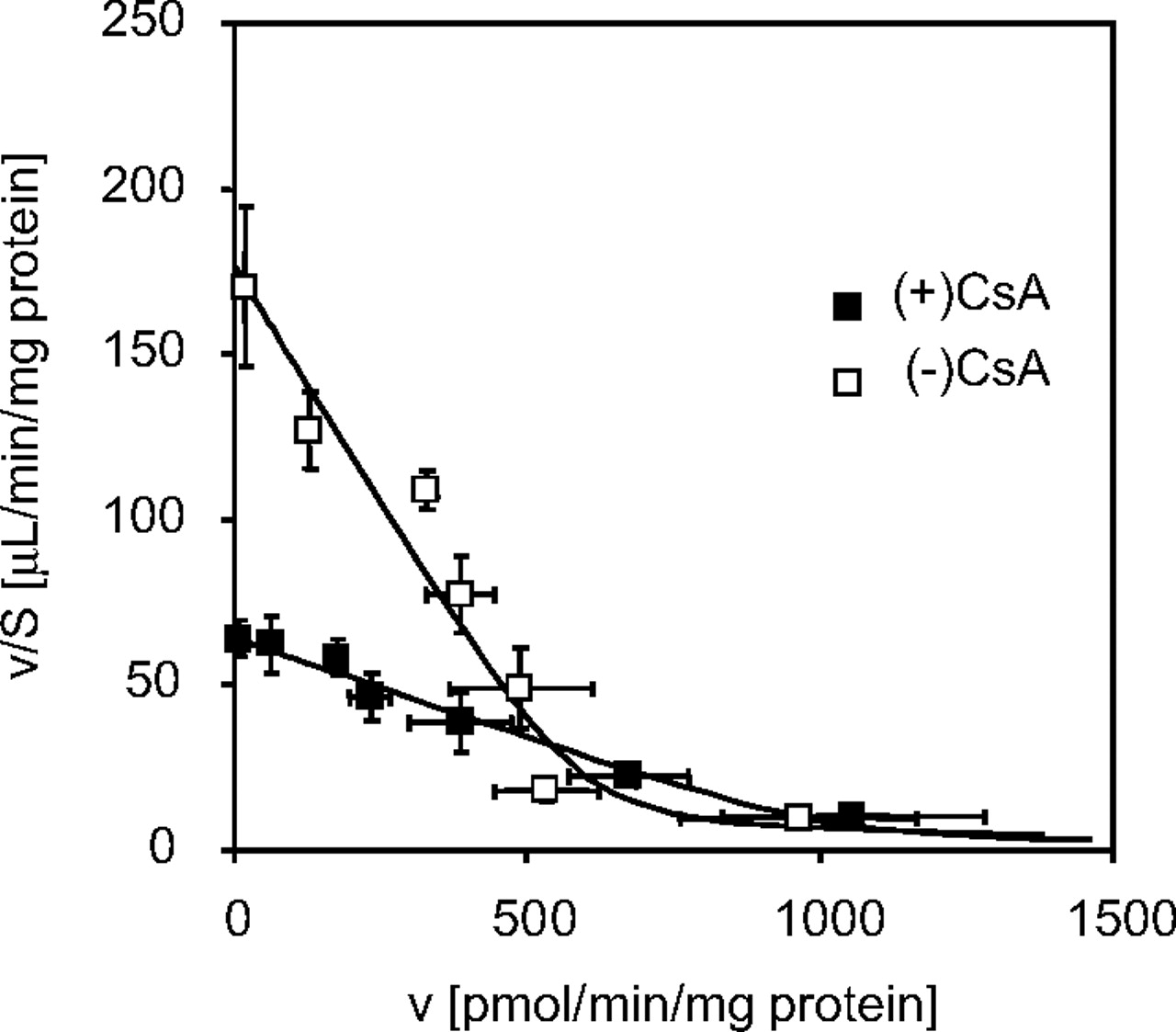
Eadie-Hofstee plot for the uptake of [3H]BSP in isolated hepatocytes prepared from CsA-treated or control rats. Isolated hepatocytes were prepared from rats at 21 h after subcutaneous administration of 15 mg/kg CsA or vehicle, and uptake of [3H]BSP was examined. Eadie-Hofstee plots for the uptake of [3H]BSP in isolated hepatocytes prepared from CsA-treated (▪) or vehicle-treated control (□) rats are shown. ——, fitted lines. Each point represents the mean ± S.E. (n = 9 from three independent cell preparations).
