


Abstract
Ramelteon is a melatonin receptor agonist used as a treatment for insomnia. It is subject to a remarkably large drug-drug interaction (DDI) caused by fluvoxamine coadministration, resulting in a more than 100-fold increase in exposure. The objective of this study was to determine whether the DDI could be estimated using in vitro metabolism data. Ramelteon was shown to undergo hydroxylation in human liver microsomes to eight metabolites via six pathways. The main routes of metabolism included hydroxylation on the ethyl side chain and the benzylic position of the cyclopentyl ring, as assessed through enzyme kinetic measurements. Hydroxylation at the other benzylic position was observed in human intestinal microsomes. Ramelteon metabolism was catalyzed by CYP1A2, CYP2C19, and CYP3A4 as shown through the use of recombinant human cytochrome P450 enzymes and specific inhibitors. In liver, CYP1A2, CYP2C19, and CYP3A4 were estimated to contribute 49, 42, and 8.6%, respectively, whereas in intestine only CYP3A4 contributes. The in vitro data were used to estimate the magnitudes of DDI caused by ketoconazole, fluconazole, and fluvoxamine. The DDIs caused by the former were reliably estimated (1.82-fold estimated versus 1.82-fold actual for ketoconazole; 2.99-fold estimated versus 2.36-fold actual for fluconazole), whereas for fluvoxamine it was underestimated (11.4-fold estimated versus 128-fold actual). This suggests that there may be a limit on the magnitude of DDI that can be estimated from in vitro data. Nevertheless, the example of the fluvoxamine-ramelteon DDI offers a unique example wherein one drug can simultaneously inhibit multiple enzymatic pathways of a second drug.
The need to use more than one drug simultaneously in one patient (polypharmacy) can arise for two main reasons: 1) some diseases call for the use of more than one drug for effective treatment (e.g., cancer chemotherapy); and 2) some patients, especially the elderly, have more than one condition, with each requiring its own drug treatment. However, the simultaneous use of more than one drug can also lead to drug-drug interactions (DDIs), in which the exposure to one agent can be altered by the other. This can potentially lead to undesired side effects, more severe safety concerns, and even decreased efficacy in some cases. The understanding that has been achieved regarding the role of the cytochrome P450 (P450) enzyme superfamily and the roles of specific P450 enzymes in the clearance of drugs have led to an ability to leverage in vitro drug metabolism data in the prediction of DDIs (Houston and Galetin, 2008; Huang et al., 2008; Obach, 2009). Considerable effort is expended in the search for new pharmacotherapies to design molecules that will be less likely to cause or be subject to DDI.
Among the large number of drugs available, there is a small subset well established as perpetrators of DDIs known to cause large (i.e., >5-fold) increases in exposures to other drugs. Examples include ketoconazole, itraconazole, ritonavir, clarithromycin, and paroxetine, among others. These perpetrators of DDI are either potent inhibitors or mechanism-based inactivators of specific P450 enzymes, and their dosing regimens used in therapy yield concentrations in vivo that are high enough to have a profound effect on their target P450 enzyme. Whereas dose and potency drive the potential magnitude of DDI caused by a perpetrator, the magnitude of the interaction for a victim drug is driven mostly by the fraction of the clearance of the drug that is mediated by the affected enzyme. For example, if 80% of the clearance of a victim drug is mediated by a single enzyme and if that enzyme were completely inhibited by administration of a perpetrator drug, then the largest increase in exposure would be 5-fold. This is explained by the Rowland-Matin equation, which relates the magnitude of DDI to inhibitory potency (Ki), concentration of the perpetrator ([I]), and the fraction of the victim drug that is cleared by the affected enzyme (fCL) (eq. 1) (Rowland and Matin, 1973):

In most cases, the victim drugs have multiple clearance pathways, and even if one pathway were to be completely inhibited, there would be remaining pathways of clearance operating that would dampen the overall effect. Thus, observations of DDI of greater than 10-fold are infrequent.
Ramelteon (Fig. 1), introduced into clinical practice in 2005, is an agonist of the melatonin receptors MT1 and MT2 and is used to treat insomnia. Within the product label, a DDI study is briefly described in which coadministration of fluvoxamine leads to a 190-fold increase in the AUC of ramelteon (Rozerem; Takeda Pharmaceuticals, Osaka, Japan), which perhaps represents the largest DDI recorded to date for any drug. The mechanism underlying this massive DDI is not described, with the exception that ramelteon is described as being metabolized by CYP1A2, and it is known that fluvoxamine can cause DDI with other drugs known to be cleared by this enzyme, such as theophylline (Rasmussen et al., 1997; Yao et al., 2001; Orlando et al., 2006) and tizanidine (Granfors et al., 2004).

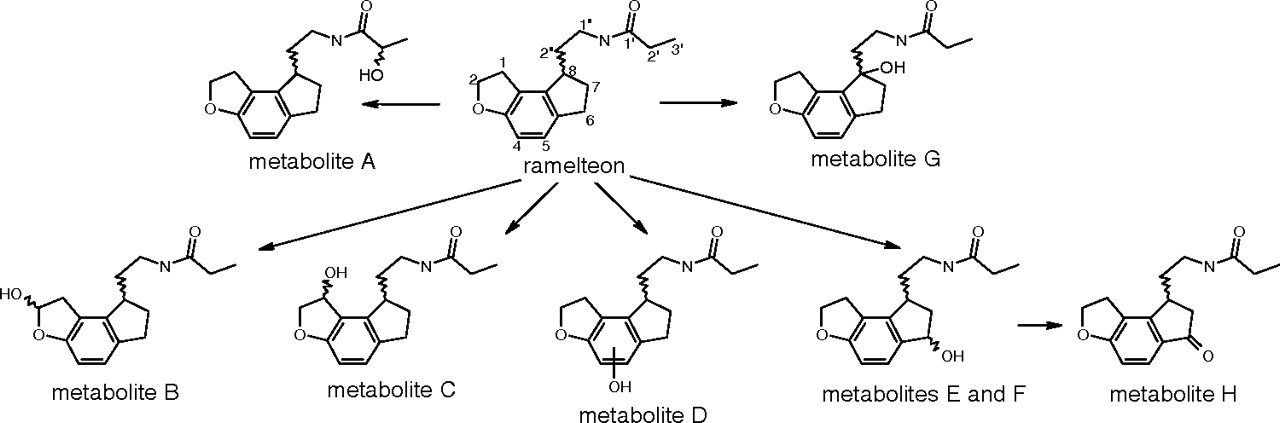
In vitro metabolic scheme for ramelteon. The numbering of positions on ramelteon refers to positions described under Results.
However, despite the fact that fluvoxamine is known to inhibit CYP1A2, whether this is actually the mechanism underlying its effect on ramelteon is not known. According to the Rowland-Matin equation, the fCL value for CYP1A2 for ramelteon would need to be very close to unity (i.e., 0.995), with fluvoxamine causing complete inhibition of CYP1A2 to elicit a 190-fold increase in exposure. However, it is also known that fluvoxamine is a potent inhibitor of other P450 enzymes, such as CYP2C19 (Christensen et al., 2002; Yasui-Furukori et al., 2004). Fluvoxamine can also mildly affect drugs cleared by CYP3A4 (e.g., buspirone; Lamberg et al., 1998), even though its intrinsic inhibitory potency for CYP3A4 is less than that for other P450 enzymes. This is likely the result of an effect of very high concentrations of fluvoxamine in the gut causing an impact on drugs subject to high first-pass intestinal extraction catalyzed by CYP3A4. Thus, it is possible that the DDI for ramelteon caused by fluvoxamine is the result of simultaneous inhibition of multiple enzymes, and it could be the case that ramelteon metabolism is carried out by these same enzymes that are sensitive to fluvoxamine. To be able to successfully extrapolate in vitro inhibition data to the in vivo fluvoxamine-ramelteon DDI, as complete a picture of ramelteon clearance pathways as is possible must be obtained. Data on the clearance pathways of ramelteon in human are scant, and it has been stated that it is primarily cleared by P450-mediated metabolism with less than 0.1% of the drug excreted unchanged (Karim et al., 2006; Rozerem Product Label, http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=search.label_approvalhistory). The objectives of the present study were to determine the in vitro metabolite profile of ramelteon, to quantitatively determine the enzymes involved in each metabolic pathway, and to attempt to extrapolate the in vivo DDI from in vitro data using various modeling approaches.
