


Abstract
To assess the feasibility of using sandwich-cultured human hepatocytes (SCHHs) as a model to characterize transport kinetics for in vivo pharmacokinetic prediction, the expression of organic anion-transporting polypeptide (OATP) proteins in SCHHs, along with biliary efflux transporters, was confirmed quantitatively by liquid chromatography-tandem mass spectrometry. Rifamycin SV (Rif SV), which was shown to completely block the function of OATP transporters, was selected as an inhibitor to assess the initial rates of active uptake. The optimized SCHH model was applied in a retrospective investigation of compounds with known clinically significant OATP-mediated uptake and was applied further to explore drug-drug interactions (DDIs). Greater than 50% inhibition of active uptake by Rif SV was found to be associated with clinically significant OATP-mediated DDIs. We propose that the in vitro active uptake value therefore could serve as a cutoff for class 3 and 4 compounds of the Biopharmaceutics Drug Disposition Classification System, which could be integrated into the International Transporter Consortium decision tree recommendations to trigger clinical evaluations for potential DDI risks. Furthermore, the kinetics of in vitro hepatobiliary transport obtained from SCHHs, along with protein expression scaling factors, offer an opportunity to predict complex in vivo processes using mathematical models, such as physiologically based pharmacokinetics models.
Introduction
Hepatobiliary elimination is a primary elimination route for many endogenous and exogenous xenobiotics. Hepatic uptake and efflux transporters, located on sinusoidal and canalicular membranes, respectively, contribute to the vectorial transport of drugs and their metabolites from systemic circulation to bile (Meier et al., 1997; Kullak-Ublick et al., 2000). Two classes of hepatic uptake transporters, sodium-dependent and sodium-independent transporters, coexist on the sinusoidal membranes of hepatocytes with overlapping substrate specificities. For example, the sodium-dependent transporter, sodium-taurocholate cotransporting polypeptide, has been shown to transport the organic anion-transporting polypeptide (OATP) substrates atorvastatin and rosuvastatin (Ho et al., 2006; Choi et al., 2011). OATP1B1/OATP1B3, organic anion transporter 2, and organic cation transporter 1 are expressed specifically in the liver and transport structurally diverse substrates in a sodium-independent manner. In the last decade, significant advances toward the prediction of in vivo new molecular entity clearance using in vitro models have been well documented. Indeed, in vitro human liver microsomes and isolated hepatocytes have been shown to be important tools in drug discovery and development to confidently predict in vivo human drug metabolism for compounds predominantly eliminated by cytochrome P450 (Obach, 1999; McGinnity et al., 2004). However, human pharmacokinetic prediction remains very challenging for compounds where drug transporters are involved in the clearance mechanism. In 2010, the International Transporter Consortium (ITC) recommended decision trees using in vitro systems to assess the risk of in vivo transporter mediated drug-drug interactions (DDIs) (Giacomini et al., 2010). For OATP transporters, the investigation cascade is initiated by the criteria for active hepatic uptake if hepatic clearance is an important route of elimination (e.g., >0.3 of total clearance) (Giacomini et al., 2010). However, the models and the extent of active hepatic uptake that would trigger the investigation cascade remain undetermined, and suitable in vitro tools to assess active or passive hepatic uptake need to be validated further. We hypothesized that clinically relevant OATP-mediated DDI is associated with significant active uptake in vitro.
Recently, a physiologically based pharmacokinetic model, in which physiological compartments representing organs or tissues are connected with blood flow, was developed to predict in vivo clearance and time profiles of drug elimination using in vitro models, such as suspension hepatocytes and canalicular membrane vesicles (Watanabe et al., 2009). The approach is accepted generally as a useful tool to predict tissue concentration, DDI, and effects of enzyme or transporter genetic polymorphisms on drug exposure. Practically, parameters for distinct clearance processes, including passive diffusion, transporter-mediated hepatic uptake (sinusoidal), metabolism, and transporter-mediated efflux (canalicular), into the bile could be determined in vitro and used to predict in vivo human pharmacokinetics.
The sandwich-cultured hepatocyte model, built by culturing hepatocytes between two layers of gelled matrix in a sandwich configuration, forms a bile canalicular network and provides three-dimensional orientation and proper localization of hepatobiliary transporters that mimic in vivo conditions and allow vectorial transport of xenobiotics (Bi et al., 2006). Turncliff et al. (2006) evaluated the hepatobiliary disposition of metabolites of terfenadine generated in sandwich-cultured rat hepatocytes (SCRHs). The advancement of in vitro models for clearance prediction in humans has further reflected an increase in the mechanistic understanding of the hepatic vectorial elimination process (Kotani et al., 2011). For example, through the use of liquid chromatography-tandem mass spectrometry (LC-MS/MS) quantification methods, the expression of biliary efflux transporters in SCRHs was determined and used to improve the extrapolation from in vitro to in vivo for the compounds excreted from bile (Li et al., 2009, 2010). Meanwhile, a decrease in functional uptake in SCRHs was observed, raising concerns for its suitability as a model for active hepatic uptake evaluations (Li et al., 2010; Kotani et al., 2011). However, unlike SCRHs, on the basis of our in-house, unpublished results and recently published data (Kotani et al., 2011), active uptake functions in sandwich-cultured human hepatocytes (SCHHs) are well maintained, and the system appears promising as a single in vitro model for the evaluation of candidate drug uptake and biliary excretion. The objectives of this research are threefold: first, confirm the maintenance of hepatic-specific uptake transporter function and expression in SCHHs; second, optimize the experimental conditions needed to define active hepatic uptake and biliary excretion to obtain in vitro parameters that could be used as inputs for a mathematical, physiologically based pharmacokinetic model for in vivo prediction (Jones et al., 2012); and third, obtain in vitro cutoff values for active uptake in SCHHs that would be appropriate to trigger the clinical investigations recommended by the ITC.