Inhibitory Effect of CsA on the Uptake of [3H]BSP in Primary Culture of Rat Hepatocytes. An inhibitory effect of CsA on the uptake of [3H]BSP in rat hepatocytes was observed in primary culture of rat hepatocytes. CsA inhibited the uptake of [3H]BSP in a concentration-dependent manner (Fig. 6). The IC50 was estimated to be 0.126 ± 0.047 μM (computer-estimated data) in hepatocytes cultured without CsA. On the other hand, pretreatment with CsA reduced the apparent IC50 values. They were estimated to be 0.0583 ± 0.0214 and 0.0422 ± 0.0149 in hepatocytes, which were cultured with CsA for 20 and 60 min, respectively.
Discussion
We have shown previously that CsA is an inhibitor of human OATP1B1 and OATP1B3 and rat Oatp1a1 (Shitara et al., 2003, 2004). Indeed, CsA has been reported to cause clinically relevant drug-drug interactions by inhibition of OATP1B1. Among OATP1B1 inhibitors, CsA is reported to produce drug-drug interactions with a large number of drugs in clinical practice, and it causes marked increases in their plasma concentrations. Thus, in the present study, we investigated the inhibition of Oatp family transporters in rats by CsA using BSP as a probe substrate.
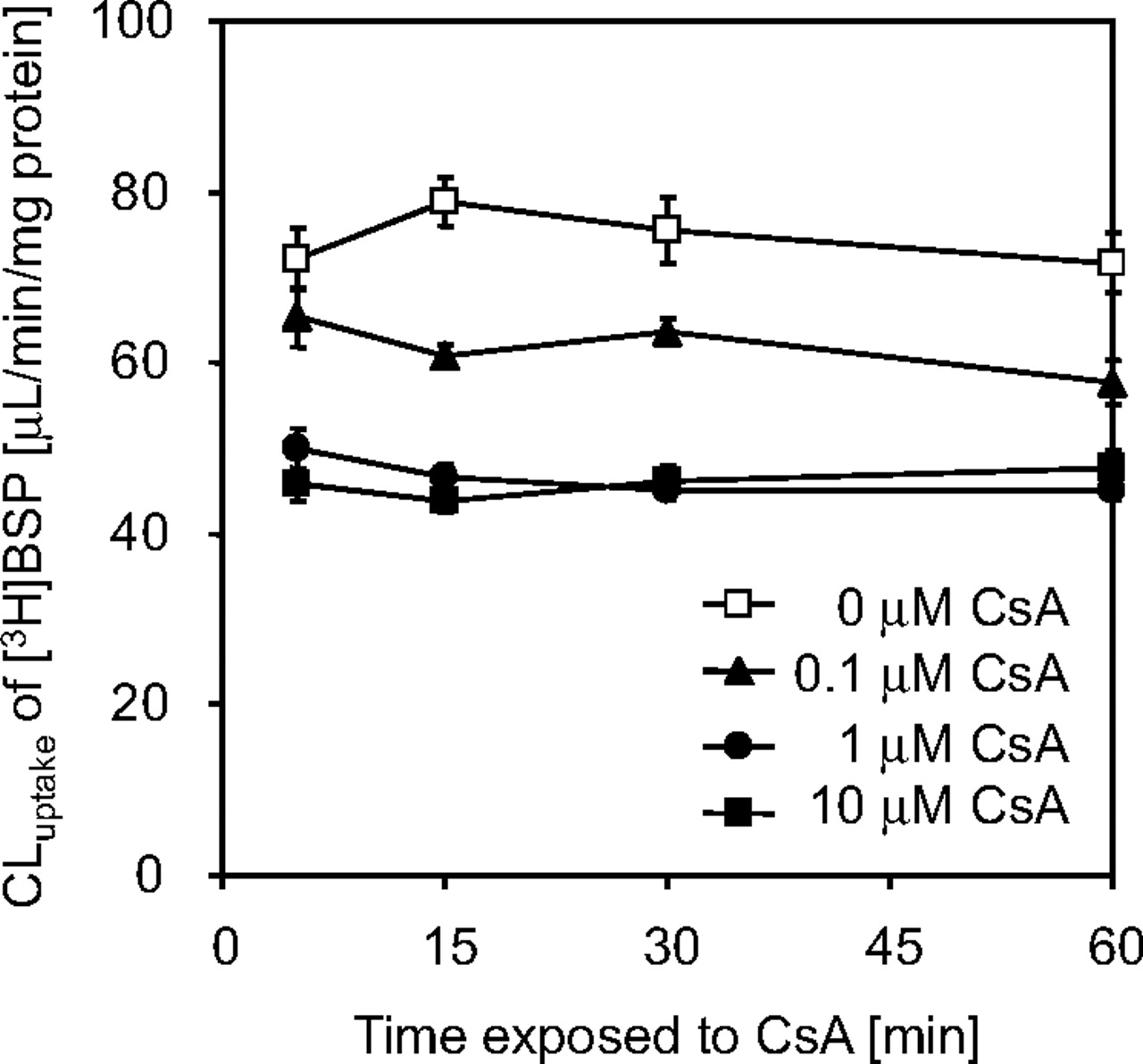
Uptake of [3H]BSP in primary cultures of rat hepatocytes after preincubation with CsA. Uptake of [3H]BSP was examined in primary cultures of rat hepatocytes after preincubation with 0 to 10 μM CsA. Uptake studies were performed after removal of CsA from the incubation buffer. Uptake of [3H]BSP was plotted to the preincubation time with CsA. Each point represents the mean ± S.E. (n = 12 from four independent cell preparations).

The inhibitory effect of CsA on the uptake of [3H]BSP in primary cultures of rat hepatocytes after preincubation with CsA. The inhibitory effect of CsA on the uptake of [3H]BSP in primary cultures of rat hepatocytes was examined. Hepatocytes were exposed to different concentrations of CsA for 0 (□), 20 (▴), or 60 min (▪), subsequently followed by inhibition studies by the same concentrations of CsA. Each point represents the mean ± S.E. (n = 9 from three independent cell preparations).
