


Abstract
The potential of substrates and modifiers of CYP3A4 to show differential effects, attributed to the existence of multiple binding sites, confounds the straightforward prediction of in vivo drug-drug interactions from in vitro data. A set of in vitro interaction studies was performed in human lymphoblast-expressed CYP3A4 involving representatives of two CYP3A4 subclasses, midazolam (MDZ) and testosterone (TST); a distinct subgroup, nifedipine (NIF); and its structural analog, felodipine (FEL). Mechanistic insight into the interaction of each pair of substrates was provided by employing a range of multisite kinetic models; most were subtypes of a generic two-site model, but a three-site model was required for TST interactions. The complexity of the inhibition profiles and the selection of the kinetic model with appropriate interaction factors were dependent upon the kinetics of substrates involved (hyperbolic, substrate inhibition, or sigmoidal for MDZ/FEL, NIF, and TST, respectively). In no case was a simple reciprocity seen between pairs of substrates. The interaction profiles observed between TST, MDZ, NIF, and FEL involved several atypical inhibition features (partial, cooperative, concentration-dependent loss of characteristic homotropic behavior) and pathway-differential effects reflecting an 80-fold difference in Ki values and a δ factor (defining the alteration in the binding affinity in the presence of a modifier) ranging from 0.04 to 2.3. The conclusions from the multisite kinetic analysis performed support the hypothesis of distinct binding domains for each substrate subgroup. Furthermore, the analysis of intersubstrate interactions strongly indicates the existence of a mutual binding domain common to each of the three CYP3A4 substrate subclasses.
Substrate-dependent effects (Kenworthy et al., 1999; Stresser et al., 2000; Wang et al., 2000; Lu et al., 2001), pathway-differential effects (Shou et al., 2001a; Galetin et al., 2002), and heteroactivation (Ludwig et al., 1999; Tang et al., 1999; Ngui et al., 2000; Kenworthy et al., 2001) are all phenomena observed in in vitro drug-drug interaction studies that are not commonly incorporated into in vitro-in vivo scaling and prediction procedures. The above, and other non-Michaelis-Menten kinetic properties, are frequently linked with CYP3A4 (Kenworthy et al., 2001; Shou et al., 2001b; Tang and Stearns, 2001; Galetin et al., 2002), but recent studies indicate atypical kinetics for some other human enzymes, namely CYP2C9 (Hutzler et al., 2001b), UDP-glucuronosyltransferase 1A1 (Williams et al., 2002), and 2B7 (Stone et al., 2003). The complexity of effects seen in the homotropic situation when more than one molecule of the same substrate is present at the active site (Shou et al., 1999; Lin et al., 2001) is increased for heterotropic interactions involving two different substrates (Ueng et al., 1997; Korzekwa et al., 1998). Heterotropic effects (either activation or inhibition) require a more elaborate approach involving either the simultaneous binding of two different substrates to the active site (Shou et al., 1994, 2001b) or possibly an effector site (Ueng et al., 1997; Domanski et al., 2001; Kenworthy et al., 2001; Galetin et al., 2002), and hence there is an increase in the number of enzyme complexes formed.
The significance of nonstandard Michaelis-Menten data in vitro, and their correlation with the in vivo situation, remains ambiguous (Atkins et al., 2002); to date, there are few confirmatory studies in vivo (Tang et al., 1999; Egnell et al., 2003), and in certain cases, their relevance could be questioned (Hutzler et al., 2001a; Ngui et al., 2001). The introduction of maximum clearance as an alternative for intrinsic clearance (Houston and Kenworthy, 2000) represents one attempt to introduce autoactivation into the in vitro-in vivo scaling strategy. The dependence of clearance on substrate concentrations below the Ks is associated with positive cooperativity and indicates the possibility of clearance underestimation in rapid in vitro screening procedures based on only one substrate concentration (Houston and Kenworthy, 2000). In terms of predicting drug-drug interactions in vivo, the use of multiple substrates in vitro at various substrate concentrations is recommended to explore the range of possible consequences of a heterotropic interaction (Kenworthy et al., 1999).
The selection of CYP3A4 substrates employed for in vitro testing is mainly based on three distinct CYP3A4 subgroups, first identified via a number of statistical tests, including cluster analysis, by Kenworthy et al. (1999) and substantiated by others (Stresser et al., 2000; Lu et al., 2001). A recent collation (Galetin et al., 2002) of CYP3A4 interactions from our laboratory (Houston and Kenworthy, 2000; Kenworthy et al., 2001; Galetin et al., 2002) and elsewhere (Wang et al., 2000; Ngui et al., 2001, Shou et al., 2001a) has highlighted the value of multisite interaction factors to rationalize the range of atypical Michaelis-Menten kinetic effects observed. Here we expand this approach by performing a multisite kinetic analysis of mutual interactions of the three most commonly used CYP3A4 substrates (Yuan et al., 2002): midazolam (MDZ1), testosterone (TST), and nifedipine (NIF). In addition to their role as representative prototypes of the CYP3A4 substrate subgroups, MDZ, TST, and NIF show distinctive kinetic properties namely, hyperbolic, sigmoidal, and substrate inhibition, respectively. Felodipine (FEL), a structural analog to NIF, was included in these studies for further evaluation and possible inclusion in the NIF distinct CYP3A4 subgroup.