Materials and Methods
Reagents and Hepatocytes.
High-performance liquid chromatography (HPLC)-grade acetonitrile, water, and methanol were purchased from Honeywell Burdick & Jackson (Muskegon, MI) and EMD Chemicals, Inc. (Gibbstown, NJ), respectively. Hanks' balanced salt solution (HBSS) was purchased from Invitrogen (Carlsbad, CA). Rosuvastatin, atorvastatin, pitavastatin, valsartan, fluvastatin, pravastatin, cerivastatin, buprenorphine, and midazolam were obtained from Sequoia Research Products Ltd. (Oxford, UK). Rifamycin SV (Rif SV) was purchased from Sigma-Aldrich (St. Louis, MO). Ammonium acetate was obtained from Mallinckrodt Baker, Inc. (Phillipsburg, NJ). [3H]Taurocholate (TC) (4.6 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). The bicinchoninic acid protein assay kit was purchased from Thermo Fisher Scientific (Waltham, MA). The ProteoExtract native membrane protein extraction kit was purchased from Calbiochem (San Diego, CA). Trypsin was purchased from Promega (Madison, WI). Matrigel (phenol red free) and collagen-coated 24-well plates were obtained from BD Biosciences (San Jose, CA). In Vitro GRO-HT, In Vitro GRO-CP, and In Vitro GRO-HI hepatocyte media were purchased from Celsis In Vitro Technologies, Inc. (Baltimore, MD). Cryopreserved human hepatocytes (lot Hu4165) were purchased from CellzDirect (Durham, NC).
SCHHs.
Cryopreserved hepatocytes were thawed and plated as described previously (Bi et al., 2006). In brief, hepatocytes were thawed in a water bath at 37°C and then immediately poured into 50 ml of prewarmed In Vitro GRO-HT medium in a conical tube. The cells then were centrifuged at 50g for 3 min and resuspended at a concentration of 0.85 × 106 cells/ml in In Vitro GRO-CP medium. Cell viability was determined by a trypan blue exclusion assay. On day 1, hepatocyte suspensions were seeded in collagen-coated 24-well plates in a volume of 0.5 ml/well. After incubation overnight at 37°C, the hepatocytes were overlaid with 0.5 ml of In Vitro GRO-HI medium with Matrigel (0.25 mg/ml). The In Vitro GRO-HI medium was refreshed every 24 h.
Extraction and Digestion of Membrane Protein.
At day 5 after culture, SCHHs were detached from cell culture plates and washed with HBSS. Along with suspension hepatocytes, the membrane protein fraction of hepatocytes was extracted as described previously (Li et al., 2008) using the ProteoExtract native membrane protein extraction kit (Calbiochem). In brief, hepatocyte pellets were homogenized in extraction buffer I of the kit containing a protease inhibitor cocktail followed by incubation at 4°C for 10 min with gentle rocking. The homogenate was centrifuged at 16,000g for 15 min at 4°C. The supernatant containing cytosolic proteins was discarded, and the pellets were resuspended in extraction buffer II of the kit containing a protease inhibitor cocktail. After 60 min of incubation at 4°C with gentle rocking, the suspension was centrifuged at 16,000g for 15 min at 4°C. The protein concentration in the membrane fractions was determined using the BCA protein assay kit.
LC-MS/MS Quantitative Measurement of OATP1B1, OATP1B3, and OATP2B1 Transporters.
Proteotypic peptides and stable isotope label (SIL) peptides of OATP1B1, OATP1B3, and OATP2B1 were selected (Table 1) and synthesized as surrogate analytes for the corresponding protein quantification by LC-MS/MS (Li et al., 2008). LC-MS quantification for human OATP proteins and two of six peptides identified (NVTGFFQSFK and SSPAVEQQLLVSGPGK) also were reported by Ji et al. (2012). The digestion conditions were optimized by our previous report (Balogh et al., 2012). In brief, 80 μg of the membrane fraction of the protein was reduced with 6 mM dithiothreitol, alkylated with iodoacetamide in 25 mM ammonium bicarbonate digestion buffer containing 10% sodium deoxycholate monohydrate, and then digested by trypsin (the final concentration of sodium deoxycholate monohydrate during digestion was 1%). At the end of the digestion, samples were acidified with an equal amount of 0.2% formic acid in water with SIL internal standards and centrifuged at 14,000g for 5 min. The supernatants were transferred to a new plate and dried. The samples were reconstituted with 0.1% formic acid in water and analyzed by LC-MS/MS.
Target peptides and multiple reaction monitoring transitions monitored for human OATP proteins
m/z is the mass-to-charge ratio of the ion.
The calibration curve was prepared using the synthetic proteotypic peptide with a fixed concentration of each SIL peptide as the internal standard. Sample quantification was conducted by coupling a triple-quadrupole mass spectrometer (API4000; Applied Biosystems, Foster City, CA) to a high-performance LC system (SLC-10A; Shimadzu, Kyoto, Japan) and HTS PAL autosampler (LEAP Technologies, Carrboro, NC). A Kinetex 2.6-μm C18 100 Å column (3.0 × 100 mm) (Phenomenex, Torrance, CA) was used for peptide chromatography. A linear gradient elution program was used to achieve chromatographic separation with mobile phase A (0.1% formic acid in HPLC-grade water) and mobile phase B (0.1% formic acid in acetonitrile). A sample volume of 10 μl was injected onto the LC column at a flow rate of 0.5 ml/min. The parent-to-product transitions for the proteotypic peptide generally represent the doubly charged parent ion to the singly charged product ions for each peptide (Table 1). Data were processed by integrating the appropriate peak areas for the analyte peptides and the SIL internal standard peptides in Analyst 1.4.2 (Applied Biosystems).
