


Abstract
Intestinal first-pass metabolism may contribute to low oral drug bioavailability and drug-drug interactions, particularly for CYP3A substrates. The current analysis predicted intestinal availability (FG) from in vitro metabolic clearance and permeability data of 25 drugs using the QGut model. The drug selection included a wide range of physicochemical properties and in vivo FG values (0.07–0.94). In vitro clearance data (CLuint) were determined in human intestinal (HIM) and three liver (HLM) microsomal pools (n = 105 donors) using the substrate depletion method. Apparent drug permeability (Papp) was determined in Caco-2 and Madin-Darby canine kidney cells transfected with human MDR1 gene (MDCK-MDR1 cells) under isotonic conditions (pH = 7.4). In addition, effective permeability (Peff) data, estimated from regression analyses to Papp or physicochemical properties were used in the FG predictions. Determined CLuint values ranged from 0.022 to 76.7 μl/min/pmol of CYP3A (zolpidem and nisoldipine, respectively). Differences in CLuint values obtained in HIM and HLM were not significant after normalization for tissue-specific CYP3A abundance, supporting their interchangeable usability. The FG predictions were most successful when Papp data from Caco-2/MDCK-MDR1 cells were used directly; in contrast, the use of physicochemical parameters resulted in significant FG underpredictions. Good agreement between predicted and in vivo FG was noted for drugs with low to medium intestinal extraction (e.g., midazolam predicted FG value 0.54 and in vivo value 0.51). In contrast, low prediction accuracy was observed for drugs with in vivo FG <0.5, resulting in considerable underprediction in some instances, as for saquinavir (predicted FG is 6% of the observed value). Implications of the findings are discussed.
CYP3A enzymes represent the principle drug-metabolizing system in the small intestine, accounting for approximately 80% of total cytochrome P450 (P450) content (Lin et al., 1999; Paine et al., 2006). Although the total amount of CYP3A expressed in the human small intestine represents approximately 1% of the hepatic estimate (Paine et al., 1997), considerable drug extraction occurs during absorption of orally administered drugs (Hall et al., 1999; Galetin et al., 2008; Gertz et al., 2008a). This is due to the relatively high enterocytic drug concentration and the considerably lower blood flow to the intestine in comparison to the liver that allows prolonged exposure to the intestinal metabolizing enzymes. The contribution of intestinal first-pass metabolism has been shown indirectly for a number of CYP3A drugs administered both intravenously and orally in the absence and presence of inhibitors/inducers (Galetin et al., 2010). Such studies have allowed delineation of the relative roles of the liver and intestine and are particularly abundant for midazolam and to a lesser extent for cyclosporine, tacrolimus, alfentanil, and nifedipine.
The ability to predict the intestinal first-pass metabolism of drugs is of considerable importance for the assessment of oral clearance and the drug-drug interaction potential of CYP3A substrates. In both cases prediction models are very sensitive to the accuracy of the FG estimate, and the contribution of intestinal first-pass metabolism cannot be ignored. Two indirect methods have been proposed to estimate FG in vivo, namely use of plasma concentration-time profiles after either oral and intravenous administration or in the presence and absence of grapefruit juice. Both methods have several assumptions that may lead to potential bias in the FG estimates (Galetin et al., 2008; Gertz et al., 2008a).
In addition, in silico approaches have been proposed to estimate FG. These are based on the incorporation of drug permeability and metabolism data and enterocytic blood flow together with zonal and cellular heterogeneous distribution of metabolic enzyme and efflux/uptake transporters along the length of the intestine (Ito et al., 1999; Tam et al., 2003; Badhan et al., 2009; Jamei et al., 2009). In contrast to complex physiologically based models, a “minimal” QGut model has been proposed. This model allows prediction of FG using in vitro drug clearance and permeability (Chalasani et al., 2002; Rostami-Hodjegan and Tucker, 2002; Yang et al., 2007) and accounts for either permeability or perfusion as the rate-limiting process in the small intestine (see Materials and Methods) (eq. 2). However, the suitability of this model has yet to be demonstrated for a broad range of drugs. Yang et al. (2007) investigated the FG prediction success of the QGut model using in vitro clearance and permeability data collated from a variety of sources; although results were promising, this study did not allow a comprehensive assessment of the QGut model. In addition, a systematic assessment of the predictive utility of different in vitro systems to generate either clearance or permeability data as input parameters in this model is currently lacking.
The aim of this study was to investigate several aspects of the use of the QGut model to predict FG from in vitro data. First, a comparison was made between human intestinal and liver microsomes in their capacity to assess the metabolic drug clearance of a large number of structurally diverse drugs using a range of commercially available microsomal pools. The current analysis involves 25 drugs with differing physicochemical, metabolic, and permeability properties (Tables 1 and 3). These drugs also show differing extents of intestinal first-pass metabolism in vivo with FG values ranging from 0.07 to 0.94 for lovastatin and alprazolam, respectively. The suitability of these in vitro clearance data to predict both intravenous and oral clearance (incorporating the available in vivo FG values) was explored. Second, the use of permeability data [Papp (A-B)] generated under standardized conditions in both MDCK-MDR1 and Caco-2 cell lines was investigated, together with effective permeability (Peff), as permeability input parameters in the QGut model. The latter parameter was estimated either from regression analysis of the Caco-2 and MDCK-MDR1 data or from physicochemical parameters. Finally, the value of using midazolam as a calibrator within the QGut model was investigated given the abundance of information on this drug both in vivo and in vitro. On the basis of these analyses, recommendations on the suitability of the various in vitro input parameters in the QGut model and associated prediction accuracy are discussed.
Materials and Methods
Prediction of Intestinal Availability.
The CLuint values were scaled by the total amount of intestinal CYP3A to an intestinal intrinsic clearance, CLint,g (eq. 1). The total CYP3A content used in the current analysis was 70.5 nmol (Paine et al., 1997). The FG values were predicted using the QGut model (Chalasani et al., 2002; Rostami-Hodjegan and Tucker, 2004) as defined in eq. 2. QGut represents a hybrid parameter of enterocytic blood flow and drug permeability as defined by eq. 3 (Yang et al., 2007). FG predictions from the QGut model were compared with in vivo FG estimates obtained either from intravenous/oral or grapefruit interaction data (Galetin et al., 2008; Gertz et al., 2008a) and are summarized in Table 5. For indinavir, the FG value was estimated from intravenous/oral data reported by Yeh et al. (1999).
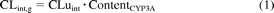
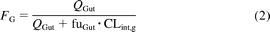
 where FG represents intestinal availability, Qent represents mucosal blood flow (liters per hour), CLuint represents unbound intrinsic clearance (microliters per minute per picomole CYP3A), fuGut represents fraction unbound in the enterocytes, and CLperm represents permeability clearance (liters per hour), the product of intestinal surface area and either apparent (nanometers per second) or effective permeability (micrometers per second).
where FG represents intestinal availability, Qent represents mucosal blood flow (liters per hour), CLuint represents unbound intrinsic clearance (microliters per minute per picomole CYP3A), fuGut represents fraction unbound in the enterocytes, and CLperm represents permeability clearance (liters per hour), the product of intestinal surface area and either apparent (nanometers per second) or effective permeability (micrometers per second).