Materials and Methods
In Vitro Metabolite Profiling of Ramelteon.
The metabolite profile for ramelteon (10 μM) was determined in pooled human liver microsomes (1.0 mg/ml), pooled human intestinal microsomes (1.0 mg/ml), and heterologously expressed recombinant human P450 enzymes (50 pmol/ml). Incubations were carried out in 1.0 ml of KH2PO4 (0.1 M, pH 7.5) containing 3.3 mM MgCl2 and 1.3 mM NADPH for 30 min, followed by addition of 5 volumes of CH3CN. The precipitated material was removed by spinning in a centrifuge at 1700g, and the supernatant was evaporated in vacuo. The residue was reconstituted in 0.2 ml of 1% HCOOH in 10% CH3CN, spun in a microfuge at 14,000 rpm, and the clarified supernatant was analyzed by high-performance liquid chromatography/mass spectrometry (HPLC/MS) as described below.
Biosynthesis and Isolation of Ramelteon Metabolites.
Ramelteon (50 μM) was incubated with rat (0.5 mg/ml) and dog (0.5 mg/ml) liver microsomes, 1.3 mM NADPH, and 3.3 mM MgCl2 in 0.1 M KH2PO4, pH 7.5 (40 ml). The incubation was carried out at 37°C open to air for 1 h, followed by addition of CH3CN (40 ml), and removal of precipitated protein by spinning in a centrifuge at 1700g for 5 min. The supernatant was subject to vacuum centrifugation for 1.5 h, addition of 0.1% HCOOH to a total volume of 100 ml, and the material spun in a centrifuge at 40,000g for 30 min. The supernatant was applied to a Varian, Inc. (Palo Alto, CA) Polaris C18 column (4.6 × 250 mm; 5-μm particle size) through a Jasco (Easton, MD) pump at 0.8 ml/min. The column was moved to a gradient HPLC/MS system described below. Fractions were collected every 20 s, and fractions containing individual metabolites were pooled followed by evaporation of the solvent in vacuo for subsequent NMR analysis for structure elucidation and determination of the concentration.
Enzyme Kinetic and Inhibition Incubations.
Initial kinetic experiments were conducted to establish a protein concentration and an incubation time that provide linear reaction velocity. Incubation mixtures were composed of pooled human liver microsomes (0.5 mg/ml), pooled human intestinal microsomes (1.0 mg/ml), recombinant CYP1A2 (12.4 pmol/ml), CYP2C19 (7.9 pmol/ml), or CYP3A4 (9.2 pmol/ml) in 0.1 M KH2PO4, pH 7.5, containing 3.3 mM MgCl2 and 1.3 mM NADPH. The incubation volume was 0.1 ml. In substrate saturation experiments, ramelteon concentrations ranged from 1 to 300 μM, and in inhibition experiments a concentration of 10 μM was used. Incubations were commenced with the addition of NADPH and carried out for 15 min at 37°C open to the air. Incubations were terminated with the addition of 0.1 ml of 1% HCOOH in CH3CN containing 0.5 μM flunitrazepam as an internal standard. The mixtures were spun in an Eppendorf AG (Hamburg, Germany) microfuge at 14,000 rpm for 4 min. Supernatants were transferred to HPLC vials, and 0.08 ml was injected. The effect of inhibitors was also examined on ramelteon first-order consumption using a substrate concentration of 1 μM, microsomal protein at 2 mg/ml, and incubation times of 0, 5, 15, and 30 min.
Microsome Binding.
Ramelteon (10 μM) was mixed with pooled human liver microsomes (range of protein concentrations, 0.03 to 2.0 mg/ml) in 0.1 M KH2PO4, pH 7.5, containing MgCl2 (3.3 mM) and subjected to equilibrium dialysis. Dialysis was carried out for 4.5 h at 37°C in a Spectrum apparatus (Spectrum Industries, Los Angeles, CA) rotated at 20 rpm. SpectraPor membranes (no. 4; Spectrum Industries) with a molecular mass cutoff of 12 to 14 kDa were used. After the dialysis, the resulting samples were analyzed for ramelteon using the HPLC/MS method described below.
HPLC/MS.
The HPLC/MS system consisted of a Thermo Fisher Scientific (Waltham, MA) Surveyor quaternary pump, autoinjector, and diode array UV/visible detector coupled to a Thermo Fisher Scientific LTQ ion trap mass spectrometer. The UV/visible detector scanned over a range of 200 to 400 nm. The mass spectrometer was operated in the positive ion mode, with ionization conditions adjusted to optimize the signal for the protonated molecular ion for ramelteon (m/z 260). Source parameters included source potential, 3.9 kV; capillary voltage, 9 V; sheath gas flow, 20; and capillary temperature, 325°C. The collision energy was set at 22 for second- and third-stage ion fragmentation. Mobile phase components were 0.1% HCOOH in water (solvent A) and CH3CN (solvent B). Additional mass spectral fragmentation data were obtained by infusing solutions of isolated metabolites into a Thermo Fisher Scientific Orbitrap mass spectrometer to obtain exact mass data. Ionization and fragmentation conditions were similar to above, with the instrument operated in Fourier transform mass spectrometry mode at a resolution setting of 30,000.
Three different conditions were used, depending on the experimental endpoint. For metabolite profiling, metabolite purification, and structure elucidation, the system consisted of a Varian, Inc. Polaris C18 column (4.6 × 250 mm; 5 μm) held at 0.8 ml/min 10% B for 5 min. This was followed by a linear gradient to 60% B at 45 min, rinsing of the column at 90% B for 5 min, and re-equilibration to initial conditions for 10 min. The effluent was split with ∼60 μl/min introduced into the source of the mass spectrometer. The mass spectrometer was operated in the data-dependent scanning mode, over a range of m/z 200 to 400.
For quantitation of ramelteon metabolites, the system consisted of a Varian, Inc. Polaris C18 column (4.6 × 250 mm; 5 μm) held at 0.8 ml/min 25% B for 5 min. This was followed by a linear gradient to 49.5% B at 19 min and an immediate increase to 75% B, held for 2 min, followed by re-equilibration at initial conditions for 4 min. The effluent was split with ∼60 μl/min introduced into the source of the mass spectrometer. The following mass transitions were monitored: metabolite A, m/z 276→258 (18.4 min); metabolites B and C, m/z 258→204 (15.6 and 14.9 min, respectively); metabolite D, m/z 276→220 (13.2 min); metabolites E and F, m/z 258→159 (10.6 and 9.2 min, respectively); metabolite G, m/z 258→185 (12.2 min); metabolite H, m/z 274→256 (12.5 min); and internal standard, m/z 314→268 (22.6 min). Quantitation was accomplished using a standard curve ranging from 0.01 to 10 μM, using 1/x weighting. The standards used were those biosynthesized and isolated, with quantitation of the mother stock solutions being done with quantitative proton NMR spectroscopy.