Determination of Hepatic Uptake in SCHHs.
SCHHs were rinsed twice with 0.5 ml of regular HBSS buffer at 37°C or Ca2+/Mg2+-free HBSS buffer containing 1 mM EGTA, and the buffer then was replaced with fresh regular HBSS buffer or Ca2+/Mg2+-free HBSS buffer containing 1 mM EGTA. The disruption of bile canalicular network was achieved by preincubation with Ca2+/Mg2+-free HBSS containing 1 mM EGTA buffer for 10 min. For the determination of the effects of various inhibitors on rosuvastatin uptake in SCHHs, parallel incubations were conducted in the presence of the uptake transporter inhibitors Rif SV (100 μΜ), cyclosporine A (CsA) (10 μΜ), and gemfibrozil (30 μΜ). SCHH uptake was initiated by the addition of 500 μl containing the substrate at the concentration indicated in the figure legends, with or without inhibitors. Reactions were terminated at the designated time points by quickly washing the hepatocytes three times with ice-cold HBSS buffer. The cells were lysed with either 0.5% Triton X-100 (radiolabeled compounds) or 100% methanol containing the internal standard (nonradiolabeled compounds). Samples were analyzed by LC-MS/MS.
LC-MS/MS Analysis of Probe Substrates.
LC-MS/MS analysis of probe substrates was conducted with an API4000 triple-quadruple mass spectrometer (Applied Biosystems) coupled with a turbo ion spray interface in positive ion mode and connected with a SLC-10A LC system (Shimadzu) and 215 autosampler (Gilson, Inc., Middleton, WI). The mass spectrometer was controlled by Analyst 1.4.2 software (Applied Biosystems). The autosampler was independently controlled by Gilson 735 software and synchronized to Analyst via contact closure. The HPLC method consisted of a step gradient with 25-μl samples loaded onto a 1.5 × 5 mm Showadenko (Tokyo, Japan) ODP 13-μm particle size column using 95% 2 mM ammonium acetate/5% 50:50 methanol/acetonitrile. Incubation samples and standard curve samples were eluted with 10% 2 mM ammonium acetate in 50:50 methanol/acetonitrile. Peak area counts of analyte compounds and internal standards were integrated using DiscoveryQuant Analyze as an add-on to Analyst 1.4.2. The obtained values underwent data analysis by averaging the analyte area divided by the internal standard analyte for each concentration.
Data Analysis.
The apparent in vitro biliary clearance (CLbile) (Liu et al., 1999) and percentage active uptake were determined by eqs. 1 and 2, respectively. The hepatic uptake of test compounds was estimated from the initial uptake phase (0.5–1.5 min), whereas biliary excretion was assessed at 10 min. The data represent the results from a single study run in triplicate or duplicate, and a minimum of two experiments were performed. The standard deviation or coefficient of variation is listed in the legend of the figure or table.
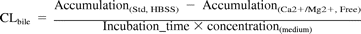

Results
Protein Expression of OATP1B1, OATP1B3, and OATP2B1 in Suspension Hepatocytes and SCHHs.
Because functional active uptake has been shown previously in SCHHs both in house and in the literature (Kotani et al., 2011), in the present study, the protein expression of OATP1B1, OATP1B3, and OATP2B1 in SCHHs was measured by LC-MS/MS and compared with that in suspension hepatocytes. Peptides proteolytically released from the target OATP proteins were quantified using external synthetic peptide calibration curves. The peptide fragments were monitored using multiple reaction monitoring (Table 1). Sample preparation, digestion, and detection limits of OATP protein quantification were evaluated previously (Balogh et al., 2012). As shown in Table 2, although OATP1B3 and OATP2B1 expression levels were reduced to approximately half of that in suspension hepatocytes, OATP1B1 expression in SCHHs was slightly higher (1.5-fold) than that found in suspension hepatocytes. The results provide support for OATP-mediated active hepatic uptake in SCHHs, which indicates that SCHHs could be a suitable tool for the assessment of active OATP uptake. In addition, the protein expression obtained by LC-MS/MS measurement could be integrated into a mathematical model as a component of scaling factors for in vivo extrapolation from in vitro.
Quantification of OATP1B1, OATP1B3, and OATP2B1 in suspension hepatocytes and SCHHs of lot Hu4165
The protein expression of OATP1B1, OATP1B3, and OATP2B1 in SCHHs was measured by LC-MS/MS and compared with that in suspension hepatocytes.
Inhibition of Active Uptake in SCHHs.
The active component of hepatic uptake can be determined by the total hepatic uptake (PSuptake) minus the passive diffusion (PSpassive) in an in vitro hepatocyte model. In general, passive diffusion in hepatocytes can be obtained by either coincubating with an OATP inhibitor or conducting the uptake experiment at low temperature (e.g., at 4°C). We reported previously that the lack of uptake might be an artificial effect of a rigid cell membrane at 4°C and that the uptake in the hepatocyte model determined at 37 and 4°C might not truly reflect the functional active uptake (Kimoto et al., 2011). To select a suitable inhibitor blocking carrier-mediated active hepatic uptake in SCHHs, time-dependent accumulation of rosuvastatin, a known substrate of OATP proteins, was investigated in the presence or absence of known OATP inhibitors, Rif SV (100 μM), CsA (10 μM), and gemfibrozil (30 μM). As depicted in Fig. 1, rosuvastatin was transported actively into SCHHs, and the uptake was linear up to 1.5 min. It is worthwhile to note that a positive y intercept was obtained by extending the trend line of rosuvastatin uptake. Nonspecific binding on the surface of hepatocytes has been proposed to contribute the intercept and was not included here for the calculation of the initial uptake rate. The initial uptake rates of rosuvastatin estimated from 0.5 to 1.5 min were inhibited by all three OATP inhibitors, Rif SV, CsA, and gemfibrozil, by 95, 80, and 78%, respectively. The active uptake of rosuvastatin was inhibited almost completely in the presence of Rif SV at a concentration of 100 μΜ.