Twenty-one hours after the subcutaneous administration of CsA, the plasma AUC of BSP was 2.89-fold increased with the initial plasma concentration being markedly increased (Fig. 1a). An increase in the initial plasma concentration, i.e., the reduced initial distribution volume, is commonly observed under inhibition or dysfunction of hepatic uptake transporters for their substrate drugs (Mück et al., 1999; Shitara et al., 2006; Zaher et al., 2008). BSP is mainly excreted into the bile and, thus, the reduction in the plasma clearance can be explained by reduced biliary excretion (Fig. 1b). The LUI study showed that the hepatic uptake of BSP was reduced in CsA-treated rats in vivo although the reduction in the hepatic uptake in the LUI study (Fig. 2) (>54.0% of control) was smaller than the reduction in its hepatic clearance estimated in the in vivo study (Fig. 1) (34.0% of control). This result suggests that the interaction between BSP and CsA is at least partly explained by the reduced hepatic uptake.
It should be noted that the reduction in the hepatic uptake of BSP evaluated by LUI study was sustained for at least 3 days. Three days after the subcutaneous administration of CsA, its total blood concentration decreased to approximately 0.2 μM, and thus its unbound concentration should be less than 0.02 μM, considering the plasma unbound fraction of CsA to be approximately 10%. On the other hand, in our pilot study, we estimated the IC50 of CsA to be 0.1 to 0.3 μM (data not shown), which is similar to the IC50 value of CsA for the hepatic uptake of [14C]cerivastatin (0.20 μM) (Shitara et al., 2004). Thus, the unbound concentration of CsA at 3 days after its administration is not high enough to cause a marked reduction in the hepatic uptake by competitive or noncompetitive inhibition. Its inhibition on the hepatic uptake of BSP should be explained by other mechanisms.