The fraction unbound in the enterocytes was assumed to be 1 (Yang et al., 2007) and an average enterocytic blood flow (Qent) of 18 l/h was used for the predictions (Granger et al., 1980). Use of either fu in plasma or blood as an alternative to fuGut = 1 resulted in complete loss of prediction success and FG values approaching 1 for all drugs investigated. When effective permeability was used to predict FG, an intestinal surface area of 0.66 m2 (intestine treated as a tube as Peff accounts for the surface area magnifications of the fold of Kerkering, the villi, and the microvilli) was used to estimate permeability clearance (Yang et al., 2007). If apparent permeability was used to estimate permeability clearance, an intestinal surface area of 200 m2 was used.
Midazolam as QGut Calibrator.
To define a maximal value for QGut that approaches mucosal blood flow, the use of midazolam as a calibrator was investigated. Midazolam was selected because this drug is characterized by high apparent permeability. The QGut value of midazolam was estimated from the mean in vitro CLint,g data determined in this study and the mean FG value in vivo was determined from 16 intravenous/oral studies (Galetin et al., 2008) by rearranging eq. 2. The assumptions were that enterocytic binding is negligible (fuGut = 1) and that midazolam in vitro clearance is representative of its in vivo clearance. The FG in vivo for midazolam ranged from 0.49 to 0.70 (Galetin et al., 2008) and the CLint,g ranged from 7.30 to 28.7 l/h (Table 1). The coefficient of variation on the mean QGut value of midazolam was assessed by taking into account the variability of in vivo data (FG) or both in vivo and in vitro clearance data.
Comparison of CLuint corrected for CYP3A abundance in liver and intestine for one HIM and three HLM pools
Data obtained for 22 CYP3A4 substrates represent the individual clearance and the average clearance of all HIM and HLM batches together with the associated coefficient of variation. The clearance values determined in the HLM 1 pool represented 75% (65, 84, 95% CI) of the clearance determined in the HLM 2 pool and 56% (50, 62, 95% CI) of the clearance determined in the HLM 3 pool, whereas the HLM 2 pool represented 77% (72, 82, 95% CI) of the HLM 3 pool. Clearance values for triazolam, alprazolam, and quinidine were taken from Galetin and Houston (2006).
Determination of In Vitro Clearance.
The in vitro clearance data were determined by depletion in three liver and one human intestinal microsomal pools in 0.1 M phosphate buffer (pH 7.4) containing 10 mM MgCl2, 7.5 mM isocitric acid, 1.2 units/ml isocitric acid dehydrogenase, and 1 mM NADP. The human liver microsomal pools, HLM 1 (n = 22), HLM 2 (n = 50), and HLM 3 (n = 33), were purchased from BD Gentest (Woburn, MA); HLM 2 (n = 50) was kindly provided by Pfizer (Pharmacokinetics, Dynamics and Metabolism Department, Sandwich, Kent, UK). The human intestinal microsomal pool (n = 10) was purchased from XenoTech, LLC (Lenexa, KS), and microsomes were prepared by the elution method to ensure high enzyme activity, as reported previously (Galetin and Houston, 2006).
The final substrate concentration was 10-fold below the reported Km values in the literature for the drugs investigated. For indinavir and saquinavir, a substrate concentration of 0.1 μM was used. The drug was added from methanol stock solution, resulting in a final concentration of organic solvent in the incubation of 0.1% (v/v). Clearance incubations were prepared as replicates of two in Eppendorf tubes at 37°C and 900 rpm in an Eppendorf Thermomixer. The metabolic reaction was initialized by adding warm NADP solution to warm incubation mixture, and samples were taken at six designated time points within 60 min. To monitor non-P450-dependent loss of drug over the incubation time additional samples were prepared in the absence of NAPD. The metabolic reaction was terminated by addition of an equal volume of ice-cold acetonitrile containing the internal standard, samples were centrifuged at 1000g for 20 min at 4°C in a Mistral 3001 centrifuge (MSE, London, UK), and a 150-μl supernatant was removed from each Eppendorf vial and transferred to MVA glass vials (VWR International, Leicestershire, UK) before analysis on the LC-MS/MS system. Samples were embedded in the calibration curves upon LC-MS/MS analysis. CLint data were obtained for all drugs in the dataset, with the exception of alprazolam, quinidine, and triazolam; for these CYP3A substrates data were taken from a previous in-house study (Galetin and Houston, 2006).
Microsomal Binding.
Nonspecific binding values of drugs to microsomal protein (fuinc) were experimentally determined at different microsomal protein concentrations in HLM 1 using the microdialysis method (Gertz et al., 2008b). In addition to reported experimental values in Gertz et al. (2008b), fuinc values for alfentanil, atorvastatin, cisapride, lovastatin, methadone, nisoldipine, rifabutin, sildenafil, trazodone, and zolpidem were determined at 0.1, 0.5, and 1.0 mg/ml microsomal protein concentrations over 8 h. The fuinc values were fitted in Grafit 5.0.10 (Erithacus Software Limited, Surrey, UK) against protein concentrations to determine the binding constant (Ka). The drug-specific Ka values are summarized at http://www.pharmacy.manchester.ac.uk/capkr/. For cyclosporine, the extent of nonspecific binding was predicted (Hallifax and Houston, 2006).
CLuint was calculated using eq. 4. Clearance values were corrected for the different organ abundance of CYP3A in liver and intestinal microsomes: 50 and 155 pmol of CYP3A/mg of protein for the small intestine and the liver, respectively (Paine et al., 1997; Rostami-Hodjegan and Tucker, 2007).
 where CLuint is unbound intrinsic clearance (microliters per minute per picomole CYP3A), k is the depletion rate constant (minute−1), V is the initial incubation volume (milliliters), proteinmicrosomal is the initial amount of protein (milligrams), and abundanceCYP3A is the abundance of CYP3A (picomoles of CYP3A per milligram of protein).
where CLuint is unbound intrinsic clearance (microliters per minute per picomole CYP3A), k is the depletion rate constant (minute−1), V is the initial incubation volume (milliliters), proteinmicrosomal is the initial amount of protein (milligrams), and abundanceCYP3A is the abundance of CYP3A (picomoles of CYP3A per milligram of protein).
Testosterone 6β-Hydroxylation Activity.
CYP3A enzyme activities of the HLM and HIM pools were determined at a testosterone concentration of 250 μM (equivalent to Vmax), monitoring 6β-hydroxytestosterone formation. Incubation conditions were the same as those used in the depletion assay, and organic solvent content was 0.3% (v/v) methanol. Microsomal protein concentrations for the HLM and HIM pools were 0.25 and 0.50 mg/ml, respectively. On each occasion, three samples were taken after a 5-min incubation. Aliquots of 100 μl were removed into Eppendorf vials with 100 μl of ice-cold acetonitrile containing 1 μM progesterone as the internal standard. Activity was determined on three separate occasions to account for interday variability. Testosterone 6β-hydroxylation activities were (mean ± S.D.) 3.38 ± 0.323, 4.48 ± 0.218, and 6.09 ± 0.195 nmol/min/mg for HLM pool 1, 2, and 3, respectively, and 1.84 ± 0.173 nmol/min/mg for HIM.
Prediction of Intravenous and Oral Clearance.
Oral and intravenous clearance values, fup values, and blood/plasma distribution ratios (Rb) were collated from the literature for all compounds investigated (Table 2). References for all clinical studies considered are available at http://www.pharmacy. manchester.ac.uk/capkr/. When multiple clinical studies were available, mean clearance values and associated 95% credible intervals were calculated by meta-analyses using fixed or random effects models in WinBUGS (version 1.4.3; available at http://www.mrc-bsu.cam.ac.uk/bugs/), assuming log-normal distribution of CLi.v. and AUCoral. Criteria to exclude studies from analysis were non-white populations, nonlinear dose-AUC response, AUC reported over an insufficiently long time-course, analytical method inappropriate to determine the concentration of the drug of interest adequately, and studies performed in elderly or patient populations.