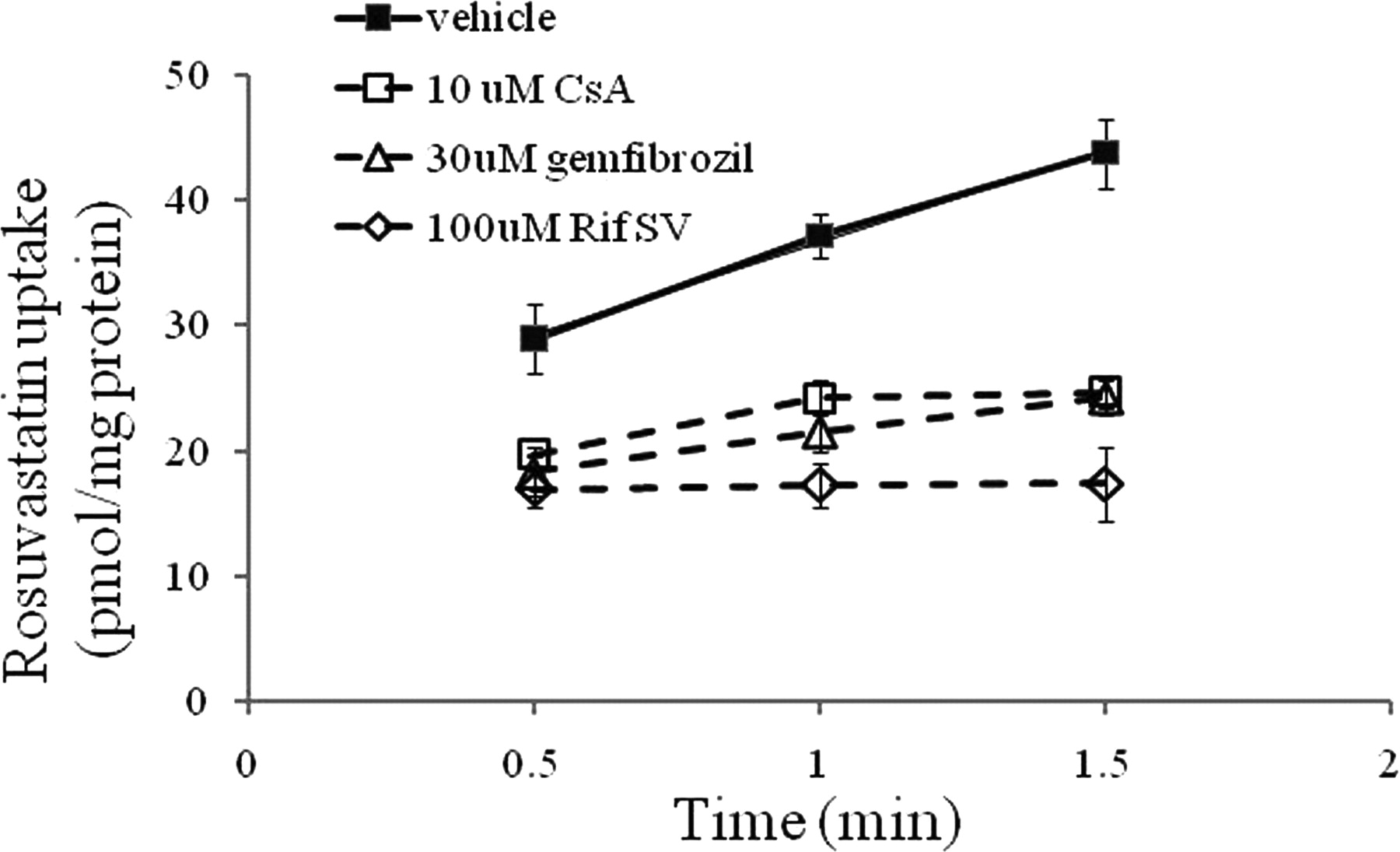
Inhibitory effects of Rif SV, CsA, and gemfibrozil on rosuvastatin uptake in SCHHs. The uptake of rosuvastatin (1 μM) was measured at 37°C in the presence and absence of Rif SV (100 μM), CsA (10 μM), and gemfibrozil (30 μM). Data are presented as mean ± S.D.
Hepatic Uptake and Biliary Excretion for Compounds That Are the Substrates of Phase I/II Metabolizing Enzymes or Hepatic Uptake Transporters in SCHHs.