Although there have been no reports of the inhibitory effect of CsA metabolites on the hepatic uptake transporter(s), we examined the blood concentration of CsA, including its metabolites in rats. As shown in Figure 2b, a higher concentration of metabolites (open symbols minus closed symbols) other than CsA itself was detected in the blood for 120 h after its administration. As of now, there has been no study determining the inhibitory effect of CsA metabolites on hepatic uptake transporters; thus, their contribution to the long-lasting inhibition of transporters observed in the present in vivo studies cannot be ruled out. In addition, Tanaka et al. (2000) reported that CsA is highly distributed to many tissues; thus, we examined the liver concentration of CsA (Fig. 2c). In the present study, CsA was highly accumulated in the liver. The liver/blood concentration ratio of CsA [(nanomoles per gram of liver)/(nanomoles per milliliter of blood)] was 5.37 at 6 h after the administration of CsA, and it increased to 10.5 at 120 h. A high concentration of CsA in the liver may modulate the transporter function although further studies are required.
We observed the expression levels of Oatp1a1 and Oatp1b2, which are responsible for the uptake of BSP (Jacquemin et al., 1994; Hagenbuch et al., 1996; Cattori et al., 2000). Their mRNA expression levels in the liver and protein expressions in the plasma membrane were not changed in CsA-treated rats (Fig. 3). This finding suggests that altered expression and localization of transporters are not involved in the drug-drug interaction with CsA. However, in our method, transporters in the plasma membrane may possibly include internalized ones and, thus, the possibility of their altered localization cannot be fully excluded. In addition, intrahepatic GSH, which is a driving force of Oatp1a1 (Li et al., 1998), was not changed. These results support the fact that CsA directly suppresses the activity of hepatic uptake transporters for BSP.
The uptake of BSP in isolated hepatocytes prepared from CsA-treated rats was decreased, although uptake studies were performed after removal of CsA from the incubation buffer (Fig. 4). This result supports the present LUI studies, which show that CsA inhibits the hepatic uptake of BSP for a long time. The reduced uptake of BSP was also examined in primary cultures of rat hepatocytes by a short-term preincubation with CsA (Fig. 5). The reduction depends on the concentration of CsA. In the present study using primary cultured hepatocytes, even after exposure to >1 μM CsA, approximately 60% of the uptake of BSP remained unaffected. It is possibly due to nonspecific binding of BSP to the culture plates, which causes a larger portion of BSP uptake to be apparently unaffected by CsA compared with using isolated hepatocytes. Taking this possibility into consideration, the present result suggests that the transporter-mediated uptake of BSP was almost completely diminished by exposure to >1 μM CsA.
Similar to the in vivo studies, CsA was highly accumulated in hepatocytes after its exposure. After a 20-min exposure of CsA to hepatocytes, 22.4 ± 0.5, 119 ± 4, and 541 ± 49 pmol/mg protein of CsA was associated to hepatocytes (n = 6 from two independent cell preparations) when exposed to 0.1, 1, and 10 μM CsA, respectively, although CsA in the incubation buffer was removed (data not shown). This high concentration of CsA was retained for at least 60 min even after incubation in CsA-free KHB at 37°C, which may partly explain its long-lasting inhibitory effect on the transporter function. The amount of CsA associated with hepatocytes was not significantly different between 20- and 60-min exposures.
The reduction in the uptake of BSP was attributed to the alteration in Km rather than Vmax (Fig. 4). That result means that CsA altered the affinity of BSP to the transporter, but not the capacity of the transporter, although it is hard to reach this conclusion because of the large variation of the present data (Fig. 4). This result was matched with the fact that no alteration was observed in the expression levels of Oatp1a1 and Oatp1b2 (Fig. 3).