Several distinctive types of CYP3A4 interactions are reported here for MDZ, TST, NIF, and FEL (e.g., cooperative and partial inhibition, pathway-differential effects, concentration-dependent positive and negative homotropy), and their association with specific multisite interaction factors is defined. Furthermore, additional kinetic evidence for the existence of mutual and distinct substrate-binding domains for particular substrate subgroups within the CYP3A4 active site is presented.
Materials and Methods
Chemicals. TST, 6β-hydroxytestosterone (6β-HTS), NIF, MDZ, NADP, and isocitric dehydrogenase were purchased from Sigma-Aldrich (Poole, Dorset, UK). Oxidized NIF (OX NIF) and MDZ metabolites were obtained from Ultrafine Chemicals (Manchester, UK). FEL and pyridine metabolite (FEL pyridine) were gifts from Astra (Hässle, Mölndal, Sweden). UK-58,790 was obtained from GlaxoSmithKline (The Frythe, Welwyn, Hertfordshire, UK). All other reagents and solvents were of high analytical grade. Microsomes from human lymphoblastoid-expressed CYP3A4 with coexpressed NADPH-cytochrome P450 reductase (CYP3A4/OR) were obtained from BD Gentest (Woburn, MA).
Incubation Conditions. Interaction studies were performed at incubation times and protein concentrations within the linear range for each individual substrate. Microsomes from human B-lymphoblastoid cells containing recombinant human CYP3A4/OR were suspended in phosphate buffer (0.1 M, pH 7.4). The final incubation volume was 0.2 ml, containing 47 to 111 pmol of P450/ml. Samples were preincubated for 5 min in a shaking water bath at 37°C, and each reaction was initiated with an NADPH-regenerating system (1 mM NADP+, 7.5 mM isocitric acid, 15 mM magnesium chloride, and 0.2 unit of isocitric dehydrogenase). The substrates (concentration ranged from at least ½Km to 2Km) were added to each incubation in either methanol or phosphate buffer depending on the solubility. Neither of the substrates showed significant microsomal binding (<10%). The final concentration of methanol in incubation media was ≤0.5% (v/v). The range of modifier concentrations applied was from 0.5 to 100 μM in most studies. The reaction was terminated by 0.1 ml of ice-cold methanol. Samples were then centrifuged at 13,400g for 5 min and analyzed by high-pressure liquid chromatography-UV or liquid chromatography-tandem mass spectrometry as described earlier (Galetin et al., 2002).
Data Analysis. The kinetic parameters for each substrate alone and in the presence of an inhibitor were obtained from untransformed data by nonlinear least-squares regression using GraFit 4 (Erithacus Software Ltd., Horley, Surrey, UK). In the case of FEL and MDZ, the Michaelis-Menten equation with the weighting factor of 1/y was used for preliminary kinetic analysis. Preliminary analysis of NIF kinetic data was carried out assuming single-site Michaelis-Menten kinetics with substrate inhibition (Houston and Kenworthy, 2000). Kinetic parameters Vmax, S50, and Hill coefficient (n) were calculated from untransformed data using the Hill equation for initial analysis of TST kinetics. In addition to the Hill equation, a two-site model (eq. 3; Kenworthy et al., 2001) was also used for the preliminary analysis of the TST data in the presence of increasing concentrations of the modifiers. The changes in kinetic parameters observed in the presence of various modifiers were significance tested using analysis of variance.
Further data analysis to provide a more detailed model of the molecular events was based on the application of various steady-state and rapid equilibrium multisite kinetic approaches. Two- and three-site models and the corresponding equations derived assume the existence of particular substrate-binding domains within the active site. Various interaction factors are defined in order to characterize the effect of a certain modifier. There are more factors involved in a heterotropic than a homotropic interaction due to the increased number of enzyme complexes and binding sites involved and a possible overlap between the sites for substrate and modifier.
The kinetic models applied assume rapid equilibrium, i.e., the rate at which ES/SE complex dissociates is much faster than the rate of product formation (Segel, 1975). In all the cases (apart from pathway-differential effects and substrate inhibition kinetics), two substrate-binding sites were assumed to be identical, with no distinguishable difference between ES and SE conformations. Each complete data set (n = 20–30) in the presence and absence of the modifier was fitted to the rate equations for various multisite kinetic models using GraFit. The least number of parameters, lowest standard errors of the parameter estimates, and consistency with kinetic properties of both the substrate and modifier represent the principal criteria for the selection of a certain model. Goodness of fit was determined by comparison of statistical parameters (χ2 and Akaike information criterion values) between the models and a reduction in the standard errors of the parameter estimates. Kinetic parameter estimates generated from different multisite kinetic models were used to simulate velocity curves for metabolite formation. A major advantage of this type of kinetic analysis is the ability to simultaneously fit all the data covering the range of modifier concentrations, in contrast to the preliminary analysis (e.g., Hill plots) in which individual fits are obtained for each specific concentration of modifier. The enzyme complexes that are involved in metabolite formation are in the numerator of all the equations, whereas the denominator contains all the enzyme complexes present (Segel, 1975).