For quantitation of ramelteon, a Phenomenex (Torrance, CA) phenyl-hexyl column (4.6 × 50 mm; 3 μm) was used. Initial mobile phase composition was held for 0.5 min at 50% B at a flow rate of 0.8 ml/min, followed by a linear gradient to 90% B at 2 min, held at 90% B for 0.5 min, and re-equilibration to initial conditions for 1.5 min. The effluent was split with ∼60 μl/min introduced into the source of the mass spectrometer. The following mass transitions were monitored: ramelteon, m/z 260→204; and internal standard, m/z 314→268. Retention times for ramelteon and internal standard were 2.2 and 2.6 min, respectively.
NMR Spectroscopy.
All the NMR spectra were recorded on a Bruker Avance 600 MHz controlled by TopSpin version 2.0 and equipped with a 5-mm cryo-TCI probe (all by Bruker BioSpin Corporation, Billerica, MA). Ramelteon and isolated metabolites were dissolved in 0.20 ml of dimethyl sulfoxide (DMSO)-d6 “100%” (Cambridge Isotope Laboratories, Inc., Andover, MA). All the spectra were referenced using residual DMSO-d6 (δ = 2.49 ppm relative to tetramethylsilane, δ = 0.00). One-dimensional spectra were recorded using a sweep width of 7500 Hz and a total recycle time of 7 s. Four dummy scans preceded the 40-scan acquisition. The resulting time-averaged free induction decays were transformed using an exponential line broadening of 1.0 Hz to enhance signal to noise. All the two-dimensional data were recorded using the standard pulse sequence provided by the manufacturer. NMR spectra and interpretation for ramelteon and its metabolites can be found in the supplemental data.
NMR Quantitation.
Concentrations of metabolite solutions in d6-DMSO were determined by comparison of proton resonance signals versus a computer-generated single frequency reference signal, which was based on a standard 10 mM solution of maleic acid using NMR-SIM version 5.0 (Bruker BioSpin Corporation) (G. S. Walker and T. F. Ryder, manuscript in preparation). Transformed NMR spectra were phase-corrected and manually baseline-corrected. Standard concentrations for metabolites ([C]metabolite) were calculated using eq. 2:
 in which [C]reference signal is the concentration represented by the computer-generated signal for one proton as calibrated from the 10 mM maleic acid reference solution, Areference signal is the integrated peak area for that signal, n is the number of protons in the resonance signal used for the metabolite, and Ametabolite is the integrated peak area for that metabolite signal.
in which [C]reference signal is the concentration represented by the computer-generated signal for one proton as calibrated from the 10 mM maleic acid reference solution, Areference signal is the integrated peak area for that signal, n is the number of protons in the resonance signal used for the metabolite, and Ametabolite is the integrated peak area for that metabolite signal.
Enzyme Kinetic and Inhibition Data.
Enzyme kinetic parameters and IC50 values were generated using SigmaPlot (version 9; Systat Software, Inc., San Jose, CA). The most appropriate enzyme kinetic model was selected based on the visual inspection of the data plotted on Eadie-Hofstee plots, followed by nonlinear regression of the reaction velocity versus substrate concentration data to the Michaelis-Menten equation (eq. 3):
 or Michaelis-Menten equation with a second, nonsaturable component [CLint(2)] (eq. 4):
or Michaelis-Menten equation with a second, nonsaturable component [CLint(2)] (eq. 4):
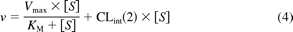
For some reactions, saturation kinetics were not observable, so the data were fit to a straight line with a slope equal to the intrinsic clearance, CLint.
Prediction of In Vivo Pharmacokinetics from In Vitro Data.
The prediction of in vivo clearance was performed using the well stirred model of hepatic extraction (eq. 5):

The value for CL′int was scaled from in vitro intrinsic clearance data using scaling factors of 45 mg of microsomal protein/g of liver and 20 g of liver/kg b.wt. Hepatic blood flow (Qh) was 21 ml/(min · kg), and binding values were obtained from published information (see Results).
The value for intestinal extraction (Fg) of an oral dose of ramelteon was estimated using eq. 6:
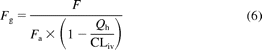 in which F represents the absolute bioavailability value, Fa is the fraction absorbed, and fraction evading hepatic extraction is the function of aforementioned hepatic blood flow, Qh, and intravenous blood clearance (CLiv).
in which F represents the absolute bioavailability value, Fa is the fraction absorbed, and fraction evading hepatic extraction is the function of aforementioned hepatic blood flow, Qh, and intravenous blood clearance (CLiv).
Fg was also estimated from scaling the in vitro CL′int,gut from intestinal microsomes (eq. 7):
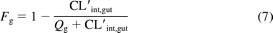 using a gut plasma flow (Qg) of 3.5 ml/(min · kg). The CL′int,gut value was estimated by using the hepatic CL′int generated from recombinant CYP3A4 divided by a factor of 94 to account for the amount of CYP3A4 in the intestine relative to the liver. Values of 3 mg of intestinal microsomes/g of tissue and 30 g of tissue/kg b.wt. were used in scaling from intestinal in vitro CL′int (Chiba et al., 1997).
using a gut plasma flow (Qg) of 3.5 ml/(min · kg). The CL′int,gut value was estimated by using the hepatic CL′int generated from recombinant CYP3A4 divided by a factor of 94 to account for the amount of CYP3A4 in the intestine relative to the liver. Values of 3 mg of intestinal microsomes/g of tissue and 30 g of tissue/kg b.wt. were used in scaling from intestinal in vitro CL′int (Chiba et al., 1997).
The prediction of DDI for three simultaneous clearance pathways when the inhibitor and substrate are coadministered orally was done using eq. 8:

The first term represents the impact on hepatic intrinsic clearance, which in this case is the impact on the three enzymes CYP1A2, CYP2C19, and CYP3A4. The second term is the impact on intestinal first-pass extraction caused by inhibition of CYP3A4. Values for Ki for ketoconazole, fluconazole, and fluvoxamine were those previously determined and successfully used in the prediction of DDI for more than 100 drugs (Obach et al., 2006).
Results
Identification of Metabolites of Ramelteon in Human Liver Microsomes.
An HPLC/UV trace of a ramelteon metabolism incubation mixture is in Fig. 2. Eight metabolites were observed: seven arising from hydroxylation reactions and the eighth from a subsequent dehydrogenation reaction. The metabolic scheme is shown in Fig. 1.

HPLC/UV traces (290 nm) of ramelteon (10 μM) metabolic incubations in pooled human liver microsomes.
Ramelteon.
The ion trap mass spectrum and NMR spectral data for ramelteon are shown in Supplemental Fig. S1. The protonated molecular ion was observed at m/z 260 along with a sodiated ion at m/z 282. Fragmentation of m/z 260 yielded m/z 204 (loss of propenal), followed by further fragmentation to m/z 187 (loss of ammonia) and m/z 159 (loss of ethylamine). The following were the observed 1H NMR chemical shift assignments [δ (ppm)]: 7.79 (1H, t broad), 6.89 (1H, d), 6.52 (1H, d), 4.51 (1H, m), 4.42 (1H, m), 3.16 (1H, m), 3.12 (1H, ov), 3.09 (2H, ov), 3.06 (1H, m), 2.79 (1H, m), 2.65 (1H, m), 2.17 (1H, m), 2.06 (1H, q), 1.93 (1H, m), 1.72 (1H, m), 1.43 (1H, m), and 0.99 (3H, t); 13C NMR [δ (ppm)]: 172.8 qC, 158.9 qC, 143.4 qC, 132.2 qC, 122.7 CH, 122.2 qC, 106.5 CH, 70.6 CH2, 41.5 CH, 36.8 CH2, 32.8 CH2, 31.2 CH2, 29.9 CH2, 28.5 CH2, 27.9 CH2, and 10.1 CH3. Heteronuclear single quantum correlation (HSQC) and heteronuclear multiple-bond correlation spectroscopy spectra were used to determine 13C chemical shifts.