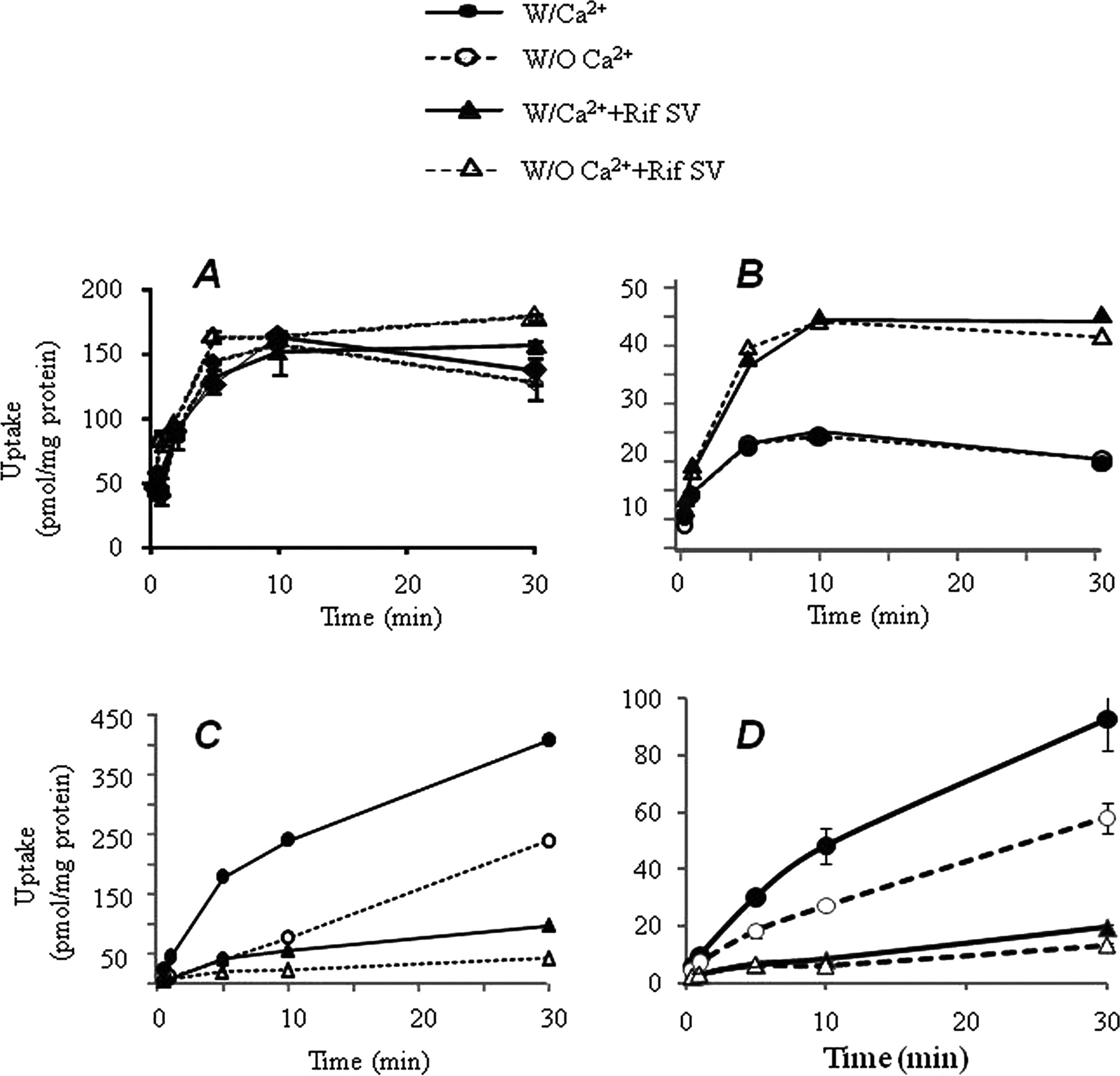
As mentioned above, rosuvastatin uptake in SCHHs was nearly abolished by coincubation with 100 μΜ Rif SV. The results agree with previous reports that Rif SV (100 μM) totally blocks OATP functions in OATP-transfected cell lines (Vavricka et al., 2002). To determine whether Rif SV also affects metabolizing enzymes, biliary excretion, and active hepatic uptake by transporters other than OATPs, initial uptake and biliary excretion of various compounds were tested further in SCHHs. Buprenorphine and midazolam, which are metabolized by UGT1A1 and CYP3A4, respectively, were used as compounds that penetrate the hepatocytes via passive diffusion. TC was selected as a probe substrate of sodium-taurocholate cotransporting polypeptide transporters. Rosuvastatin as a known OATP substrate was tested again in SCHHs to characterize initial uptake and biliary excretion kinetics. In addition, rosuvastatin and TC are excreted from bile through biliary efflux transporters, such as multidrug resistant protein 2, breast cancer resistant protein, or bile salt exporting pump (Ito et al., 2010). The uptake (PSuptake and PSpassive) and biliary excretion (CLbile) in SCHHs of the compounds are shown in Fig. 2 and Table 3. As expected, Rif SV (100 μΜ) inhibited approximately 93 and 83% of the uptake rate of rosuvastatin and TC in SCHHs, respectively. After a decrease of uptake into SCHHs caused by the inhibition of Rif SV, biliary excretion of rosuvastatin and TC also were decreased substantially (Table 3). Although significant increases in the intracellular concentrations of buprenorphine or midazolam in SCHHs were detected in the presence of Rif SV (Fig. 2), the initial uptake rates of the compounds were not altered significantly. The intracellular accumulation of buprenorphine or midazolam by Rif SV might be due to the inhibition of CYP3A4 and UGT1A1 activities in SCHHs. Because no biliary excretion was detected for buprenorphine and midazolam (Table 3) in both the absence and the presence of Rif SV, the results revealed that Rif SV did not change the elimination profile by switching elimination pathways between metabolizing enzyme-mediated clearance and transporter-mediated biliary excretion.

Hepatic uptake and biliary excretion of several compounds in SCHHs. The hepatic uptake was investigated at 37°C in the presence and absence of Rif SV (100 μM) or in the buffer with or without Ca2+/Mg2+. A, midazolam (1 μM); B, buprenorphine (0.2 μM); C, TC (1 μM); D, rosuvastatin (1 μM). Data are presented from single studies run in duplicate or triplicate. A minimum of two experiments were performed on different days to verify coefficient of variation <15%.
Comparison of initial rate of hepatocyte uptake in the presence or absence of Rif SV
Substrates were incubated in the presence or absence of the inhibitor Rif SV (100 μM) for passive and active uptake or in the buffer with or without Ca2+ for in vitro biliary clearance (CLbile). Data are presented from single studies run in duplicate or triplicate. A minimum of two experiments were performed on different days to verify coefficient of variation <15%.