Use of the Multisite Kinetic Approach. Many authors seem uncertain about how to deal with cooperativity/unusual kinetics, and the literature contains numerous examples of standard Michaelis-Menten hyperbolic curves forced through data that clearly show atypical kinetic features. In some of these cases, the insufficient number of data points rules out any meaningful selection of an alternative model. We have found that multisite models provide a valuable insight into complex CYP3A4 interactions (Kenworthy et al., 2001; Shou et al., 2001b; Galetin et al., 2002), once certain practical issues of dealing with the data that cannot be described by the Michaelis-Menten model are addressed.
For the purposes of the mechanistic studies, recombinant systems have proved to be a better defined and more controlled system in comparison to human liver microsomes. However, concentrations of accessory proteins (e.g., OR and cytochrome b5) can differ considerably between these two systems (Venkatakrishnan et al., 2000), and their lack/addition, as well as membrane lipid composition and ionic strength of the in vitro matrix, may also affect CYP3A4 catalytic activity.
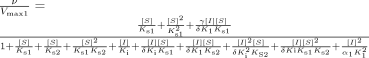
According to the kinetic properties of the substrate, variations of the generic two-site model (Galetin et al., 2002) were applied to rationalize the inhibition profiles obtained for NIF, FEL, and MDZ. These two-site kinetic models accommodated a range of effects and varied in the number and type of corresponding interaction factors, associated with either binding affinity (α, δ) or rate of product formation (β, γ) (Fig. 1A). The assumption of a fast release of products (Segel, 1975) was the rationale for considering the metabolism of each substrate molecule independently. However, the kinetic properties of the effector, alterations in substrate and/or modifier binding affinity (α, δ), and catalytic efficiency upon effector binding (γ) were also considered.

Multisite kinetic equilibria models.
A, a generic two-site model for CYP3A4 interactions and the corresponding equation. Interaction factors associated with the changes in binding affinity (Ks/Ki) are as follows: α-homotropic substrate cooperativity, δ-heterotropic cooperativity, and αI-inhibitor cooperative binding. Interaction factors associated with the changes in catalytic rate constant (Kp) are as follows: β (SES) and γ (MES). B, a kinetic model for an enzyme that binds substrate cooperatively and where the inhibitor eliminates the sigmoidicity. C, three-site kinetic model with two distinct substrate-binding sites (S1 and S2) and an effector site (pathway-differential effect of a modifier).
The initial step in the selection of a model and the relevant interaction factors (α, β, γ, or δ) is highly dependent on the kinetics of the substrate (hyperbolic, substrate inhibition, or sigmoidal), as illustrated in Scheme 1 by the values of α and β. The net effect of the increased binding affinity in the presence of another substrate (δ < 1) can range from heteroactivation to inhibition depending on the corresponding γ value (changes in the rate of metabolite formation). In contrast, partial inhibition is typically characterized by the decreased affinity of a second inhibitor molecule for a binding site in the presence of another substrate.

Summary of interaction factors from multisite kinetic models.
The possibility of enzyme-product complex formation, which would lead to reduced enzyme availability for the substrate interaction and decreased rate of the reaction (Narasimhulu et al., 1998), could be an issue of concern. However, these complexes were not included in the total sum of the metabolically productive complexes in the model derivation, in order to keep the modeling procedure relatively simple.
Positive Cooperative Inhibition. Cooperative inhibition profiles result from binding of a second inhibitor molecule in a cooperative manner to the enzyme active site, indicated by the steeper slope of IC50 plots (>1) compared with the standard one-site type of inhibition. This phenomenon is analogous to the positive cooperativity observed for some CYP3A4 substrates (e.g., TST), as the binding affinity of the second inhibitor molecule increases in the presence of the first. The enhanced extent of inhibition with increasing inhibitor concentrations is characterized by the changes in Ki value by the factor αi, where αi < 1.
In addition to alterations in the binding affinity, cooperative inhibition
profiles can be attributed to changes in the product formation
(Kp) by the factor γ, with a decrease in the overall
rate of the reaction when γ < 1 (eq. 1). In cases where no changes to
the effective catalytic rate constant are observed (γ = 1), the
corresponding equation can be simplified by eliminating γ.

The opposite effect, decreased binding affinity of the second inhibitor molecule in the presence of the first (negative cooperativity, αI > 1), is associated with partial inhibition, where full inhibition is not achieved at high inhibitor concentrations.
Negative Cooperativity and Partial Inhibition. Partial inhibition is
characterized by incomplete inhibition, even at saturating concentrations.
Competitive and noncompetitive types could be distinguished, depending on
whether changes in either binding affinity (Ks) or product
formation (Kp) are observed in the presence of a modifier.