Mean intravenous and oral plasma clearance data, number of datasets, blood/plasma ratios, fraction unbound in plasma, and the observed and predicted intrinsic clearance values for 25 drugs investigated
Values in parentheses represent the 95% credible intervals for intravenous and oral plasma clearance for drugs for which more than one clinical study were available and 95% confidence intervals for the predicted CLint,h. References for all clinical studies are available at http://www.pharmacy.manchester.ac.uk/capkr/.
For the fixed-effects model, lnCLi.v. and lnAUCoral ∼N(μ, ω2), where AUCoral represents the dose-normalized AUC, μ represents the log-transformed mean CLi.v. or mean AUCoral, and ω represents the variance (S.D.2/N). The fixed-effects model was used for drugs for which sparse data were available; this model made no distributional assumption on ω. The mean and variance of the untransformed variables are exp(μ + 0.5 ω2) and exp(2μ + ω2)(exp(ω2) − 1), respectively. Otherwise random-effects models were used [lnCLi.v. and lnAUCoral ∼N(μ, ω2), gamma distribution of ω] or a modified random-effects model [for midazolam CLi.v. only: lnCLi.v. and lnAUCoral ∼N(μ, ω2)], accounting for the distribution of the true S.D. of study i [S.D.i ∼N(S.D.mean, i, S.D.precision, i), where ω and the true precision followed gamma distributions].
CLint,h values after intravenous and oral drug administration were obtained using eqs. 5 and 6, respectively (Pang and Rowland, 1977):
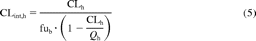
 where CLh and D/AUC represent the hepatic blood clearance obtained from mean plasma data (Table 2) after correcting for renal clearance (where applicable) and Rb. The fub represents the fraction unbound in blood, Qh represents the average hepatic blood flow of 20.7 ml/min/kg (Kato et al., 2003), D represents the oral drug dose (milligrams per kilogram), AUC represents the area under the drug concentration-time curve (milligram per minutes per milliliter), and Fa represents the fraction absorbed.
where CLh and D/AUC represent the hepatic blood clearance obtained from mean plasma data (Table 2) after correcting for renal clearance (where applicable) and Rb. The fub represents the fraction unbound in blood, Qh represents the average hepatic blood flow of 20.7 ml/min/kg (Kato et al., 2003), D represents the oral drug dose (milligrams per kilogram), AUC represents the area under the drug concentration-time curve (milligram per minutes per milliliter), and Fa represents the fraction absorbed.
Clearance data for cisapride, lovastatin, simvastatin, and terfenadine were only available after oral drug administration. Oral clearance data for cyclosporine were available for two different oral formulations (Sandimmune and Neoral) and both were used in the assessment. Rb data were not available for lovastatin, nisoldipine, and trazodone. Given the very high structural similarity between simvastatin and lovastatin, the Rb value used for lovastatin was 0.57. The Rb values of nisoldipine and trazodone were assumed to be 1. The CLint,h estimate of trazodone was not sensitive to changes in Rb, whereas nisoldipine CLint,h displayed a high sensitivity to changes in Rb. Dose-AUC response data were assessed where available to avoid any bias in CLint,h estimates from oral data. Furthermore, indinavir was excluded from the oral dataset as the high dose at which it is administered (400–800 mg) was shown to significantly reduce its systemic clearance (Yeh et al., 1999). The unbound CLuint from all HLM pools investigated were scaled using the mean microsomal recovery of 40 mg of protein/g of liver (Barter et al., 2007) and a liver weight of 21.4 g of liver/kg.
Determination of In Vitro Permeability.
Drug permeability experiments in Caco-2 and MDCK-MDR1 cells were performed at Pfizer (Pharmacokinetics, Dynamics and Metabolism Department). The permeability experiments were performed during cell passages 11 to 32 for the MDCK-MDR1 cells and 25 to 35 for the Caco-2 cells. To determine the passive permeability of drugs with efflux ratios greater than 2, the P-gp inhibitor CP-100356 (Wandel et al., 1999) was coincubated at a concentration of 10 μM. MDCK-MDR1 cells (250 μl, density 2.5 × 105cells/ml in MDCK-MDR1 cell media) were added to the apical sides of Costar HTS 24-well Transwell plates and MDCK-MDR1 cell media (1 ml) was added to the basolateral sides. Plates were incubated for 4 days in a LEEC Research Incubator (37°C under 5% CO2 in air) before cell permeability experiments. One day before the experiment, the cell media in the apical and basolateral sides of the plate were replaced by fresh media. The MDCK-MDR1 cell media consisted of Alpha MEM (500 ml), fetal bovine serum (50 ml), penicillin-streptomycin liquid (5 ml), MEM nonessential amino acids (5 ml), and l-glutamine (5 ml). In contrast, the Caco-2 cells (250 μl, density of 1.6 × 105cells/ml) were plated in BD Falcon HTS 24-well Transwell plates (BD Biosciences Discovery Labware, Bedford, MA). Caco-2 cells were ready for use after 3 weeks of incubation in a LEEC Research Incubator (37°C under 5% CO2 in air). Media were replaced on alternate weekdays by Caco-2 cell media, 250 μl and 1 ml into the apical and basolateral side, respectively. The cell media for the Caco-2 cells consisted of MEM (500 ml), fetal bovine serum (100 ml), MEM nonessential amino acids (6 ml), sodium pyruvate solution (6 ml), and l-glutamine solution (6 ml).
The incubation buffer was prepared from HEPES (2.38 g) solubilized in HBSS (500 ml) and titrated to pH 7.4 with 1 M sodium hydroxide solution. Experiments were performed under isotonic conditions at pH 7.4. Nadolol (2 μM) or Lucifer yellow (100 μM in HBSS, pH 7.4) was used as an integrity marker. Lucifer yellow-mediated fluorescence was measured on a Victor2 1420 Multilabel Reader (PerkinElmer Life and Analytical Sciences-Wallac Oy, Turku, Finland). Papp (A-B) values of nadolol or Lucifer yellow were calculated using eq. 7; Papp values <10 nm/s were an indication of an intact cell monolayer. One microliter of the drug stock solution (in dimethyl sulfoxide) was added to 10 ml of HBSS, pH 7.4, buffer containing nadolol, resulting in a final substrate concentration of 0.1 μM (final concentration of dimethyl sulfoxide 0.03%, v/v). For each plate, metoprolol and talinolol were coincubated as positive controls for passive permeability and active efflux, respectively; both were incubated at a final substrate concentration of 2 μM. The substrates and the positive controls were incubated in three separate wells in both directions. The plates were placed in a LEEC Research Incubator (37°C under 5% CO2 in air) on a shaker at 150 rpm for 2 or 2.5 h for the Caco-2 and MDCK-MDR1 cell permeability experiment, respectively. The setup and sampling were performed on a Tecan Genesis RSP 150 Robot. Samples and calibration curves were added to acetonitrile and centrifuged for 20 min at 3000 rpm before analysis on an LC-MS/MS system.
Sample Analysis.