Metabolite A.
Metabolite A eluted at 32 min and had a protonated molecular ion at m/z 276, indicating addition of oxygen to ramelteon, as well as a sodiated ion at m/z 298 (Supplemental Fig. S2). Initial fragmentation proceeded via dehydration, followed by various fragmentations of the N-ethylacrylamide portion. The tetrahydroindenofuran portion was unchanged as indicated by the presence of fragment ions at m/z 204, 187, 161, and 159. From 1H NMR data, the protons on the tetrahydroindenofuran ring remained unchanged, whereas the 2′ methylene position adjacent to the carbonyl lost one proton and the signal shifted to 3.93 ppm, and the three protons on position 3′ became a doublet. In the correlation spectroscopy (COSY) spectrum, the proton on position 2′ coupled to the protons on position 3′. This assignment was corroborated by the HSQC spectrum.
Metabolite B.
Metabolite B eluted at 29.3 min (Fig. 2) and possessed protonated and sodiated molecular ions at m/z 276 and 298, respectively, and a base peak at m/z 258, indicating facile loss of water in the ionization source (Supplemental Fig. S3). The major fragments arising from m/z 258 were m/z 202, 185, and 157, analogous to ions observed from ramelteon but missing two protons from the indenofuran ring. However, assigning the exact location of the site of hydroxylation required 1H NMR data. The hydroxylation was narrowed down to the furan portion of the ring system and the single proton on carbon-2 at 5.94 ppm and the two protons on carbon-1 at 3.22 ppm. Furthermore, metabolite B is a diasteromeric mixture caused by reversible opening and closing of the hemiacetal ring through the aldehyde, as shown by additional coupling of aromatic protons. Heteronuclear coupling was observed between protons and carbons on the hydroxydihydrofuran portion, further supporting structural assignment.
Metabolite C.
Metabolite C eluted at 28.5 min (Fig. 2) and had a sodiated molecular ion at m/z 298. A protonated molecular ion was absent, and the base peak was m/z 258, indicating facile dehydration in the ion source (Supplemental Fig. S4). Fragmentation was identical to metabolite B, suggesting that the position of hydroxylation is also on the dihydrofuran ring. The two protons on the carbon adjacent to the furan oxygen remained at 4.20 and 4.46 ppm, whereas the third at carbon-1 was now at 5.36 ppm. The COSY spectrum showed coupling between these protons, and heteronuclear coupling was consistent with the assignment.
Metabolite D.
Metabolite D eluted at 26.7 min (Fig. 2) and possessed a protonated molecular ion at m/z 276 and a sodiated ion at m/z 298 (Supplemental Fig. S5). The fragment ions at m/z 220 and 203 are analogous to those in ramelteon with an additional 16 mass units, indicating that hydroxylation is on the indenofuran ring system. NMR data indicated that metabolite D is actually a mixture of two unresolved phenol regioisomers at a ratio of 2:1. The spectral data did not permit specific assignment of which isomer was present in greater abundance.
Metabolites E and F.
Metabolites E and F eluted at 23.0 and 24.5 min (Fig. 2) and possessed a sodiated molecular ion at m/z 298 and base peak at m/z 258, indicating dehydration in the ion source (Supplemental Fig. S6). Attempts to isolate these showed that in aqueous solution they were interconverted. The major fragment ion at m/z 159 is indicative of a dehydrated indenofuran ring. Because of the interconversion, the metabolites were isolated as a diasteromeric mixture, and 1H NMR data indicated that the position of hydroxylation is at benzylic carbon-6. The methine proton on carbon-6 was observed at 4.86 and 5.01 in a 1:1 mixture, and two-dimensional COSY data were consistent with the structure assignment.
Metabolite G.
Metabolite G eluted at 25.8 min (Fig. 2) and possessed a sodiated ion at m/z 298 (Supplemental Fig. S7). Like several of the other metabolites, the base peak was at m/z 258, suggesting facile dehydration in the ion source. Fragment ions at m/z 185 and 157 indicate analogous indenofuran fragments to those seen for ramelteon but with two less protons, consistent with dehydration of a metabolite that had undergone hydroxylation on the indenofuran ring. 1H NMR data showed that the position of hydroxylation is on benzylic carbon-8 because no proton resonance existed at this position in the metabolite. The assignment was corroborated with two-dimensional COSY and HSQC coupling data.
Metabolite H.
Metabolite H eluted at 26 min (Fig. 2) and possessed a protonated molecular ion at m/z 274, 14 mass units greater than ramelteon, consistent with dehydrogenation of one of the hydroxyl metabolites (Supplemental Fig. S8). The fragment ion at m/z 256 is proposed to arise from dehydration/ring contraction of the dihydrofuran, with subsequent cleavage of the propenal and propanamide portions to yield m/z 200 and 183, respectively. NMR data showed a loss of the benzylic protons on carbon-6 along with downfield shift of the protons on carbon-7 and change in splitting consistent with the replacement of the protons on carbon-6 with a carbonyl. Furthermore, incubation of metabolites B, C, E/F, and G in liver microsomes showed that only metabolites E/F gave rise to metabolite H (data not shown).
Metabolite Profile of Ramelteon in Human Intestinal Microsomes.
An HPLC/UV trace of a ramelteon metabolism incubation mixture in pooled human intestinal microsomes is shown in Fig. 3. Two hydroxylated metabolites were observed: metabolites C and G.
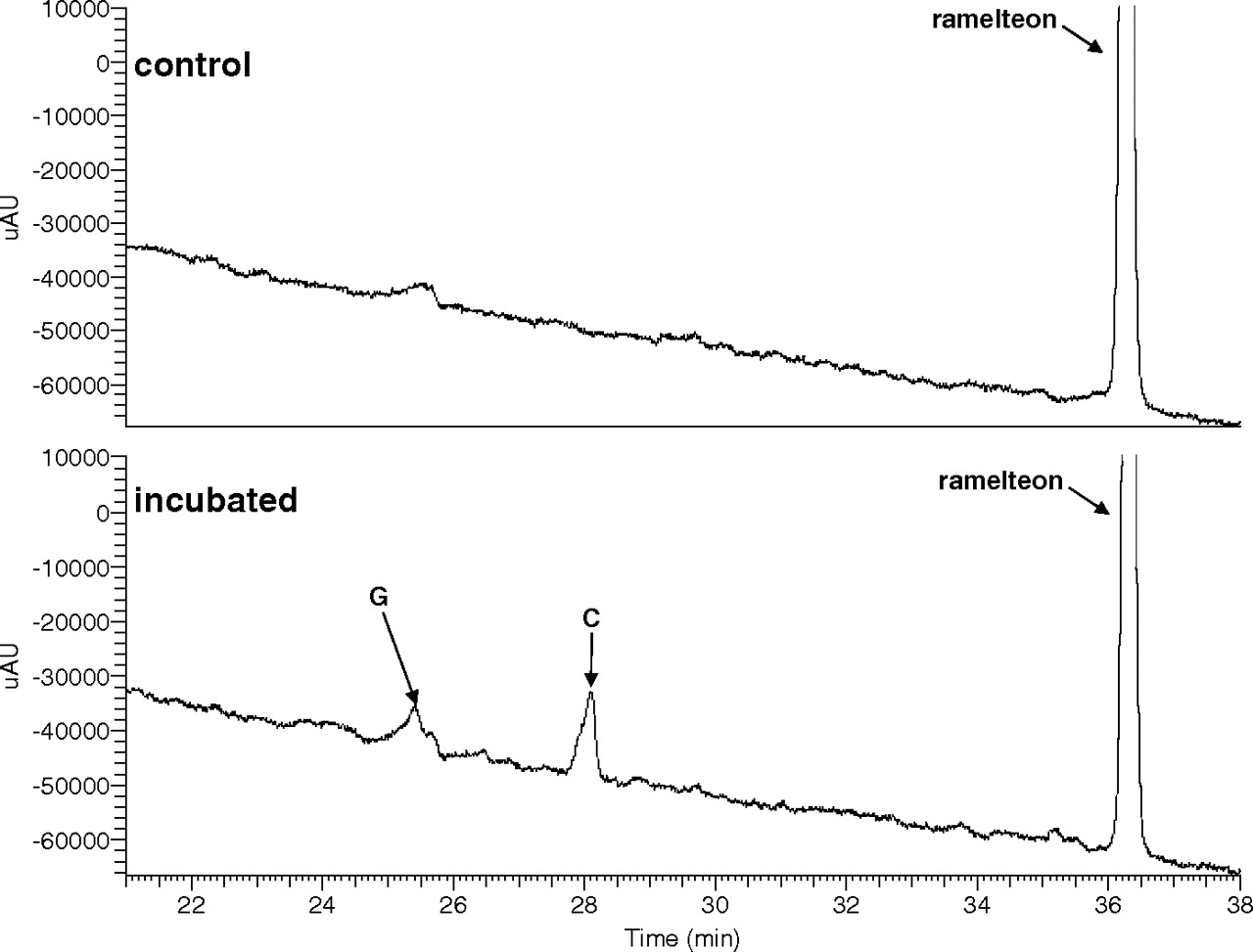
HPLC/UV traces (290 nm) of ramelteon (10 μM) metabolic incubations in pooled human intestinal microsomes.