In the present study, the mechanism for how CsA suppressed the hepatic uptake of BSP still remains to be elucidated. On the other hand, there have been some reports that phosphorylation of rat Oatp1a1 and Oatp1a4 results in suppression in their activities by short-term regulation (Glavy et al., 2000; Guo and Klaassen, 2001). In the case of rat Oatp1a1, phosphorylation reduced its activity in primary cultures of rat hepatocytes without alteration in its expression and membrane localization (Glavy et al., 2000), which seems to be similar to our result. However, in this case, the reduction is attributed mainly to the alteration in Vmax (Glavy et al., 2000), which apparently differs from the present result.
Similar to the mechanism-based inhibition (MBI) of metabolic enzymes, CsA reduces the uptake of BSP even after the inhibitor is removed. In the case of MBI, the inhibitor compound is converted to a reactive metabolite, which forms a covalent bond with the metabolic enzyme, and irreversibly inactivates it. Thus, the inhibitory effect is enhanced by the preincubation of inhibitor drugs with metabolic enzyme in the presence of cofactors (Ito et al., 1998; Lin and Lu, 1998). We examined the relationship between the extent of reduction in the hepatic uptake of BSP and preincubation time with CsA. However, preincubation time-dependent enhancement of the inhibitory effect by CsA was not observed, although it was enhanced for the first 5 min when the CsA concentration was 0.1 μM (Fig. 5). Thus, an irreversible inhibition of transporters by CsA was not fully supported by the present study.
In the case of MBI of metabolic enzymes, the IC50 value is reduced, depending on the preincubation time, and the extent of drug-drug interactions should be higher than that expected by a simple equation, 1/(1 + Iu/Ki), where Iu is the unbound concentration of inhibitor drugs at the inlet to the liver and Ki is the inhibition constant (Ito et al., 1998; Obach et al., 2007). In the present study, we performed the inhibition study with or without preincubation with CsA and compared the estimated IC50 values. The IC50 value was reduced by the preincubation (Fig. 6) although the inhibition mechanism of CsA is not the same as the MBI. It suggests that the extent of interaction caused by transporter inhibition by CsA should be higher than expected by the simple equation, 1/(1 + Iu/Ki). This suggestion means that we cannot predict the extent of drug-drug interactions by using this simple equation for this case.
Among clinically reported drug-drug interactions, most of severe cases, which cause more than a 5-fold increase in the AUC, occur by coadministration of mechanism-based inhibitors or azole antifungals, which irreversibly inhibit metabolic enzymes (Kato et al., 2008). Because CsA inhibits the hepatic uptake transporter sustainably, it may cause a severe drug-drug interaction in a clinical situation. That may be one of the reasons that CsA, in fact, causes clinically reported drug-drug interactions with a large number of OATP1B1 substrates and produces severe pharmacokinetic alterations.
In conclusion, CsA sustainably inhibits the Oatp-mediated uptake of BSP in rat liver, although the mechanism remains to be elucidated. CsA possibly causes a drug-drug interaction to a larger extent than expected because it inhibits the transporters even after it is removed. The estimated IC50 value was decreased by preincubation with CsA. Thus, CsA should be used with more caution when it is coadministered with OATP substrates.
Acknowledgments
We are grateful to Pharsight for providing us a license for WinNonlin Professional as part of the Pharsight Academic License program.
Footnotes
-
This study was supported in part by a Grant-in-Aid for Young Scientists (B) [Grant 17790130] provided by the Ministry of Education, Culture, Sports, Science and Technology of Japan; and the Japan Research Foundation for Clinical Pharmacology.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.025544.
-
ABBREVIATIONS: CsA, cyclosporin A; OATP/Oatp, organic anion-transporting polypeptide; AUC, area under the plasma concentration-time curve; P450, cytochrome P450; Mrp, multidrug resistance-associated protein; BSP, sulfobromophthalein; SD, Sprague-Dawley; PE, polyethylene; TBS-T, Tris-buffered saline containing 0.05% Tween 20; BSA, bovine serum albumin; PCR, polymerase chain reaction; RT-PCR, reverse transcription-PCR; KHB, Krebs-Henseleit buffer; LUI, liver uptake index; MBI, mechanism-based inhibition.
- Received November 5, 2008.
- Accepted March 9, 2009.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}