Quantification of samples was performed with Analyst version 1.4.1 software (Applied Biosystems, Foster City, CA). Permeability values were calculated in both directions using eq. 7; drug recovery was between 75 and 130% except where noted in Table 3. Apparent permeability (Papp) and efflux ratio (ER) data, defined as Papp (B-A)/Papp (A-B), for metoprolol and talinolol were assessed with every plate. Papp (A-B) for metoprolol >200 nm/s and an ER value for talinolol of >5 were used as an indicator for a functional cell monolayer.
 where VR represents volume of the receiver chamber, A represents surface area of the cell monolayer (0.33 cm2), C0 represents the initial substrate concentration (micromolar concentration), and dC/dt represents the change of concentration over time.
where VR represents volume of the receiver chamber, A represents surface area of the cell monolayer (0.33 cm2), C0 represents the initial substrate concentration (micromolar concentration), and dC/dt represents the change of concentration over time.
Database of Papp and efflux ratio data for the FG dataset obtained in MDCK-MDR1 and Caco-2 cells
Metoprolol Papp data were available for 11 and 9 occasions for the assessment of interday variability of Papp in MDCK-MDR1 and Caco-2 cells, respectively. Permeability data for the drugs investigated were normalized for the mean metoprolol permeability to account for interday variability. The mean ± S.D. Papp (A-B) for metoprolol in MDCK-MDR1 cells was 341 ± 92 nm/s and in Caco-2 cells was 281 ± 42 nm/s. No permeability data could be determined for nisoldipine, terfenadine, and atorvastatin, and literature values were taken.
LC-MS/MS Analysis and List of Chemicals.
Detailed information on LC-MS/MS analysis and the list of chemicals are provided at http://www.pharmacy.manchester.ac.uk/capkr/.
Regression Analysis between In Vitro and In Vivo Permeability.
A correlation between in vivo Peff and in vitro Papp (A-B) data in MDCK-MDR1 cells was investigated. Peff data were obtained in the upper jejunum for a range of compounds (Lennernäs, 2007). The corresponding Papp data for this additional set of drugs were provided by Pfizer (Pharmacokinetics, Dynamics and Metabolism Department) and were used as a training set for the regression analysis between Papp and Peff (Fig. 1). The Papp values for this training set (determined at 2 μM under isotonic conditions, pH 7.4) ranged from 2.4 to 442 nm/s for amoxicillin and antipyrine, respectively. The Peff values ranged from 0.04 to 8.7 μm/s for hydrochlorothiazide and ketoprofen, respectively. Permeability data for the training dataset are available at http://www.pharmacy.manchester.ac.uk/capkr/. Considering that the in vivo permeability of amoxicillin, amiloride, cephalexin, and cyclosporine might be facilitated by active transport, the regression analysis was performed either using all drugs (subset A: n = 24) or drugs that were characterized by passive permeability (subset B: n = 20). The regression analysis and the S.E. associated with the regression coefficient were calculated in Grafit 5.0.10. The precision of Peff predictions based on the regression analysis using subset A and B was determined. The linear correlation between Papp and Peff was weak (R2 = 0.40) for both subsets A and B. The coefficients of determination between the logPapp and logPeff were 0.61 and 0.70 if subsets A or B were used, respectively. Equations based on the regression analysis of the subset A and B are shown as eq. 8 and eq. 9, respectively.
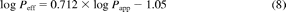
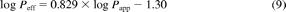 The respective slopes of the regression analysis were associated with S.E. of 17 and 15% for subset A and B, respectively. The S.E. associated with the regression equations resulted on average in a 2.2-fold deviation of the Peff predictions from unity.
The respective slopes of the regression analysis were associated with S.E. of 17 and 15% for subset A and B, respectively. The S.E. associated with the regression equations resulted on average in a 2.2-fold deviation of the Peff predictions from unity.
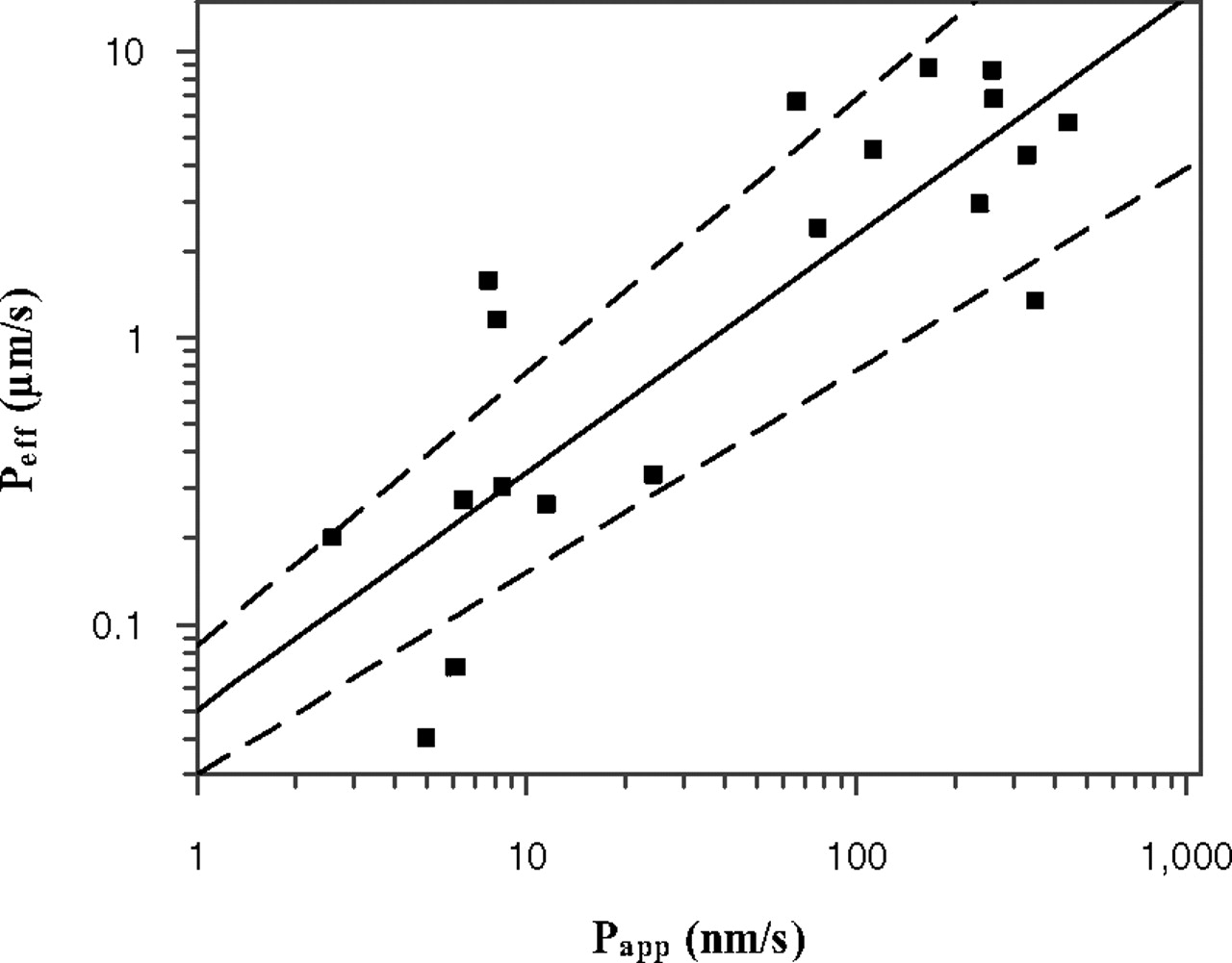
Comparison of Papp (A-B) obtained at isotonic pH 7.4 in MDCK-MDR1 cells and human Peff available from the literature (Lennernäs, 2007) for 20 passively permeable drugs. The line of best fit is described by logPeff = 0.829 × logPapp − 1.30 where the dashed lines indicate uncertainty in the line of best fit, as a consequence of S.E. associated with the parameter estimates of slope (15%) and intercept (17%).