Partial competitive inhibition (regardless of the substrate kinetic
properties) is characterized by decreased binding affinity of a second
I molecule for the binding site in comparison with the first
(δKi > Ki), analogous to the
negative cooperativity. Simultaneous presence of both S and
I at the active site and access to active oxygen enables the
complexes with I to be productive, leading to unchanged
Kp and Vmax for the reaction (γ
= 1, and it can be eliminated from the equation). In cases when partial
inhibition occurs via competition at only one binding site, the application of
a simpler model is possible, as in the case of NIF-FEL interaction (eq. 2).

Inhibition of a Substrate Showing Substrate Inhibition Kinetic Properties (Loss of Negative Homotropy). A two-site model, with only one catalytically active site, has been applied for all NIF interactions, as described previously (Galetin et al., 2002). The “substrate inhibition” site cannot be occupied until the active site is filled (sequential binding of substrate molecules). The presence of a substrate in the second binding site causes a decrease in product formation from SES, defined by the factor β (<1). Similar to all cases described by the generic two-site model, the interaction factor γ is associated with the alterations in the product formation due to the presence of an inhibitor molecule at the active site.
When γ is comparable to β, the effect of a modifier is analogous
to the binding of a second substrate molecule, and the substrate inhibition
phenomenon remains. However, at high concentrations of substrate and
inhibitor, the profile changes to a hyperbolic curve due to dominance of the
nonproductive S(EI) complex.

Heterotropic Inhibition of a Substrate Showing Sigmoidal Kinetics (Loss
of Positive Homotropy). Derived from a three-site model described
previously for the interactions of quinidine (QUI) and TST
(Galetin et al., 2002), the
model presented in Fig. 1B
describes the inhibition of substrates showing sigmoidal kinetics, in which
the inhibitor eliminates substrate cooperativity. In the absence of the
inhibitor, the substrate binds cooperatively with an interaction factor
α (<1). However, the interaction between two substrate-binding sites
resulting in an increase in the affinity of the vacant substrate sites is
prevented in the presence of the inhibitor. The increased affinity of SE/ES
and SES complexes for the inhibitor molecule is defined by an alteration in
the Ki value by the factor δ (<1) (eq. 4). At the
same time, the enzyme complexes containing both substrate and inhibitor
molecules are not productive; hence, the Vmax could be
driven to zero values at very high inhibitor concentrations.

Pathway-Differential Effects. In addition to describing interactions
for substrates with sigmoidal kinetic properties, the three-site kinetic model
approach is more appropriate for elucidating the phenomenon of
pathway-differential effects. Equation 5 is derived for 1′-OH MDZ
formation using a three-site model with two distinct substrate-binding sites;
ES1 is preferable for 1′-OH MDZ (defined by
Ks1 and Kp1) and S2E for
4-OH MDZ formation (Ks2 and Kp2)
(Fig. 1C). In contrast to a
similar model applied for the effect of QUI on MDZ
(Galetin et al., 2002), no
interaction between the two occupied MDZ binding sites is assumed. Competition
between TST and MDZ for the mutual site is characterized by the inhibition of
1′-OH MDZ formation. At the same time, binding of TST molecules to a
distinct effector site alters the kinetic properties of the substrate site
preferential for the 4-OH formation, stimulating the metabolite formation
(λKp2 > Kp2).
Results
The kinetic properties of the four CYP3A4 substrates selected showed hyperbolic (MDZ, FEL), substrate inhibition (NIF), and sigmoidal (TST) characteristics. Secondary metabolism was minimal throughout the course of the study, and less than 10% substrate depletion was observed. The short incubation times employed for MDZ (2.5 min) minimized any enzyme inactivation in regard to its mechanism-based inhibition behavior recently reported by Khan et al. (2002). A range of substrate-dependent differences is observed in the inhibitory potency that prevents any consistent rank ordering. The lack of relationship between the Ks for the particular probes and their respective Ki values is shown in Fig. 2.

Binding affinity constants for MDZ, TST, NIF, and FEL in lymphoblast-expressed CYP3A4 when incubated alone and in various combinations.
The Ks values represent the mean values of the Ks derived for a particular substrate for all the interactions studied. Multisite kinetic models applied for their derivation are stated in the data analysis for each interaction.
TST activated NIF oxidation in lymphoblast-expressed CYP3A4 (36% of control value 2.59 pmol/min/pmol of P450 at a 10 μM substrate concentration). Similarly, in the TST-FEL interaction, slight activation of FEL metabolism occurred at low substrate concentrations, changing to inhibition at higher concentrations of both substrate and modifier; the effects did not exceed 20% of the control value. No further modeling of the TST effect on NIF/FEL metabolism was performed.
Inhibition was observed for the 10 other interactions and was characterized by changes either in the binding affinity or in the product formation or a combined effect, defined by following multisite interaction factors (see Tables 1, 2, 3):
-
Alterations in the binding affinity and effect on Ks and Ki: a) δ < 1, increased binding affinity for the formation of ISE/ESI/ISES complexes (as seen for the NIF/FEL/MDZ effect on TST, the FEL effect on NIF, and the MDZ effect on FEL); or b) δ > 1, nonfavorable formation of ISE/SEI/ISES resulting in partial inhibition (TST effect on MDZ and NIF effect on FEL).