Metabolite Profile of Ramelteon in Recombinant Human P450 Enzymes.
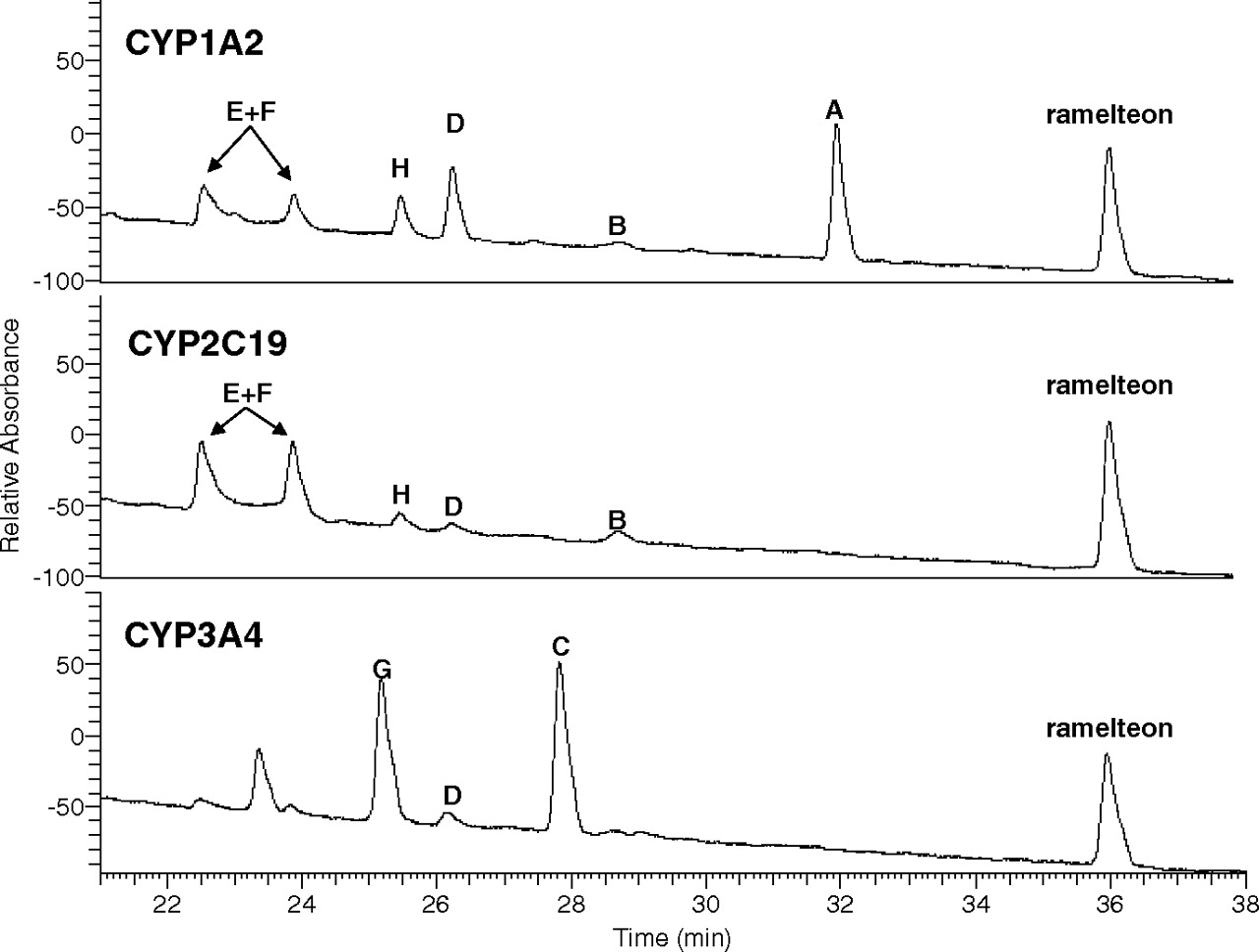
The metabolism of ramelteon was examined in heterologously expressed recombinant human P450 enzymes CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP3A5. Metabolism was observed in incubations of CYP1A2, CYP2C19, and CYP3A4 (Fig. 4). In CYP1A2 incubations, metabolite A was present in greatest abundance, with B, D, E, F, and H also readily observed. In CYP2C19, the major metabolites were E and F, with B, D, and H also observable. In CYP3A4 incubations, metabolites C and G were major, and metabolite D was also observable. An additional peak was observed at ∼23.5 min possessing a molecular ion at m/z 274, but it was not further characterized because it was observed in neither liver nor intestinal microsomal incubations. No ramelteon metabolism was observed to be catalyzed by CYP1A1, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2D6, or CYP2E1. A small amount of generation of metabolites C, D, and G was observed in CYP3A5, but substantially less than CYP3A4.

HPLC/UV traces (290 nm) of ramelteon (10 μM) metabolic incubations in recombinant human CYP1A2, CYP2C19, and CYP3A4.
Enzyme Kinetics of Ramelteon Metabolism in Pooled Human Liver Microsomes, Pooled Human Intestinal Microsomes, and Human CYP1A2, CYP2C19, and CYP3A4.
The enzyme kinetics of ramelteon metabolism to metabolites A through H in pooled human liver microsomes are listed in Table 1, and the substrate saturation plots are depicted in Fig. 5. The greatest intrinsic clearance was observed for formation of metabolites A, E, and F, which accounted for 85% of total intrinsic clearance. Michaelis constants for five of the metabolites were in the range of 17 to 39 μM, whereas these values were more than 10-fold greater for metabolites C and G. Formation of metabolite H did not show saturation, precluding estimation of a KM value.
Enzyme kinetic data for ramelteon metabolites in pooled human liver and intestinal microsomes
Values are mean (S.E.) of triplicate determinations.

Enzyme kinetic plots for the metabolism of ramelteon in pooled human liver microsomes.