In addition to data from MDCK-MDR1 generated in the present study, correlation between Peff and Papp in Caco-2 cells (pH 7.4) was already available for 24 drugs from previous studies (Sun et al., 2002), as shown in eq. 10. Regression equations from both cell lines (eq. 9 for MDCK-MDR1 and eq. 10 for Caco-2) were used for the prediction of Peff and consequently FG for drugs in the current study; their prediction success and application in the QGut model were assessed.
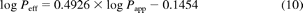
Prediction of Peff from Physicochemical Data.
In addition to in vitro data, the physicochemical parameters, hydrogen bond donors, and polar surface area were collated for all drugs investigated on http://pubchem.ncbi.nlm.nih.gov/. With use of eq. 11, Peff data were predicted from these physicochemical parameters, as described previously (Winiwarter et al., 1998). The application of Peff values using this approach for the prediction of FG was assessed in comparison to other permeability approaches described above.
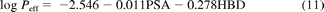 where PSA is polar surface area and HBD is hydrogen bond donor.
where PSA is polar surface area and HBD is hydrogen bond donor.
Bias and precision in estimating CLint,h and FG were calculated as geometric fold error (gmfe) in eq. 12 and root mean squared error (rmse) (units of the parameter investigated) in eq. 13 (Sheiner and Beal, 1981; Fahmi et al., 2008). The gmfe does not allow over- and underpredictions to cancel each other out and indicates therefore an absolute deviation from the line of unity.
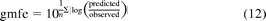
 where n is number of observations.
where n is number of observations.
Results
In Vitro Clearance.
In vitro clearance data were obtained in pooled human intestinal microsomes (n = 10) and three liver microsomal pools (in total, n = 105 donors). The CLuint values covered 4 orders of magnitude and ranged from 1.10 to 3840 μl/min/mg protein for zolpidem and nisoldipine, respectively, for the HIM pool. In the HLM pools, CLuint values span over a similar range, from 10.9 to 9790 μl/min/mg protein for zolpidem and saquinavir, respectively. The correlation between the HIM and the mean HLM clearance was strong (R2 = 0.98). The CLuint values (expressed per milligram of protein) determined in HIM represented between 32 and 47% of the average HLM CLuint. However, tacrolimus and midazolam showed higher than average CLuint values in HIM representing 63 and 79% of the HLM clearance, respectively.
A direct comparison of CLuint obtained in HLM and in HIM after the correction for the tissue-specific abundance of CYP3A is shown in Table 1. Corrected CLuint values in the HIM pool ranged from 0.022 to 76.7 μl/min/pmol of CYP3A for zolpidem and nisoldipine, respectively, and from 0.106 to 48.1 μl/min/pmol of CYP3A in the HLM pools for zolpidem and saquinavir, respectively. Figure 2 illustrates comparison of the CLuint values of the drugs investigated normalized for the population CYP3A abundance in the intestine and liver. Good agreement between the estimates was observed with 50% of the clearance values within 1.5-fold and 14% outside 2-fold of unity. The most pronounced discrepancy in the HLM and HIM clearance was observed for midazolam (CLuint in HIM represented 246% of mean CLuint in HLM) and zolpidem (CLuint in HIM represented 21% of mean CLuint in HLM). After correction for tissue-specific CYP3A abundance, the CLuint determined in the HIM pool represented 100 to 144% of the CLuint in the HLM pools and was not statistically different at a significance level of 5% (Student's t test).

Comparison of CLuint (microliters per minute per picomole of CYP3A) from HLM (mean ± S.D., n = 3) and HIM for 22 drugs. Dashed line indicates a bias of 1.55-fold deviation from unity.
The CLuint data obtained from the three different liver pools were compared and found to be significantly different (p < 0.05). The highest clearance values were determined in the HLM 3 pool, whereas clearance in HLM pools 1 and 2 represented 56 to 75% of the CLuint determined in this pool. The microsomal activity toward 6β-hydroxytestosterone was determined in all microsomal pools and ranged from 1.84 to 6.09 nmol/min/mg in the HIM and the HLM batch 3, respectively. The activity determination for each microsomal batch was associated with low interday variability (≤10%). The coefficient of variation between the activities of the different HLM batches was 29%, comparable to the 31% average interbatch variability in clearance for the drugs investigated. Most of the CLuint variability between different microsomal pools could be attributed to the differences in 6β-hydroxytestosterone activity (R2 = 0.99). For midazolam, nifedipine, nisoldipine, and tacrolimus only 49 to 73% of the changes in clearance could be attributed to differences in testosterone 6β-hydroxylation activity.
Prediction of Hepatic Intrinsic Clearance from Intravenous and Oral Data.
A large body of clinical clearance data after intravenous drug administration was collated for the drugs investigated, with up to 30 clinical studies investigating 469 individuals available for midazolam (Table 2). The systemic plasma clearance of midazolam was estimated to be 6.16 ml/min/kg (5.64, 6.72, 95% credible interval) using meta-analysis of literature data, which corresponded to an CLint,h of 440 ml/min/kg (364, 533, 95% credible interval). In total, the database consisted of 21 drugs, because no intravenous clearance data were available for cisapride, lovastatin, simvastatin, and terfenadine, with a range of in vivo systemic plasma clearance, varying from 0.76 to 28.3 ml/min/kg for the intravenous data of alprazolam and buspirone, respectively (Table 2). The fup ranged from 0.3 to 36% for nisoldipine and indinavir, respectively, and the CLh ranged from 4 to ∼100% of Qh for tacrolimus and buspirone, respectively. Overall, 11 of the investigated drugs showed CLh ≥50% of Qh. The highest blood clearance values were observed for buspirone, indinavir, and saquinavir (all >80% of Qh).
A moderate correlation existed between the observed and the predicted logCLint,h values when the well stirred liver model was used (R2 >0.65); 43 and 76% of the predictions were within 2- and 5-fold of unity, respectively. The most significant CLint,h underpredictions (≤20% of observed) were noted for atorvastatin (2.5%), buspirone (7.6%), repaglinide (7.9%), felodipine (10%), and sildenafil (20%). The use of the average HLM clearance data and a mean microsomal recovery value of 40 mg/g resulted in a median underprediction of 19%, a 3.1-fold bias (gmfe), and a precision (rmse) of 4140 (Fig. 3A). The bias was notably decreased with increasing microsomal CYP3A4 activity of the different liver pools; however, the precision of CLint,h predictions was not affected by the use of different HLM pools.

Comparison of predicted and observed CLint,h values. A, comparisons of observed and predicted CLint,h values from intravenous data obtained with the well stirred model for 21 drugs. B, comparison of observed and predicted CLint,h values from oral data after correction for in vivo FG (corresponding values listed in Table 5). Dashed lines represent the observed prediction bias of 3.1- and 2.9-fold deviation from unity for A and B, respectively, and error bars indicate the S.D. from the in vitro clearance experiments. Outliers identified represent the following: 1, rifabutin; 2, tacrolimus; 3, zolpidem; 4, sildenafil; 5, repaglinide; 6, atorvastatin; 7, buspirone; 8, felodipine; 9, terfenadine; 10, lovastatin; and 11, simvastatin.