Hepatic Uptake in SCHHs for the Compounds That Undergo Clinically Significant DDIs with OATP Inhibitors.
As noted above, SCHHs have been characterized as a suitable model to assess in vitro hepatic uptake (active and passive) and biliary excretion. To evaluate the risk of in vivo OATP-mediated DDIs from the in vitro SCHH model, a literature review was conducted to compile the OATP substrates that undergo significant DDIs in the clinic [>2-fold increase in the area under the curve (AUC)] when coadministered with OATP inhibitors. The substrates are listed in Table 4, and we conducted uptake and biliary excretion assays in SCHHs to determine the in vitro active uptake (PSactive) and CLbile. To avoid potential saturation of active uptake transporters, the substrate concentrations selected either were below the OATP Km known from literature reports or a lack of saturation was confirmed by our preliminary experiments. As indicated in Table 4, initial uptake rates into SCHHs for the compounds ranged from 2 to 45 μl · min−1 · mg protein−1. Rif SV inhibitable active hepatic uptake ranged from 55 to 84% in SCHHs for the compounds reported to undergo OATP-mediated DDIs. Although the Rif SV inhibitable active uptake (percentage) did not correlate with the AUC changes that are observed in the clinic (Fig. 3A), these compounds were actively taken into hepatocytes to an extent of >50% (Fig. 3A). However, an increase of passive diffusion into hepatocytes tended to diminish the fold increase in AUC change caused by OATP inhibitors (Fig. 3B), suggesting that OATP-mediated DDI risk could be low for highly permeable compounds.
OATP-related DDIs and hepatic active uptake in SCHHs
Clinical substrates were incubated in the presence and absence of the inhibitor Rif SV (100 μM) for passive and active uptake or in the buffer with or without Ca2+ for in vitro biliary clearance (CLbile). Data are presented as the mean of duplicates or as the mean (S.D.) of three to five experiments.
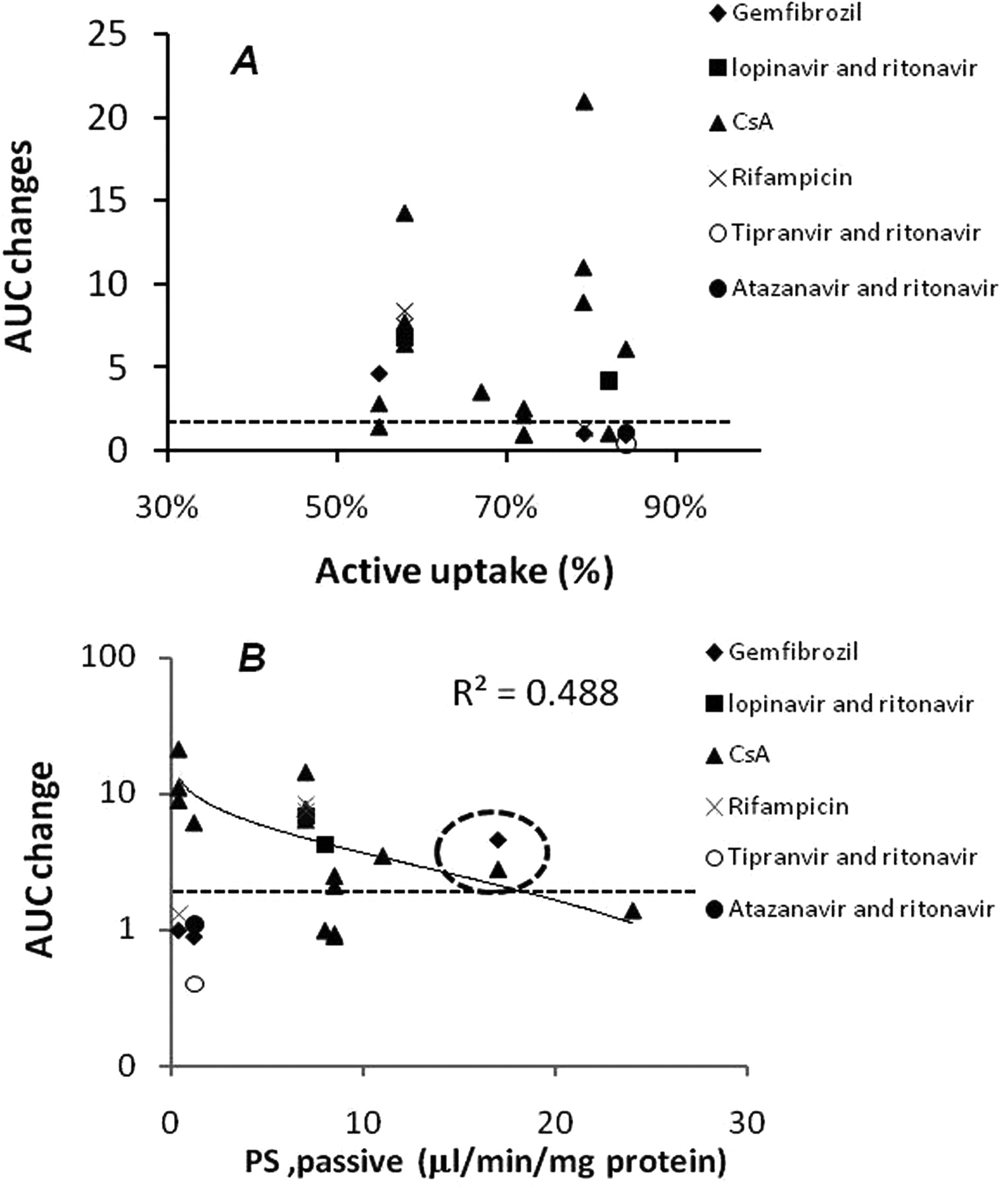
The correlation between AUC fold changes with OATP inhibitors observed in the clinic and the active uptake (A) or passive diffusion (PSpassive) (B) obtained from SCHHs with Rif SV (100 μM) inhibition. Dashed circle, AUC changes of cerivastatin with CsA or gemfibrozil. Dashed line, 2-fold AUC changes for clinical relevance.