-
Alterations in the rate of metabolite formation and effect on Kp: a) γ < 1, less productive complexes with I (NIF/FEL effect on MDZ resulting in cooperative inhibition and MDZ/FEL effect on NIF), or b) γ = 0, nonproductive complexes (interactions with TST as a substrate).
Kinetic parameters for the in vitro effect of various CYP3A4 substrates on midazolam in human lymphoblast-expressed CYP3A4, generated by multisite kinetic model approach (mean ± S.E.) The kinetic parameters were determined using multisite kinetic models (n = 20-30) defined in footnotes a and b.
Kinetic parameters for the in vitro effect of various CYP3A4 substrates on NIF/FEL in human lymphoblast-expressed CYP3A4, generated by multisite kinetic model approach (mean ± S.E.) The kinetic parameters were determined using generic two-site model (n = 20-30) defined in footnotes a, b, and c.
Kinetic parameters for the in vitro effect of various CYP3A4 substrates on testosterone in human lymphoblast-expressed CYP3A4, generated by multisite kinetic model approach (mean ± S.E.) The kinetic parameters were determined using kinetic model (n = 25-30) defined by equation 4, β (Kp from SES) = 2. Vmax is equivalent to 2Kp[E]t, in which [E]t is the total enzyme concentration (Segel, 1975).
To systemically approach the various kinetic phenomena observed and link the observations with particular interaction factors, four distinct types of inhibition (A-D) and a pathway-differential effect (E) have been identified.
A. Cooperative Competitive Inhibition—Exemplified by the NIF-MDZ and FEL-MDZ Interactions. The enhanced inhibition of MDZ 1′-hydroxylation observed with increasing inhibitor (FEL, NIF) concentrations and the steeper slopes of the IC50 plots (>1) at high S and I concentrations indicate the cooperative binding of the inhibitor (Fig. 3, A and B, respectively). Preliminary kinetic analysis, applying a one-site model, suggested a competitive nature of the inhibition, demonstrated by a 4- and 17-fold (p < 0.05) increase in the Km value for 1′-OH MDZ metabolic pathway in the presence of NIF and FEL, respectively. Although the simple one-site competitive inhibition model generated a satisfactory fit for the range of low I concentrations, it did not predict the enhancement of the inhibition observed with increasing concentrations of I.

Examples of cooperative competitive inhibition.
A and B, 1′-OH MDZ formation in the presence of increasing concentrations of FEL and NIF, respectively. Slopes increasing from 1.3 to 5.2 (FEL) and 0.6 to 1.4 (NIF) over the 5 to 50 μM substrate concentration range, consistent with cooperative binding of a modifier. C, correlation of FEL and NIF inhibitory effects on MDZ 1′-hydroxylation at various substrate concentrations. The dashed line represents the line of unity. Data points represent the mean of duplicate determinations.
Ki values obtained from the generic two-site model are similar for FEL and NIF (Table 1). A correlation plot of the inhibitory effects of NIF and FEL at various MDZ concentrations (Fig. 3C) is consistent with these findings. The binding affinity of the second FEL molecule is higher in the presence of the first (α = 0.24). However, this cooperativity in binding of FEL molecules contrasts with the Michaelis-Menten kinetics displayed by this substrate (Eriksson et al., 1991; Galetin et al., 2002). In addition, the enhanced inhibition can be attributed to a significant decrease in product formation in the presence of a modifier, defined by a low γ value (0.4). The formation of the IES complex is less favorable, described by altered values for the MDZ binding constant (δKs > Ks).
NIF substrate inhibition kinetic properties and sequential binding to the active site are incorporated in the generic two-site model. The enhancement of 1′-OH MDZ inhibition at higher NIF concentrations, also manifested by changes to Vmax, is analogous to the binding of the second NIF molecule to the active site causing substrate inhibition. Minimal interaction between NIF molecules is observed (α = 0.9), but the substantial effect on product formation (γ = 0.2) correlates well with the observed cooperativity in IC50 plots (Fig. 3B).
B. Partial Inhibition—Exemplified by the TST-MDZ and NIF-FEL Interactions. Similar to previously reported studies with other substrates, erythromycin (Wang et al., 1997), triazolam (Schrag and Wienkers, 2001), and terfenadine (Wang et al., 2000), TST also partially inhibited MDZ 1′-OH pathway (Fig. 4A), while activating 4-hydroxylation (described below). The IC50 plots obtained over the range of substrate (MDZ) concentrations (5–20 μM) and the 5-fold increase in IC50 values observed (114–537 μM) are consistent with a competitive type of inhibition. However, a slope <1, the high plateau of uninhibited activity at high I concentrations, and a δ interaction factor of 2.2 (Table 1) are associated with partial inhibition. The competitive nature of the observed interaction is confirmed in a 3-fold increase in MDZ Km value (4.2–11.4 μM, p < 0.05) and no alterations in Vmax values, even at high TST concentrations (100 μM).

Examples of partial inhibition.