A substantial number of clinical studies were available after oral administration; for most drugs investigated data from ≥3 clinical studies were used in the meta-analyses. For midazolam, 14 separate studies with 262 individuals in total were considered in the analysis. The oral clearance of midazolam was estimated to be 24.2 ml/min/kg (20.5, 28.5, 95% credible interval), which corresponded to an CLint,h of 402 ml/min/kg (340, 473, 95% credible interval). Clinical studies with an oral dose of midazolam exceeding 10 mg were excluded from the analysis because of a possible nonlinear response in midazolam AUC. For the entire database, the oral clearance values ranged from 0.99 to 3440 ml/min/kg for alprazolam and saquinavir, respectively (Table 2). The contribution of the small intestine to oral clearance was incorporated using the FG values listed in Table 5, as shown in eq. 6. Consequently, CLint,h values ranged from 3.22 to 16,800 ml/min/kg for alprazolam and terfenadine, respectively. After correction for the drug-specific FG values, the CLint,h values estimated from oral data corresponded to 94% of the intravenous estimate on average. Particular differences between intravenous and oral estimates were apparent for indinavir (where oral CLint,h represented 7% of intravenous data) and sildenafil (18%). For indinavir, this was attributed to enzyme saturation/inhibition at the high dose at which this drug is generally administered, and the data were subsequently excluded from the analysis.
The predictability of oral clearance from in vitro data generated in the current study was investigated. Figure 3B displays the comparison of the predicted to the observed CLint,h from oral data; 38 and 65% of the predictions were within 2- and 5-fold, respectively. The median prediction success was 93% of the observed values (interquartile range 33–204%). Accounting for FG decreased the overall degree of underpredictions from oral data; however, considerable underprediction persisted for atorvastatin (1.9% of observed), buspirone (4.7%), repaglinide (13.4%), felodipine (13.8%), and terfenadine (17.5%). Significant overprediction was apparent for rifabutin (6-fold), simvastatin, and lovastatin (7- and 8-fold, respectively). An improvement in the CLint,h prediction success relative to the analysis performed without the FG was apparent in the decrease in bias by 25% and marked increase in precision (32,600 versus 4030 without and with FG incorporated, respectively). The resulting bias and precision of the CLint,h prediction from FG corrected oral clearance data were highly comparable to the predictions of CLint,h from intravenous data.
In Vitro Permeability.
The Papp and the ER data were determined across MDCK-MDR1 and Caco-2 cells for the drugs investigated (Table 3). The permeability values ranged from 6 to 398 nm/s for cyclosporine and buspirone, respectively, in the MDCK-MDR1 cells and from 4 to 324 nm/s for saquinavir and midazolam, respectively, in the Caco-2 cells. Six drugs represented P-gp substrates in both cell lines (ER >2), namely cyclosporine, indinavir, quinidine, rifabutin, saquinavir, and tacrolimus. The ER in the MDCK-MDR1 cells increased in the following rank order: quinidine < tacrolimus < rifabutin < indinavir < cyclosporine < saquinavir and ranged from 4.4 to 75. A similar rank order of increasing ER was evident in the Caco-2 cell system and was as follows: tacrolimus < rifabutin < quinidine < cyclosporine < indinavir < saquinavir with the ER ranging from 2.1 to 57 (Table 3). Coincubation with CP-100356 inhibited drug efflux mediated by P-gp and the resulting permeability was taken to represent the passive permeability of the drugs in the respective cell line, assuming no additional transport mechanisms. A relatively small proportion of drugs investigated displayed low permeability (≤100 nm/s): 18 and 31% in the MDCK-MDR1 and Caco-2 cell systems, respectively. The Papp data in the MDCK-MDR1 cell system were subject to considerable interday variability (27% based on 11 observations); the variability in the Caco-2 cells was 15% (based on nine observations). The Papp values of the drugs investigated across both cell systems displayed a high linear correlation (R2 = 0.79). In general, Papp values in the Caco-2 cell system were lower than those generated in MDCK-MDR1 cells, but the absolute differences were minor. The Papp values determined in Caco-2 cells represented on average 66 to 89% (95% CI) of MDCK-MDR1 cell values. The relationship between Papp determined across Caco-2 and MDCK-MDR1 cells was best described by the following equation:

QGut Calibration.
Midazolam represents a drug with high apparent permeability, and therefore its QGut value is expected to approach Qent. Thus, midazolam QGut provides the possibility to normalize the QGut values of other drugs estimated from in vitro data. The choice of midazolam as a calibrator was foremost based on the large body of available in vivo studies reporting simultaneous intravenous and oral midazolam pharmacokinetic data, as well as midazolam CLuint values determined in four different microsomal systems. The QGut value of midazolam was estimated from the weighted mean of the in vivo FG data and the in vitro clearance data determined in the present study (Table 1). The QGut value of midazolam was estimated as 16.6 l/h. This value was associated with considerable uncertainty (coefficients of variation were 31% when the variability of in vivo FG estimates alone was accounted for and 61% when combined variability of the in vivo FG estimate and the in vitro CLuint data was accounted for). This value approached the Qent value of 18 l/h used in this study, suggesting no permeability limitations and supporting the use of midazolam as a QGut calibrator.
Predictions of Intestinal Availability.
Permeability and in vitro clearance data were used for FG predictions using the QGut model by eight different methods (Fig. 4). First, Papp data determined across Caco-2 and MDCK-MDR1 cell monolayers were used directly. In addition, Peff data were estimated from the regression analysis to Papp values from either Caco-2 or MDCK-MDR1 and from physicochemical properties. In the latter approaches, QGut data were investigated before and after calibration with midazolam QGut. The results of the prediction bias and accuracy of different approaches for the entire dataset and for the subset of drugs with in vivo FG <0.5 are summarized in Table 4. The best FG prediction success was apparent from the direct input of Papp data as shown by the lowest bias and interquartile range of all approaches investigated (Table 4, 1). The use of permeability data obtained in the different cell lines investigated in the current study resulted in minor differences in FG prediction success. In contrast, the input of the Peff data from the regression analysis to Papp data resulted in a larger bias regardless of the cell line used (Fig. 4A; Table 4); however, this underprediction trend was corrected after adjustment for midazolam QGut. The analysis has also indicated that the use of these empirical regression equations for drugs with Papp<10 nm/s is problematic, as highlighted by considerable scatter in this area (Fig. 2). The use of polar surface area and hydrogen bonding potential resulted in the most biased FG predictions and significant underprediction of FG (p < 0.05). Adjustment for midazolam as a calibrator of QGut had a negligible impact on the FG predictions from physicochemical properties and the underprediction trend remained (Fig. 4; Table 4, 4).

Prediction success of FG using eight different permeability inputs in the QGut model. The average CLuint data from HLM and HIM were used. A, log(predicted FG/observed FG) of the complete dataset of 25 drugs. B and C, prediction success of drugs with in vivo FG values above (n = 14) and below 0.5 (n = 11), respectively. Zero indicates unity and negative and positive values represent under- and overpredictions, respectively; *, significant underpredictions (p < 0.05). cal., denotes calibration for midazolam QGUT; PSA, polar surface area; HBD, hydrogen bond donor.