Discussion
Adverse clinically significant DDIs represent major challenges in drug discovery and development. In the last two decades, preclinical in vitro or in vivo models have been used to effectively predict human pharmacokinetics. However, the predictions were successful for the compounds that are mainly eliminated renally or via cytochrome P450-metabolizing enzymes (Obach, 2009). Predicting hepatic transporter-mediated clearance continues to be challenging due to large interspecies differences in hepatic transporter homology or expression and a lack of validated in vitro tools (Lai, 2009). Hepatic drug elimination is composed generally of a series of processes that include: entrance into hepatocytes via passive diffusion and active uptake mediated by hepatic transporters; metabolic elimination through phase I and/or phase II enzymes; and excretion to bile and/or back to systemic circulation by the efflux transporters. To assess the processes involved in hepatic uptake and biliary excretion for in vivo prediction, the development of an in vitro model that mimics the complexity of the hepatic transport system has become imperative.
Human hepatocyte in vitro models are widely accepted as a valuable tool to investigate drug metabolic liability and gene induction and toxicity, as well as serve as effective screening tools for new molecular entities. The cellular polarity that allows vectorial transport in vivo is disrupted rapidly when cells are isolated from the intact organ. Therefore, the restoration of the bile canalicular network in the cultured hepatocyte model is desired to investigate the vectorial transport of drug candidates. SCHHs have been accepted generally as a good model to aid in the prediction of biliary clearance in humans (Bi et al., 2006). After the previous efforts to understand the expression of hepatobiliary efflux transporters in SCHHs (Li et al., 2009), in the present study, we report, for the first time, the maintenance in SCHHs of hepatic uptake transporters, OATP1B1, OATP1B3, and OATP2B1, as measured by LC-MS/MS. This provides molecular evidence to support SCHHs as a model to obtain functional hepatic uptake parameters for in vivo prediction. Moreover, to better define the uptake clearance in SCHHs, ideally an inhibitor that can block all of the known hepatic uptake transporters with a minimum effect on passive diffusion and biliary excretion is needed. To meet these criteria, we measured rosuvastatin uptake in SCHHs coincubated with several known OATP transporter inhibitors. Under the concentrations applied, these inhibitors can completely block OATP1B1 or OATP1B3 activity in OATP gene overexpression systems (Vavricka et al., 2002; Yamazaki et al., 2005; Letschert et al., 2006). As a result, Rif SV (100 μΜ) was shown to completely knock down rosuvastatin uptake in SCHHs. The inhibitor selected provided the ability to assess the sum of active hepatic uptake contributed by OATP transporters expressed in hepatocytes. In contrast, Rif SV (100 μΜ) had no effect on passive diffusion of buprenorphine and midazolam into hepatocytes. Rif SV also increased the intracellular accumulation of buprenorphine and midazolam through the inhibition of metabolizing enzymes, such as UGT1A1 and CYP3A4. These inhibitory effects were minimized through the optimization of experimental conditions by calculating the initial uptake rate to avoid the artificial effect on hepatic uptake caused by the inhibitory effects of Rif SV on metabolizing enzymes. Moreover, reduced biliary excretion is observed after the decreases in hepatic uptake. Because intracellular accumulation of the compounds was not observed (Fig. 2; Tables 3 and 4) with or without Ca2+, we speculate that reduced biliary excretion was caused by the inhibitory effects on hepatic uptake. However, direct inhibitory effects on hepatobiliary efflux transporters still remain to be investigated further, because Rif SV appears as to be an inhibitor of efflux transporters, including bile salt export pump (Wang et al., 2003).
Three isoforms (OATP1B1, OATP1B3, and OATP2B1) are considered to play a pivotal role in the hepatic uptake of xenobiotics and endogenous compounds on the sinusoidal membrane of hepatocytes (Giacomini et al., 2010). The OATPs transport drugs from a wide range of therapeutic classes, including 3-hydroxymethylglutaryl-coenzyme A reductase inhibitors (statins), angiotensin II receptor antagonists (e.g., olmesartan and valsartan), angiotensin-converting enzyme inhibitors (enalapril and temocaprilat), the H1 receptor antagonist fexofenadine, and the endothelin receptor antagonist bosentan (Giacomini et al., 2010). Because the OATP proteins are poorly conserved evolutionarily, as demonstrated by a lack of human orthologs in rodents, extrapolation of in vivo human pharmacokinetics from rodent models remains limited. The ITC recommends that the clinical investigation cascade should be initiated when active hepatic transport is involved in the liver clearance pathway and outlines decision trees for in vitro evaluation of hepatic uptake using a hepatocyte model to predict the potential risk of clinical DDIs. The investigation cascades are considered to be similar to in vitro studies of drug metabolism and interaction (Giacomini et al., 2010). As a result, developing in vitro models that can predict OATP-mediated DDIs is an efficient and inexpensive approach that could reduce or eliminate the need for further clinical investigation. However, the extent of in vitro active hepatic uptake necessary to trigger the clinical assessment of DDI was not provided (Giacomini et al., 2010). In addition, the performance of in vitro hepatocyte models should be evaluated through retrospective analysis to increase confidence in the outcomes. With this in mind, a literature review was conducted, and the compounds with reported OATP-mediated clinical DDIs were tested in our optimized SCHH model. As expected, active hepatic uptake of the compounds with clinically significant OATP DDIs was observed. The contribution of the active portion to overall hepatic uptake was high, being 55 to 84% of the total uptake rate. Because hepatocyte lot differences in transporter expression exist, it is important that we confirmed OATP expression in SCHHs and compared active hepatic uptake in multiple hepatocyte lots (Supplemental Table 1). These results suggest that 50% active hepatic uptake in SCHHs could be the in vitro cutoff to trigger the clinical risk assessment of hepatic transporter-mediated DDIs.