A, inhibition of MDZ 1′-hydroxylation by TST. B, inhibition of FEL oxidation by NIF, comparison of partial versus pure competitive inhibition at high S concentration (100 μM). In contrast to competitive inhibition, partial or negative cooperative inhibition shows a limiting plateau at high concentrations of an I, the level determined by the value of the interaction factor δ.
Partial inhibition may be a result of either partial interactions at both sites or via competition at only one binding site. An example of the latter type is the effect of NIF on FEL pyridine formation, over the 5 to 100 μM FEL concentration range. In this case, NIF partially shields only one of the FEL sites from the active oxygen, allowing the application of a simpler kinetic model (eq. 2) to generate a Ki value of 33 μM and a δ factor of 2.1 (Table 2). Figure 4B illustrates another way of distinguishing partial from pure competitive inhibition by plotting the rates of metabolism at fixed substrate concentrations (e.g., 100 μM FEL) in the presence of increasing inhibitor concentrations (0.5–500 μM NIF). Unlike the competitive inhibition situation (velocity of the reaction minimized at high concentrations of an I), partial or negative cooperative inhibition results in a limiting plateau (ES and ESI are still productive), the level determined by the value of the interaction factor δ.
C. Inhibition of a Substrate with Substrate Inhibition Kinetics (Loss of Negative Homotropy)—Exemplified by the Effect of MDZ/FEL on NIF. Reduced product formation at high NIF concentrations, associated with substrate inhibition, is defined by the interaction factor β < 1 (Fig. 1A). In the presence of low MDZ concentrations, this phenomenon is still observed, as the rate of NIF product formation from IES complex is analogous to that from SES, due to comparable values of interaction factors γ and β (0.44 and 0.41, respectively). However, at higher S and I concentrations, the nonproductive I complex (S(EI)) dominates, changing the shape of the profile into a hyperbolic type (Fig. 5A). An analogous phenomenon occurs with NIF in the presence of FEL (β and γ of 0.44 and 0.56, respectively). Comparable inhibitory potency of FEL and MDZ based on Ki comparison (see Table 2) was not expected from the Ks values for these substrates (Fig. 2). This discrepancy and the higher inhibitory potency of FEL are more evident when the alterations in binding affinities for FEL/MDZ in the presence of NIF are considered (lower value of δ = 0.62 for FEL in comparison to MDZ).

Eadie-Hofstee plots for OX NIF and 6β-HTS formation in the presence of FEL and NIF, respectively.
A, loss of negative homotropy due to dominance of the nonproductive S(EI) complex. B, loss of positive homotropy [e.g., effect of NIF on TST at concentrations of 1 μM (•), 5 μM (□), 10 μM (▪), and 50 μM (▵) of the modifier and control (○)]. The interaction between TST substrate-binding sites is prevented at NIF concentrations >5 μM. Data points represent the mean of duplicate determinations.
D. Inhibition of a Substrate with Sigmoidal Kinetics (Loss of Positive Homotropy)—Exemplified by the NIF/FEL/MDZ Interactions with TST. The effect of NIF/FEL on TST 6β-hydroxylation was substrate/modifier concentration-dependent with no effect at a low TST (10 μM) concentration and 1 μM FEL, achieving full inhibition in the 50 to 200 μM TST concentration range. In the case of MDZ, similar inhibitory potency was observed (IC50 = 4.4 μM) regardless of the TST concentration studied.
The initial analysis by a two-site model (eq. 3; Kenworthy et al., 2001) shows a decrease in Vmax values to 14, 21, and 28% of control values in the presence of 50 μM FEL, NIF, and MDZ, respectively. Positive cooperativity in TST binding is defined by the interaction factor α < 1 (Kenworthy et al., 2001; Galetin et al., 2002). Depending on the affinity of a modifier for the active site, the possible overlap with TST binding sites, or the binding to a separate effector site, interactions can affect TST cooperativity. The extent of the decrease in Ks values (2.5- to 4.8-fold) and the increase in αKs values (9- to 38-fold) are consistent for these three modifiers. At concentrations above 5 μM NIF, FEL, and MDZ appear to prevent the interaction between two TST substrate-binding sites and reduce the cooperativity, as seen in the linear shape of Eadie-Hofstee plots (Fig. 5B).
Table 3 shows the kinetic parameters for the effect of NIF, FEL, and MDZ on TST, generated from the multisite model by a simultaneous fit to eq. 4. The increased affinity of ES/SE/SES complexes for the I is defined by low values of δ (0.04, 0.08, and 0.14 for FEL, MDZ, and NIF, respectively). In all three studies, Vmax values were minimized at high I concentrations, as a result of the formation of the metabolically nonproductive ESI, SEI, and SESI complexes. The binding of an effector (NIF, FEL, or MDZ) at two sites is consistent with the previously described effects of QUI and haloperidol (HAL) on these compounds when investigated as CYP3A4 substrates (Galetin et al., 2002).