Description of bias (gmfe) and percentage within 1.5-fold of unity for predictions of the FG for either total set of 25 drugs or for a subset of 11 drugs for which in vivo FG < 0.5
Different permeability approaches were used as follows: 1, Papp (A-B) data from Caco-2 (data in parentheses: MDCK-MDR1); 2, Peff data from correlation to Papp (A-B) from Caco-2 (data in parentheses: MDCK-MDR1); 3, Peff data from correlation to Papp (A-B) from Caco-2 (data in parentheses: MDCK-MDR1) calibrated for midazolam QGut; and 4, Peff data from correlation to in silico data (data in parentheses: calibrated for midazolam QGut).
Figure 4B illustrates the high degree of prediction accuracy for drugs with in vivo FG values >0.5. In contrast, a subset of 11 drugs with in vivo FG <0.5 is comparatively poorly predicted, and the degree of imprecision is considerably increased in comparison to drugs with low to moderate intestinal extraction (Fig. 4C; Table 4). Consistent over the entire set, the direct input of Papp data resulted in the lowest bias and interquartile range in FG predictions. For indinavir, significant underprediction was observed regardless of permeability parameter input. The choice to perform permeability experiments under isotonic conditions might have biased the Papp (A-B) values for certain drugs in the dataset, because the intestinal pH in the duodenum and jejunum is <7 (Fallingborg et al., 1989). Considering that a significant impact of permeability to FG predictions was only apparent at drug permeability <100 nm/s, the chosen in vitro conditions might have biased subsequent FG predictions of indinavir and saquinavir (Papp <10 nm/s) and to a minor extent quinidine (Papp <100 nm/s in Caco-2 cells).
Figure 5 and Table 5 illustrate the comparison between predicted and observed FG values based on Papp data from both cell lines; each outlier is identified by a number. Overprediction was apparent for rifabutin (1), atorvastatin (2), buspirone (3), and tacrolimus (4). The average predicted tacrolimus FG was associated with a high degree of variability (indicated by the large error bars) driven by the highly variable in vitro clearance of this drug. Underprediction was observed for simvastatin (5), saquinavir (6), terfenadine (7), felodipine (8), and indinavir (9). The degree of underprediction was dependent on the use of permeability input and was particularly high for indinavir (17–33% of observed), saquinavir (5–12%), and terfenadine (16–29%). The QGut value, a hybrid parameter reflecting both drug permeability and mucosal blood flow, ranged from 2.4 to 16.6 l/h for saquinavir and midazolam, respectively (Table 5).

FG predictions obtained using in vitro clearance (Table 1) and Papp (A-B) data obtained in either Caco-2 (□) or MDCK-MDR1 cells (*). The dashed lines represent 1.5-fold deviation from unity, and error bars represent the S.D. associated with the FG predictions using different HLM and HIM pools. Outliers identified are the following: 1, rifabutin; 2, atorvastatin; 3, buspirone; 4, tacrolimus; 5, simvastatin; 6, saquinavir; 7, terfenadine; 8, felodipine; and 9, indinavir.
Predictions of FG and QGut using the QGut model from in vitro clearance and permeability data for 25 drugs
Data are predicted values ± S.D.
Discussion
This study has evaluated the use of various sources of metabolism and permeability data for predicting FG using the QGut model. A group of 25 structurally diverse CYP3A4 substrates was used for this investigation with corresponding FG values in vivo ranging from 0.07 to 0.94 for lovastatin and alprazolam, respectively.
In Vitro Clearance Data.
A high degree of comparability was observed between in vitro clearance from HIM and three HLM pools for the dataset investigated, as illustrated in Fig. 2. This suggests that in vitro clearance between hepatic and intestinal microsomes can be extrapolated if enzyme abundance data are available and the contribution of P450 enzyme to drug metabolism is known. The current findings support our previous work (Galetin and Houston, 2006), in which good agreement between normalized hepatic and intestinal clearances was observed for a limited number of substrates for a range of P450 enzymes. No significant difference in the hepatic and intestinal clearances seen once normalized for the tissue-specific CYP3A abundance supports their interchangeable use, as illustrated here in the QGut model. CLint data were normalized using reported population enzyme abundance data based on a meta-analysis of 241 liver samples (Rowland-Yeo et al., 2004; Rostami-Hodjegan and Tucker, 2007). Abundance data for both CYP3A and CYP3A4 in the liver (155 and 111 pmol/mg, respectively) are associated with large coefficients of variation (67–119%). In the current study, actual CYP3A4 abundance was available only for the HLM pool 2 (138 pmol/mg), which was within the reported population limits. Furthermore, the reported population abundance data are based on liver microsomes of white origin and consequently the HLM pools with a high white donor percentage should be selected to allow appropriate scaling with population estimates. In contrast to the liver data, intestinal data are characterized to a lesser extent, as the CYP3A and CYP3A4 abundance data are available from 31 individual of mixed ethnicity (Paine et al., 2006).
This study showed a very strong linear correlation between clearance values determined in different microsomal pools and their respective testosterone 6β-hydroxylation activity. The differences observed in microsomal enzyme activity between the pools contributed considerably to the prediction success of clearance. Considering the aforementioned, pools with small donor sizes might not be representative of the true population mean. In addition, different substrate binding sites associated with the CYP3A4 enzyme probably explain this discrepancy (Galetin et al., 2003), as well as differing contributions of CYP3A4/CYP3A5 to drug clearance (Galetin et al., 2004; Huang et al., 2004) in comparison with the marker substrate testosterone (e.g., tacrolimus and saquinavir). However, although prediction bias was affected by the choice of microsomal pools, it had only a marginal effect on the precision of clearance predictions. The current analysis included three known inhibitors of CYP3A, namely indinavir, saquinavir, and verapamil (Eagling et al., 1997; Wang et al., 2004; Ernest et al., 2005). To avoid biased clearance estimates, incubations were performed at a substrate concentration below Ki (for competitive inhibitors) or KI (time-dependent inhibitors) and over a short (≤30 min) time period. No apparent inhibition was evident in the depletion plots; however, the possibility that enzyme inhibition in vitro might have biased the extrapolation of in vivo clearance cannot be ruled out.
Prediction of Hepatic Intrinsic Clearance from Intravenous and Oral Data.
A variable degree of CLint,h prediction success from microsomal data has been reported in the literature (Obach, 1999; Ito and Houston, 2005; Riley et al., 2005). The current study found a low degree of underprediction for the CYP3A4 substrates investigated. The prediction success of CLint,h from oral data was considerably improved in the current study when drug-specific FG values were incorporated; this was particularly evident for atorvastatin, buspirone, cyclosporine, felodipine, and nisoldipine. The CLint,h estimate from oral data represented 98% (79, 117, 95% CI) of the CLint,h estimate from intravenous data (when indinavir data were excluded) and the regression between both datasets was very strong (R2 = 0.96), ranging over 4 orders of magnitude. The incorporation of FG led to CLint,h overpredictions for rifabutin, simvastatin, and lovastatin, which might question the accuracy of the in vivo FG estimates for these drugs. Likewise, for indinavir poor prediction success for oral clearance was observed. This drug is administered at a high dose, resulting in a corresponding hepatic inlet concentration of 4 μM after oral administration; therefore, saturation/inhibition of systemic indinavir metabolism might occur, given its low Km (Chiba et al., 1997; Koudriakova et al., 1998) and Ki values <1 μM (Eagling et al., 1997).