The relationship among solubility, passive permeability, and effects of drug transporter and metabolizing enzymes on drug disposition has been well addressed by the Biopharmaceutics Drug Disposition Classification System (Wu and Benet, 2005). As depicted in Fig. 3B, the fold increase of AUC tended to decrease as PSpassive in SCHHs increased. The results suggest that low-permeability drugs that rely primarily on transporter uptake for entry are prone to clinical DDIs mediated by OATP transporters. When the classification system is applied, transporter effects are predicted to be minimal for high-permeability/high-solubility class 1 compounds (Wu and Benet, 2005). In the gastrointestinal space, good solubility is essential for the saturation of efflux transporters to obtain good absorption. After the compounds are absorbed (into the systemic space), high-permeability/low-solubility class 2 compounds could behave similarly to class 1 compounds, in that the high permeability can overcome the transporter effects and therefore reduce the risk of transporter-mediated DDIs by competing with drugs entering into hepatocytes. This simple categorization under the Biopharmaceutics Drug Disposition Classification System suggests that the high- versus low-permeability designation reflects the differences in freedom of access to phase I and/or phase II metabolizing enzymes within hepatocytes. In addition to the factors described here, hepatic blood flow needs to be considered, because under flow-limited conditions the impacts of inhibition of uptake transport could be reduced.
It is interesting to note that atorvastatin is categorized as class 2 in the previous report (Wu and Benet, 2005). However, atorvastatin was shown to have limited diffusion into SCHHs in the present study and could be categorized as a class 4 compound (9.3 μl · min−1 · mg−1 atorvastatin−1 versus 91 μl · min−1 · mg−1 propranolol−1; data not shown). In this regard, additional investigations are proposed to better understand the passive diffusion cutoff using a cellular uptake model (e.g., SCHHs). Some exceptions also were found in the relationship between passive permeability and AUC increase observed in the clinic. Although cerivastatin was a highly permeable compound in SCHHs (15 μl · min−1 · mg−1), significant AUC increases (4.8- and 5.6-fold) were reported when cerivastatin was coadministered with the OATP inhibitors CsA and gemfibrozil, respectively (Fig. 3B, dashed circle). Cerivastatin interacted with OATP proteins and also is metabolized extensively by CYP2C8 and CYP3A4 in liver via demethylation of the benzyl methyl ether moiety (Mück, 2000). CsA and gemfibrozil are potent inhibitors of OATP transporters, as well as inhibiting CYP metabolizing enzymes. These clinical observations could be explained by the inhibition of multiclearance pathways resulting in a sum of effects on overall hepatic clearance. Due to a lack of exposure data, it also is worthwhile to note that outliers were anticipated with the coadministration of CsA, because the inhibition of OATP transporters may have been insufficient in vivo (Fig. 3, below the dashed line). Collectively, caution should be raised in that the interplay between transporters and metabolizing enzymes could generate multilevel complexity in predicting clinical DDI from an in vitro system.
In conclusion, SCHHs maintain hepatic transporter expression and functional activity and appear to be a well characterized an in vitro model for the prediction of in vivo human pharmacokinetics. Rif SV inhibited the active uptake of OATP transporters, demonstrating that it was an OATP inhibitor useful for estimating the extent of active and passive uptake in SCHHs. The optimization of the experimental design minimized the impacts of inhibitory effects on metabolizing enzymes. Retrospective analysis for the compounds that undergo clinically significant hepatic transporter-mediated DDIs suggested that 50% or greater active hepatic uptake in SCHHs with low passive permeability would have a potential risk of clinical DDIs and that this value could serve as a cutoff to trigger the clinical investigation cascade for DDI risk assessment.
Authorship Contributions
Participated in research design: Bi, Kimoto, Jones, Barton, and Lai.
Conducted experiments: Bi, Kimoto, Sevidal, Whalen, and Zhang.
Performed data analysis: Bi, Kimoto, Sevidal, Jones, Barton, Kempshall, Whalen, Zhang, Ji, Fenner, El-Kattan, and Lai.
Wrote or contributed to the writing of the manuscript: Bi, Kimoto, Sevidal, Jones, Barton Kempshall, Whalen, Zhang, Ji, Fenner, El-Kattan, and Lai.
Acknowledgments
We thank Dr. Larissa M. Balogh for the help with OATP protein quantification and the editing of manuscript; Andrea Clouser-Roche for bioanalytical support; and Drs. Theodore E. Liston, Larry M. Tremaine, Bo Feng, Manthena V. Varma, John Litchfield, and Hendrik Neubert for helpful scientific comments and suggestions.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.ABBREVIATIONS:
- OATP
- organic anion-transporting polypeptide
- AUC
- area under the curve
- DDI
- drug-drug interaction
- HBSS
- Hanks' balanced salt solution
- HPLC
- high-performance liquid chromatography
- ITC
- International Transporter Consortium
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- Rif SV
- rifamycin SV
- SCHH
- sandwich-cultured human hepatocyte
- SCRH
- sandwich-cultured rat hepatocyte
- SIL
- stable isotope label
- TC
- taurocholate.

- Received October 28, 2011.
- Accepted March 1, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}