E. Pathway-Differential Effects for MDZ. In addition to partially inhibiting MDZ 1′-hydroxylation, TST activates the minor 4-OH pathway up to 50% at 50 μM MDZ, in a manner similar to QUI (Galetin et al., 2002). In contrast, neither the NIF nor the FEL interaction results in a pathway-differential effect. A three-site kinetic model, with two distinct substrate-binding sites (Ks1 = 5.1 ± 0.3 and Ks2 = 8.6 ± 0.3 μM; each preferable to one particular MDZ pathway), best accommodated this differential effect of TST on MDZ pathways. Competition for the mutual binding site for MDZ and TST causes the partial competitive inhibition of 1′-hydroxylation (described earlier), as a result of a decreased binding affinity of a second TST molecule (δKi - 306 μM > Ki). TST binding to the distinct effector site influences the substrate site preferential for 4-OH MDZ, stimulating this pathway by increasing the product formation (λKp2 > Kp2), without affecting the binding (Ks2).
Discussion
To advance the prediction of in vivo drug-drug interactions and to explore the relationship between the different binding domains for the CYP3A4 subgroups, we have performed a multisite analysis of the mutual interactions between four commonly used CYP3A4 substrates: MDZ, TST, NIF (subgroup prototypes), and FEL. For most of the 12 cases investigated, no mutual inhibition is observed, and the range of kinetic phenomena evident includes partial and cooperative inhibition and concentration-dependent activity profiles.
Whenever possible, the simplest kinetic inhibition model should be employed to obtain kinetic parameters, and often the inclusion of all possible enzyme complexes in the data analysis is not necessary (e.g., steric restrictions resulting in the interaction at only one site). However, adoption of one-site models for the analysis of enzymes known to exhibit atypical interactions has severe limitations. For example, a competitive inhibition model may generate a satisfactory fit for the range of low inhibitor concentrations, but the cooperativity of the inhibition at higher inhibitor concentrations cannot be predicted (e.g., the effect of FEL on 1′-OH MDZ formation). Additionally, certain data sets may show some of the features of competitive inhibition but not consistently (e.g., the effect of QUI on TST; Galetin et al., 2002). The application of simple models for interactions involving substrates with positive (testosterone, diazepam) or negative (terfenadine, nifedipine) homotropic kinetic properties is particularly problematic, and the misuse of a one-site model may lead to inaccurate estimation of kinetic parameters and failure to identify important drug-drug interactions.
Interactions at the TST Site(s). TST slightly activated the metabolism of NIF and FEL, whereas both dihydropyridines inhibited TST 6β-hydroxylation. These diametrical drug-drug interaction patterns are in agreement with other findings reported for TST (Wang et al., 2000; Kenworthy et al., 2001; Lu et al., 2001). The fact that this substrate represents the most used and recommended in vitro probe for CYP3A4 (Yuan et al., 2002) is, therefore, of concern.
Ki values for the inhibition of 6β-HTS formation by HAL, QUI (Galetin et al., 2002), NIF, FEL, and MDZ, generated by applying various multisite kinetic models, extend over a 10-fold range from 9.5 μM (NIF) to 99 μM (QUI). In most cases, the IC50 values are not in good agreement with the Ki values obtained from the multisite kinetic models, indicating the need for caution over rapid screening protocols based on IC50 plots for substrates with sigmoidal kinetic properties. In the presence of NIF, FEL, and MDZ, the binding affinity of the first TST molecule was increased, reflecting a difference between the effector site for these modifiers and one of the TST binding sites. In contrast, the interaction at the second site (defined by αKs) is of a competitive nature (increase in αKs up to 40-fold) and results in the elimination of cooperativity between the two TST binding sites. This effect suggests that the second site is a mutual binding domain for both TST and modifiers belonging to other CYP3A4 subgroups. Other modifiers like diazepam (Kenworthy et al., 2001), HAL, QUI (Galetin et al., 2002), and progesterone (A. Galetin, unpublished data) while decreasing the binding affinity for a second TST molecule do not affect the cooperativity of TST binding, indicating the occupancy of a different effector site from that of MDZ, NIF, and FEL as modifiers.
Interactions at the MDZ Site(s). The pathway-differential effect on MDZ observed with TST in the current study is not exclusive as similar effects have been reported previously for MDZ in the presence of various modifiers (Ghosal et al., 1996; Wang et al., 2000; Galetin et al., 2002). These findings and the difference in the Km values obtained for 1′- and 4-OH MDZ hydroxylation (Gorski et al., 1994; Mäenpää et al., 1998) indicate the possible existence of two distinct substrate-binding sites for MDZ.
To incorporate these differential effects for the two pathways, we have applied a three-site kinetic model with a distinct effector site; the latter is usually associated with substrates showing positive homotropic kinetic properties. In this case, two substrate-binding sites are assumed to be distinct, each generating one particular metabolite of MDZ, defined by their respective Ks and Kp values. The existence of two separate sites for MDZ was recently also indicated by site-directed mutagenesis studies (Khan et al., 2002). These authors revealed the significance of various active site residues for the regioselectivity of MDZ and indicated the partial overlap of the two MDZ binding sites.