In pooled intestinal microsomes, metabolite C had the greatest intrinsic clearance, accounting for 42% of the total, followed by metabolites G, E, and F; these four accounted for 86% of the total intrinsic clearance in the intestine. For metabolites C and G, KM values in the intestine and liver were similar, whereas KM values for the other metabolites were considerably greater. Metabolite A, which was major in liver, was barely measureable in intestine.
Enzyme kinetics were measured in the three human recombinant P450 enzymes that were shown to metabolize ramelteon (Table 2). Total intrinsic clearance was greatest for CYP2C19 [8.6 [μ]l/(min × pmol P450), followed by CYP1A2 and CYP3A4 [3.1 and 1.7 [μ]l/(min × pmol P450)]. For CYP2C19, 95% of intrinsic clearance was via formation of metabolites E and F, and KM values ranged from 12 to 36 μM. For CYP1A2, metabolites E, F, and A were the major metabolites, with these three accounting for 87% of total intrinsic clearance. KM values for CYP1A2 were mostly between 49 and 99 μM. For CYP3A4, metabolites C and G were the major metabolites accounting for 73% of intrinsic clearance. KM values for CYP3A4-catalyzed reactions were greater than those for CYP1A2 and CYP2C19.
Enzyme kinetic parameters for ramelteon metabolism in recombinant human P450 enzymes
Values are mean (S.E.) of triplicate determinations.
Inhibition of Ramelteon Metabolism by P450-Specific Inhibitors and Fluvoxamine.
The formation of metabolites A through G from ramelteon (10 μM) was measured in pooled human liver microsomes in the absence and presence of P450-specific inhibitors furafylline (20 μM; CYP1A2 inactivator), N-benzylnirvanol (20 μM; CYP2C19 inhibitor), and ketoconazole (3 μM; CYP3A inhibitor). Inhibition by fluconazole (inhibitor of CYP2C9, CYP2C19, and CYP3A) at two concentrations (30 and 80 μM) was also determined. These two concentrations were selected to be reflective of those measured in plasma and estimated to occur in tissues of first-pass after oral administration of 200 mg/day daily. The effects of these inhibitors are listed in Table 3. Furafylline showed the greatest effects on metabolites A and D, whereas ketoconazole inhibited formation of metabolites C and G the most. There were moderate effects of the inhibitors on formation of metabolites E and F. N-Benzylnirvanol had a mild effect on formation of metabolites D, F, and G. The effects of fluconazole at both 30 and 80 μM paralleled that of ketoconazole.
Effects of inhibitors of CYP1A2, 2C19 and 3A on ramelteon metabolism in pooled human liver microsomes
For metabolite formation, inhibition values are the mean of three determinations. For ramelteon consumption, measurements were duplicates. Ramelteon concentration was 10 μM when measuring metabolite formation and 1 μM when measuring ramelteon consumption.
The effect of these inhibitors was also measured when total ramelteon consumption was measured at a substrate concentration of 1 μM (Table 3). Furafylline had the largest impact (71% inhibition), followed by ketoconazole (42%) and N-benzylnirvanol (20%). (Note that the sum of these inhibition values exceeds 100%, suggesting that there is overlap in the specificities of these inhibitors.) Fluconazole caused a 20 to 25% decrease in ramelteon consumption.
Fluvoxamine was tested for its ability to inhibit the overall consumption of ramelteon at a substrate concentration of 1 μM and fluvoxamine concentrations of 2 and 40 μM. Intrinsic clearance was reduced by 76 and 92%, respectively. The effect of fluvoxamine on the generation of ramelteon initial metabolites was measured by determination of IC50 values (Table 4). Inhibition was most potent and complete for metabolite A. For metabolites B, D, E, F, and H, the potencies seemed similar to that as measured for metabolite A, but the slopes were noticeably different (Fig. 6), consistent with other enzymes besides CYP1A2 contributing to the generation of these metabolites (i.e., CYP2C19 and CYP3A4), with these enzymes being inhibited by fluvoxamine but not as potently as CYP1A2. (This would also be consistent with the relatively high S.E. values for the IC50 values; however, the data were not amenable to fitting to a more complex model containing two inhibition terms.) Finally, the inhibition of the generation of metabolites C and G by fluvoxamine had higher IC50 values (56 and 58 μM, respectively), consistent with these two metabolites being primarily generated by CYP3A4.
Inhibition of ramelteon metabolism by fluvoxamine
Values represent the mean (S.E.) of triplicate determinations. Ramelteon concentration was 10 μM.
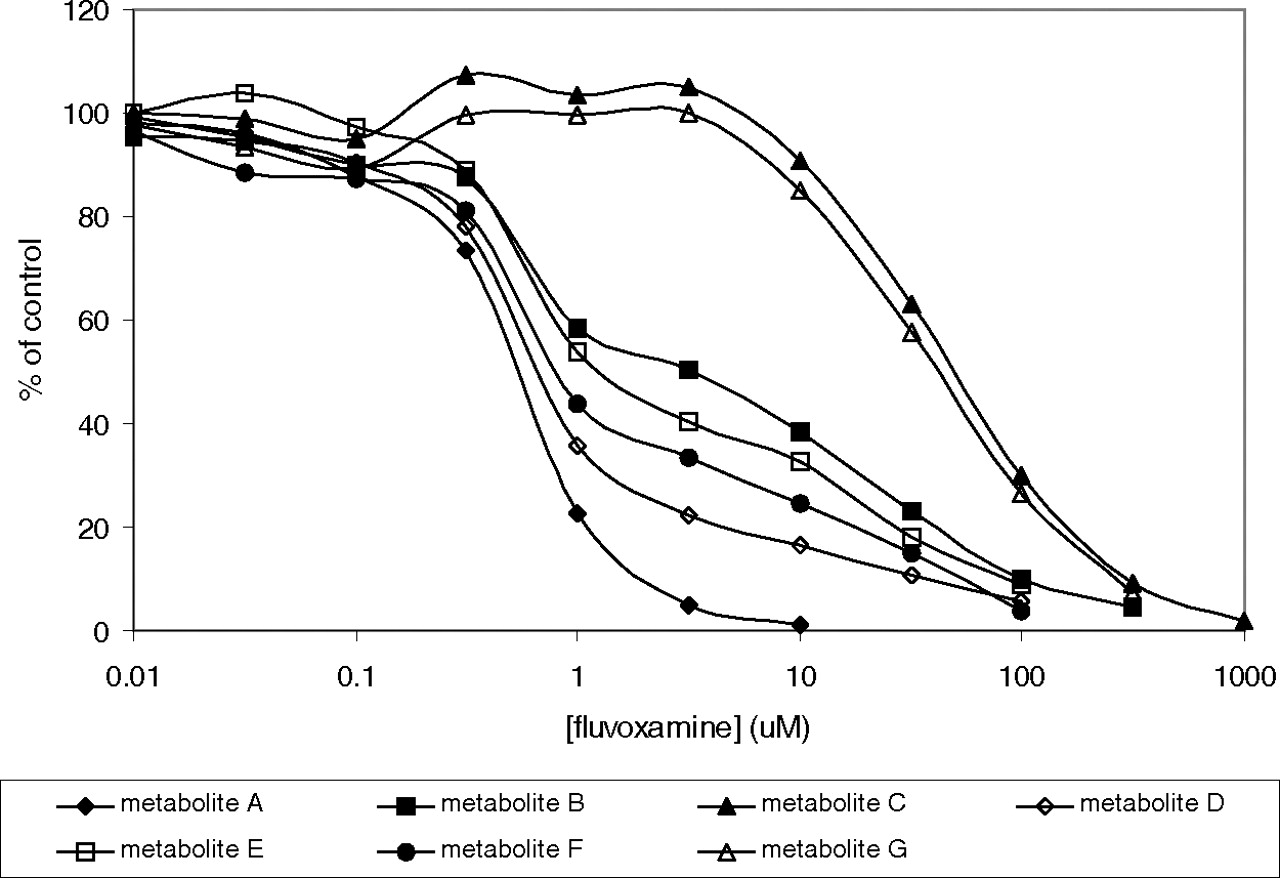
Inhibition of ramelteon metabolism by fluvoxamine in human liver microsomes.