Permeability experiments were performed in MDCK-MDR1 and Caco-2 cells at a low substrate concentration (0.1 μM) and in the presence of a P-gp inhibitor for the drugs with apparent drug efflux mediated by P-gp (ER ≥2). Permeability data were of considerable importance for the FG predictions of drugs with Papp (A-B) <100 nm/s; in contrast, if drug permeability exceeded this value, FG predictions were mainly driven by in vitro clearance because the QGut model was reduced to a perfusion rate-limited model. Because single concentrations below the anticipated luminal concentration were used in the permeability assessment, consequently an overestimation of the contribution of P-gp might have occurred. A more comprehensive in vitro assessment of P-gp-mediated transport (e.g., full kinetic profiles and differential pH to account for variability in vivo) for the P-gp substrates in the current study would be beneficial to fully understand the contribution of P-gp to intestinal first-pass metabolism.
Predictions of Intestinal First-Pass Metabolism.
The QGut model accounts for the fact that a drug with low permeability will have a longer exposure to the metabolizing enzymes in the enterocytes (Rostami-Hodjegan and Tucker, 2004; Yang et al., 2007). For drugs with in vitro permeability exceeding 100 nm/s, the hybrid function QGut (as defined in eq. 9) is reduced to Qent, resulting in simple perfusion rate-limited processes. Considering this and the large availability of in vivo intestinal first-pass metabolism data, midazolam, a highly permeable CYP3A substrate, was explored as a QGut calibrator. However, because no in vivo measure of QGut or CLint,g is available, this approch is based on the assumption that predicted CLint,g represents an adequate measure for midazolam in vivo intestinal clearance (as seen for hepatic data). The variability associated with CLint,g and FG values propagates into the estimation of QGut estimates, resulting in a considerable uncertainty associated with this parameter.
Application of the QGut model resulted in high FG prediction success for drugs with low intestinal first-pass metabolism (FG >0.5) with indinavir representing the only significant outlier (Fig. 4B). In contrast, the prediction success was considerably reduced for the subset of drugs with FG <0.5 (Fig. 4C). This trend was also observed for the prediction success of FH (data not shown) with comparable bias and imprecision (gmfe 2.9 versus 2.5 for FG and FH, respectively). In general, direct input of Papp (A-B) data from Caco-2 or MDCK-MDR1 cells resulted in the highest FG prediction success and is therefore recommended. Both cell models were considered in the present analysis because they represent common tools in the pharmaceutical industry to determine permeability of new chemical entities. In contrast, the use of a regression equation based on physicochemical properties and Peff should be avoided because it resulted in significant underprediction of FG for the current dataset. This may partly be explained by a number of drugs with very high polar surface area (>100 Å2) (e.g., indinavir, saquinavir, and tacrolimus), as the validity of the existing regression equation (Winiwarter et al., 1998) was not established for drugs with those properties. Because in vivo Peff data were available for cyclosporine and verapamil (Lennernäs, 2007), these data were used in the current FG predictions.
The FG overestimations observed for buspirone and atorvastatin are consistent with the underprediction of hepatic clearance (<8% of observed). Furthermore, for both drugs CLh approaches Qh, which impedes accurate estimation of in vivo FG, bearing in mind that this parameter is indirectly assessed from intravenous/oral data. Indeed, whereas intravenous/oral data for atorvastatin suggested an FG value of 0.24, grapefruit juice (GFJ) interaction data suggested a less extensive intestinal contribution to atorvastatin first-pass metabolism: FG, GFJ = 0.56 (Gertz et al., 2008a). In the case of tacrolimus, FG predictions were highly variable among the different microsomal pools used within this study, showing a general overprediction trend. In addition to CYP3A, tacrolimus undergoes UGT-mediated metabolism, and the underestimation of intestinal clearance might also be attributed to conjugative metabolism (Strassburg et al., 2001). However, because no absolute UGT abundance data to allow incorporation of UGT metabolism into FG predictions currently exist, the impact of this contributing pathway could not be assessed.
Underprediction of FG was observed for a number of drugs, including terfenadine, saquinavir, and indinavir. Terfenadine displayed considerable nonspecific binding even at a low protein concentration, and erroneous fuinc determination might have subsequently affected the in vitro estimate of its clearance. To minimize any issues associated with the nonspecific binding, low microsomal protein concentrations were used, and fuinc values were experimentally determined for all the drugs using microdialysis (Gertz et al., 2008b), with the exception of cyclosporine for which this value was predicted (Hallifax and Houston, 2006). In addition, the use of substrate concentrations 10-fold below Km may overestimate intestinal clearance, given the high anticipated drug concentration in the enterocytes during the absorption phase. Potential saturation of CYP3A and P-glycoprotein in vivo (e.g., saquinavir) and the region of the intestine in which drug is absorbed also need to be considered when observed FG underprediction is interpreted. Some of the substrates for which FG is underpredicted represent either time-dependent or reversible inhibitors of CYP3A (i.e., indinavir and saquinavir), which may affect their in vivo estimates of FG. Finally, one must not forget that the in vivo estimates of FG represent indirect assessments of the intestinal first-pass metabolism liable to several assumptions. In particular, for drugs with low and variable bioavailability where CLh/Qh approaches 1 (buspirone, felodipine, indinavir, lovastatin, and saquinavir), delineation of the intestinal and hepatic contribution to first-pass metabolism is virtually impossible using the intravenous/oral approach.
In conclusion, this study has comprehensively investigated the suitability of the QGut model to predict FG for drugs with differential clearance and permeability characteristics. Although drugs with low intestinal extraction were generally well predicted, the prediction success for drugs with high intestinal extraction (FG <0.5) was considerably less accurate and requires further refinement.
Acknowledgments.
We acknowledge the assistance of Dr. David Hallifax and Sue Murby with the LC-MS/MS analysis, Dr. In-Sun Nam Knutsson for guidance with the meta-analyses (University of Manchester), and Dr. Katherine Fenner, Sarah Kempshall, Rebecca Greenstreet, and Charles Malloy (Pfizer, Pharmacokinetics, Dynamics and Metabolism Department, Sandwich, UK).
Footnotes
M.G. was supported by a Ph.D. studentship from Pfizer Global Research and Development, Sandwich, Kent, UK.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.032649.
-
ABBREVIATIONS:
- P450
- cytochrome P450
- FG
- intestinal availability
- QGut
- hybrid parameter of blood flow and drug permeability
- Papp
- apparent permeability
- A
- apical
- B
- basolateral
- MDCK-MDR1
- Madin-Darby canine kidney cells transfected with multidrug resistance 1
- Peff
- effective permeability
- CLint
- intrinsic clearance
- CLint,h
- hepatic intrinsic clearance
- CLint,g
- intestinal intrinsic clearance
- CLuint
- unbound intrinsic clearance
- Qent
- enterocytic blood flow
- fu
- fraction unbound
- HLM
- human liver microsomes
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- HIM
- human intestinal microsome(s)
- Rb
- blood/plasma distribution ratio
- AUC
- area under the curve
- Qh
- hepatic blood flow
- CLh
- hepatic blood clearance
- P-gp
- P-glycoprotein
- CP-100356
- N-(3,4-dimethoxyphenethyl)-4-(6,7-dimethoxy-3,4-dihydroisoquinolin-2[1H]-yl)-6,7-dimethoxyquinazolin-2-amine
- MEM
- minimal essential medium
- HBSS
- Hanks' balanced salt solution
- ER
- efflux ratio
- gmfe
- geometric fold error
- rmse
- root mean square error
- UGT
- UDP glucuronosyltransferase.
- Received February 8, 2010.
- Accepted April 5, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}