Spectral titration analysis (Hosea et al., 2000) has suggested the existence of a higher-affinity site that overlaps with TST and MDZ and a lower-affinity binding site that overlaps with α-naphthoflavone (αNF). Distinct Km and Ki values observed for 1′- and 4-OH MDZ, and the differential effects observed in the presence of αNF, indicate the possibility of a third site, distinct from both TST and αNF. These assumptions are supported by several findings from the current multisite kinetic analysis. Interaction at the mutual site for TST and MDZ 1′-hydroxylation results in the mutual inhibition and a corresponding increase in Ks and αKs values for MDZ and TST, respectively, in the presence of each other. Partial inhibition by TST is a consequence of the higher binding affinity of MDZ than TST (αKs TST > Ks1 MDZ). The increased formation of 4-OH MDZ in the presence of higher TST concentrations and a switch from the major 1′-OH to the minor 4-OH pathway are not phenomena unique to the TST-MDZ interaction (Schrag and Wienkers, 2001; Galetin et al., 2002). No change in the MDZ binding affinity at the site preferential for 4-hydroxylation (Ks2 remains constant over the range of TST concentrations) is consistent with the hypothesis of a second distinct site.
Interactions at the NIF/FEL Site(s). The similarities in NIF and FEL metabolic pathways and the high correlation between their in vivo clearances (Soons et al., 1993) suggest that these two dihydropyridines belong to the same CYP3A4 substrate subgroup. However, a substrate-dependent effect was observed previously with HAL and QUI as modifiers (Galetin et al., 2002). NIF was more susceptible to inhibition in comparison to FEL (inhibition by both HAL and MDZ shows a difference of one order of magnitude). Differential effects of QUI (activating FEL in contrast to inhibiting NIF metabolism) indicated the possibility of different binding domains on CYP3A4 for NIF and FEL despite similarities in their chemical structures. However, these sites must be in close proximity or even overlap to a certain degree to allow simultaneous binding and access to the active oxygen on the heme. This is also indicated by their mutual inhibition and a Ki value for FEL inhibition of OX NIF formation (16.6 μM), in good agreement with the affinity of FEL for CYP3A4 (26.4 μM; Galetin et al., 2002). The presence of NIF in the active site may partially shield one of the FEL sites from the active oxygen, resulting in elimination of catalytic activity associated with that site and partial inhibition.
How Many Sites? The existence of three binding sites, one for a substrate, one for an effector, and one mutual for both, has been indicated by various approaches to the analysis of CYP3A4 atypical kinetics (Hosea et al., 2000; Kenworthy et al., 2001, He et al., 2003). The reduced cooperativity in the binding of TST molecules in the presence of NIF/FEL/MDZ, the inability of TST to inhibit the metabolism of NIF and FEL, and the ability of TST to only partially inhibit metabolism of MDZ and terfenadine (Wang et al., 2000) support the hypothesis of distinct and preferential binding domains for each substrate subgroup. However, interaction profiles observed between TST, MDZ, NIF, and FEL also indicate the existence of a mutual site for all subclasses of CYP3A4 substrates.
Site-directed mutagenesis studies have indicated that CYP3A4 substrate- and effector-binding sites are separate, but closely linked, and the residues involved in the binding of either substrate and/or effector depend on the molecules present (Domanski et al., 2001; He et al., 2003). This partial overlap of binding sites may explain certain differences in the effects of substrates apparently belonging to the same CYP3A4 subgroup [e.g., NIF and FEL interactions; the differential effect of diazepam (Kenworthy et al., 2001) and MDZ on the TST positive homotropy]. Sites for “metabolism” and “regulation” may differ for the same compound as proposed earlier for the CYP3A4 modifiers αNF (Shou et al., 2001a) and QUI (Galetin et al., 2002). The net effect depends on the particular substrate present at the active site, the possible overlap of binding domains for the substrates involved in the interaction, and the relative concentrations of both.
In conclusion, the multisite analysis presented here strongly supports the existence of one preferential binding domain for each of the three CYP3A4 substrate subgroups, indicating that extrapolation from one CYP3A4 substrate to another is only realistic within the same prototypical subgroup. Certain competitive features are apparent in the atypical interaction data set (but not consistently), and these can be attributed to the interactions at a mutual site for all CYP3A4 substrate subclasses.
Footnotes
-
↵1 Abbreviations used are: MDZ, midazolam; TST, testosterone; NIF, nifedipine; FEL, felodipine; 6β-HTS, 6β-hydroxytestosterone; OX NIF, oxidized nifedipine; QUI, quinidine; α, interaction factor for the change in binding affinity (homotropic cooperativity); β, γ interaction factors for the change in catalytic rate constant; δ, interaction factor for the change in binding affinity (heterotropic cooperativity); HAL, haloperidol; αNF, α-naphthoflavone; OR, NADPH-cytochrome P450 reductase; P450, cytochrome P450.
-
Financial support for this project was provided by GlaxoSmithKline, UK. Part of this study was presented at the European Federation for Pharmaceutical Sciences, Oct 20–23, 2002, Stockholm, Sweden.
- Received March 28, 2003.
- Accepted May 28, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}