Estimation of Ramelteon Pharmacokinetics from In Vitro Data.
The pharmacokinetics of ramelteon in humans has been briefly described (refer to Summary Basis of Approval Document, U.S. Food and Drug Administration). The total clearance after intravenous administration is listed as 916 ml/min [13 ml/(min · kg), assuming 70 kg b.wt.]. In that document, it was reported that the red blood cells contained 25% of the total ramelteon in blood, which after correction for a typical hematocrit value of 0.45 yields a blood/plasma ratio of 0.73. Converting the plasma clearance of 13 ml/(min · kg) to the more physiologically meaningful blood clearance yields a value of 18 ml/(min · kg), which approaches hepatic blood flow in humans [21 ml/(min · kg)]. Plasma protein binding was reported as 82%; correction for the blood/plasma ratio yields a free fraction in blood of 0.25.
The in vitro intrinsic clearance of ramelteon, composed of the sum of CL′int values for all eight metabolites in pooled human liver microsomes, is 110 μl/(min · mg protein). Scale-up of this value yields a CL′int value of 99 ml/(min · kg). Binding in liver microsomal incubations was determined to be negligible. Insertion of this value into the well stirred model of hepatic extraction yields an underpredicted total blood clearance value of 11.3 ml/(min · kg).
In scaling the enzyme kinetic data from the recombinant P450 enzymes, the abundance values of 52, 14, and 137 pmol of P450/mg of microsomal protein were used for CYP1A2, CYP2C19, and CYP3A4, respectively. These are additionally corrected for intersystem scaling factors (Proctor et al., 2004) determined for the lots of recombinant enzymes used in this investigation, which are based off of previously determined Vmax values using standard marker substrate activities (Walsky and Obach, 2004); these were 0.48, 0.40, and 0.41 for CYP1A2, CYP2C19, and CYP3A4, respectively. Use of these intersystem scaling factor-corrected sum of CL′int values for each of the metabolites generated for the three recombinant P450 enzymes yields a total in vitro CL′int value of 514 μl/(min · mg), which scales to an in vivo value of 462 ml/(min · kg). Insertion of this value into the well stirred model yields a value of blood clearance of 17.8 ml/(min · kg). Overall, these indicate that the in vitro enzyme kinetic data scale reasonably well to the clearance observed in vivo.
The value for intestinal extraction of an oral dose of ramelteon can be estimated from the reported absolute bioavailability value (F) of 0.018, fraction absorbed value (Fa) of 0.85, and fraction evading hepatic extraction (Fh) of 0.86, which was back-calculated from the intravenous clearance value. This yields an estimate for Fg of 0.15. Scaling the in vitro CL′int from intestinal microsomes [11.8 μl/(min · mg) protein], using scaling factors for the amount of intestinal microsomes per gram tissue and grams of tissue per kilogram body weight provided an intestinal CL′int,gut value of 1.1 ml/(min · kg b.wt). Combining with gut plasma flow yielded a value for Fg of 0.76. Assuming that CYP3A4 is the only enzyme contributing to ramelteon metabolism in the gut, and using the hepatic CL′int generated from recombinant CYP3A4 divided by a factor of 94, which accounts for the amount of CYP3A4 in the intestine relative to the liver, yielded a CL′int value for intestine of 2.2 ml/(min · kg b.wt.), which converts to a value for Fg of 0.61. Considering that coadministration of ketoconazole with ramelteon causes only a 1.4-fold increase in mean Cmax (see below) and that orally administered ketoconazole causes nearly complete inhibition of gut CYP3A4 activity, values of Fg of 0.61 or 0.76 are more in line with this effect than the value of 0.15 that is back-calculated from F, Fa, and Fh. Because CLiv is close to Qh, any error will be magnified in the back-calculated value of Fh, which could be overestimated and thus cause an underestimation of Fg.
Estimation of DDIs from In Vitro Ramelteon Metabolism Data.
The in vitro metabolism and enzyme kinetic data show that ramelteon is metabolized by CYP1A2, CYP2C19, and CYP3A4. DDI studies have been described for ramelteon for ketoconazole, fluconazole, and fluvoxamine. Thus, ramelteon affords a unique opportunity to probe the ability to predict DDI when one, two, or three enzymes involved in clearance are simultaneously inhibited. In vivo, ketoconazole inhibits CYP3A; fluconazole inhibits CYP3A and CYP2C19; and fluvoxamine inhibits CYP3A, CYP2C19, and CYP1A2. This gradation is reflected in the observation that the average DDIs, as measured by the increase in ramelteon AUC, are 1.82, 2.36, and 128 for ketoconazole, fluconazole, and fluvoxamine, respectively. Furthermore, the example of the fluvoxamine/ramelteon DDI tests the limits of the ability to predict DDI from in vitro data in an extreme case where multiple clearance pathways are substantially reduced, leaving the body with little capacity to clear the drug. The prediction of DDI for three simultaneous clearance pathways when the inhibitor and substrate are coadministered orally was done using eq. 8.
In this equation, the three enzymes shown to be involved in the metabolism of ramelteon in liver, along with an intestinal term, are included. However, it is possible that there is a fraction of ramelteon clearance that does not occur via these enzymes and tissues, which represents a limitation of the approach and the equation used for these predictions. Values for Ki were those previously determined and successfully used in the prediction of DDI for more than 100 drugs (Obach et al., 2006) (Table 5). The relative contributions of CYP1A2, CYP2C19, and CYP3A4 to the metabolism of ramelteon were estimated from the enzyme kinetic data generated above. The CL′int for each of the eight metabolites in human liver microsome was used to determine the percentage contribution of each pathway to total CL′int, and the individual contribution by each of the three enzymes to each pathway was determined separately and resummed to determine the relative contribution of each enzyme to total CL′int (Table 6). These were combined with the value of 0.61 for Fg described above.
Summary of input in vitro values for DDI prediction
Calculated contributions of CYP1A2, CYP2C19, and CYP3A4 to the generation of each ramelteon metabolite
The predicted DDIs for ramelteon caused by ketoconazole, fluconazole, and fluvoxamine are listed in Table 7. The interaction caused by ketoconazole arises largely from the impact this drug has on intestinal CYP3A4. The impact of ketoconazole on hepatic extraction of ramelteon is minimal because CYP3A4 only has a small (∼10%) contribution to ramelteon clearance, even though ketoconazole is predicted to decrease hepatic CYP3A4 activity by nearly 20-fold. The predicted DDI and actual DDI caused by ketoconazole were the same (1.82-fold). For fluconazole, both CYP3A4 and CYP2C19 are affected. As with ketoconazole, fluconazole virtually inhibits all intestinal CYP3A4 metabolism of ramelteon. The greater DDI caused by fluconazole versus ketoconazole arises by virtue of the inhibition of hepatic CYP2C19, in addition to the inhibition of hepatic CYP3A4 caused by fluconazole. The predicted DDI caused by fluconazole was 2.99-fold compared with the actual value of 2.36, a slight overprediction.
Predictions of DDIs for ketoconazole, fluconazole, and fluvoxamine on ramelteon in humans
For fluvoxamine, inhibition of all three enzymes involved in ramelteon clearance can occur. Although fluvoxamine is not a potent CYP3A4 inhibitor compared with ketoconazole, by virtue of high gut concentrations after oral administration, a considerable impact on intestinal CYP3A4 can occur. A 3.4-fold decrease in intestinal CYP3A4 is predicted with fluvoxamine dosing of 100 mg. In the liver, CYP1A2 and CYP2C19 activity will be substantially decreased by fluvoxamine, by predicted magnitudes of 94- and 10-fold, respectively. From the in vitro data, after fluvoxamine coadministration, hepatic CYP3A4 would now be expected to be the dominant clearance pathway (approximately two thirds of metabolism), with residual CYP2C19 contributing the remainder. However, although the prediction of an 11.4-fold increase in ramelteon exposure was made from the in vitro data, this is considerably less than the actual DDI of 128-fold. It is possible that the in vitro data have predicted too high a contribution of CYP2C19, at the expense of the contribution of CYP1A2, because despite causing a 10-fold decrease in CYP2C19 activity, fluvoxamine more potently inhibits CYP1A2, and the larger the contribution of CYP1A2 relative to CYP2C19, the greater the DDI will be. Nevertheless, even attempting to account for all the ramelteon metabolic pathways and all the enzymes involved in these pathways, it does not seem possible to be able to predict a greater than 100-fold DDI.
Discussion
It is highly desirable to be able to quantitatively predict DDI from in vitro inhibition data. Such data are routinely gathered during research on new drugs and in general have been successfully leveraged to predict DDI (Obach et al., 2005). To date, the effect of fluvoxamine on the pharmacokinetics of ramelteon represents the greatest magnitude for a DDI ever described, at 128-fold (or 190-fold, when using least-square averages). The mean AUC0-∞ of a single dose of 16 mg of ramelteon increased from 7.98 (±8.86) ng-h/ml to 1021 (±393) ng-h/ml when given after 4 days of 100 mg of fluvoxamine b.i.d. (Summary Basis of Approval Document, U.S. Food and Drug Administration). Thus, the fluvoxamine-ramelteon combination represents an extreme example to test whether such a large interaction could be projected from in vitro inhibition data. To attempt this, a comprehensive examination of the clearance pathways of the victim drug must be done because in theory greater than 99% of the clearance must be entirely inhibited to yield a DDI of greater than 100-fold. In this report, six metabolic pathways were shown for ramelteon in human liver microsomes. This is more than the three pathways previously reported, in which hydroxylation on the ethyl (corresponding to metabolite A), oxidation and ring opening of the furan (a downstream metabolite from metabolite B), and oxidation of the cyclopentyl ring (corresponding to metabolite H) were described previously (Karim et al., 2006). The biosynthesis and isolation of the eight metabolites and use of quantitative NMR to establish concentrations of stock solutions for these metabolites permitted determination of enzyme kinetic parameters for each ramelteon metabolic pathway.
The metabolism by CYP1A2 has been described previously (Simpson and Curran, 2008). Fluvoxamine is known to inhibit this enzyme potently in vitro and in vivo; however, to explain the >100-fold interaction CYP1A2 would need to be virtually the only enzyme involved in ramelteon clearance. This is clearly not the case because coadministration of ramelteon with ketoconazole and fluconazole both yield DDI (1.82- and 2.36-fold increases, respectively), whereas neither of these drugs is known to affect CYP1A2-cleared drugs (Naline et al., 1988; Konishi et al., 1994). However, in the present study it was shown that CYP2C19 and CYP3A4 also contribute to ramelteon metabolism. This is consistent with the effects of ketoconazole and fluconazole on ramelteon pharmacokinetics. When the enzyme kinetic data were used to estimate the fraction of ramelteon cleared by CYP2C19 and CYP3A4, and these values were used in the estimating of the DDI caused by fluconazole and ketoconazole, the estimates were close to the actual values (Table 7).
The in vitro enzyme kinetic data, the calculated contributions of CYP1A2, CYP2C19, and CYP3A4 to total ramelteon metabolism, the relative contribution of liver and intestine to ramelteon metabolism, and the inhibitory potency values for fluvoxamine were used to make an estimate of the impact of fluvoxamine on ramelteon pharmacokinetics. The estimate was 11-fold, which is a large effect, but it fell short of the actual DDI of greater than 100-fold (Table 7). The hepatic CYP1A2 activity is estimated at 1/94 of normal when fluvoxamine is administered (Table 7). If ramelteon were only cleared by CYP1A2, then this would yield an estimated 94-fold increase in exposure. However, this is not the case because CYP2C19 and CYP3A4 also contribute to ramelteon clearance. Although the prediction of 11-fold is still a remarkable magnitude of DDI, the reason(s) for the underestimate is unknown. The in vitro data suggest that hepatic CYP3A4 activity, which ordinarily contributes 8.6% of total ramelteon clearance, remains largely unaffected by fluvoxamine and would be virtually the only remaining route of ramelteon clearance. This is what limits the prediction of the fluvoxamine DDI magnitude to 11-fold. An explanation for why the fluvoxamine-ramelteon DDI is underestimated is not known. The value for in vivo hepatic concentration of fluvoxamine used was 1.63 μM, which is an estimate from the dose, absorption, free plasma fraction, and absorption rate (Obach et al., 2006). It is possible that if fluvoxamine is subject to active uptake into the liver, then the free intracellular concentration is actually higher. However, the concentration would need to be approximately 40-fold greater to explain the discrepancy, which is unlikely (Brown et al., 2007). Another possibility is that ramelteon has another as yet unknown clearance pathway that is potently inhibited by fluvoxamine, but there is no evidence to support this. Finally, fluvoxamine had been reported to show time-dependent inhibition of recombinant CYP3A4 coexpressed with cytochrome b5, with KI and kinact values of 3.7 μM and 0.05/min, respectively (Jones et al., 2007). However, in our own investigations, we did not observe time-dependent inhibition of CYP3A4 activity in human liver microsomes. It has been our observation that, despite the correlation between reversible inhibition data in liver microsomes and recombinant expressed P450 enzymes, time-dependent inhibition kinetics in these two systems do not match well (R. L. Walsky, unpublished observations), and such differences have been shown by others as well for P450 inactivators (Polasek and Miners, 2008). However, if fluvoxamine were a mechanism-based inactivator of CYP3A4, then it would be reasonably expected that the estimated DDI with ramelteon would be much higher and likely approach the observed value.
In summary, the in vitro metabolism for ramelteon in human liver microsomes, intestinal microsomes, and recombinant P450 enzymes was examined. Ramelteon shows six metabolic pathways that were catalyzed by CYP1A2, CYP2C19, and CYP3A4. The fractional contributions from each of these enzymes were determined to estimate the in vivo DDI magnitude from in vitro inhibition data. Estimations of the effects of ketoconazole and fluconazole were accurate; however, the massive >100-fold increase in ramelteon exposure caused by fluvoxamine coadministration was underestimated at 11-fold. Ramelteon is metabolized by three enzymes, all of which are inhibited by fluvoxamine, leading to a compounding of the magnitude of DDI. Thus, although a large ramelteon DDI would still be expected from the inhibition values, there may be a limit regarding how large a DDI magnitude can actually be predicted from in vitro data.
Acknowledgments.
We thank Gregory S. Walker for advice regarding the NMR spectral data and Robert L. Walsky for the determination that fluvoxamine did not show time-dependent inhibition of CYP3A.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.034009.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- DDI
- drug-drug interaction
- P450
- cytochrome P450
- AUC
- area under the curve
- HPLC/MS
- high-performance liquid chromatography/mass spectrometry
- DMSO
- dimethyl sulfoxide
- HSQC
- heteronuclear single quantum correlation
- COSY
- correlation spectroscopy.

- Received April 15, 2010.
- Accepted May 17, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}