


Abstract
New chemical entities often exhibit nonlinear pharmacokinetics (PK) profiles in experimental animals. However, the number of studies that have focused on species differences in nonlinear PK is very limited; thus, the aim of this study was to clarify the mechanism of the nonlinear PK of E2074 (2-[(2R)-2-fluoro-3-{(3r)-[(3-fluorobenzyl)oxy]-8-azabicyclo[3.2.1]oct-8-yl}propyl]-4,5-dimethyl-2,4-dihydro-3H-1,2,4-triazol-3-one), a novel sodium channel inhibitor, in rats, dogs, and monkeys. Nonlinear PK profiles with more than dose-proportional increases of Cmax and area under the plasma concentration curve were observed in all species after oral administration. The Michaelis–Menten constant (Km) values of hepatic microsomal metabolism were 7.23 and 0.41 μM in rats and dogs in vitro, respectively, which were lower than the unbound maximum plasma concentrations after oral administration in vivo, indicating that the nonlinear PK in rats and dogs was attributable to the saturation of hepatic metabolism. However, we do not believe that the saturation of hepatic metabolism was the mechanism of nonlinearity in monkeys because of the high Km value (42.44 μM) observed in liver microsomes. Intestinal metabolism was observed in monkey intestinal microsomes but not in rats and dogs, and the nonlinear PK in monkeys was diminished by inhibition of intestinal metabolism with a concomitant oral dose of ketoconazole. These results suggest that saturation of the intestinal metabolism is the potential mechanism of nonlinearity in monkeys. P-glycoprotein was not involved in the nonlinear PK profiles in any species. In conclusion, the mechanism of the nonlinear PK of E2074 is species dependent, with the saturation of hepatic metabolism in rats and dogs and that of intestinal metabolism in monkeys being the primary cause.
Introduction
In the drug discovery process, the prediction of the human pharmacokinetics (PK) profiles of new chemical entities is routinely performed at the preclinical stage. The predicted PK profile provides useful information about the clinical dosing regimen, the PK/pharmacodynamic analysis, and the estimation of safety margins. Assessment of the nonlinear PK profiles of new chemical entities and the prediction of PK profiles are important because nonlinearity occasionally results in unexpected adverse effects. For example, as phenytoin exhibits a nonlinear PK profile in the therapeutic range, a slight increase of the dose leads to remarkable escalation in plasma phenytoin levels, resulting in unexpected adverse effects (Richens, 1975; Pitner et al., 1998). Nonlinear PK profiles are often observed in preclinical animal studies, particularly in toxicologic studies, because a wide range of doses and relatively high doses are administered. This nonlinearity makes assessment of dose–toxicity and dose–efficacy relationships difficult.
Nonlinearity is caused by several different mechanisms (Ludden, 1991; Lin, 1994). Less than dose-proportional increases in exposure with oral doses could be caused by decreased drug absorption. Most drugs are lipophilic and are absorbed by passive diffusion; thus, less than dose-proportional increases in exposure could be caused by limited solubility (Chapelsky et al., 1998). Decreased absorption could also be due to the saturation of carrier-mediated intestinal transport. For example, gabapentin exhibits less than dose-proportional increases in exposure at high doses due to saturation of the active transport responsible for absorption from the intestinal lumen (L-type amino acid transporter) (Su et al., 2005). For carbapenem antibiotic (MK-826), the saturation of plasma protein binding also contributes to its nonlinear PK profile by increasing clearance, resulting in less than dose-proportional increases in exposure (Wong et al., 1999). A greater than dose-proportional increase in exposure is most likely due to decreased clearance. Phenytoin, salicylate, and theophylline are the classic examples of drugs that exhibit decreased clearance due to saturated hepatic metabolism (Ludden, 1991; Lin, 1994).
Decreased biliary clearance is reported to be another cause of nonlinear PK profiles in rats. Methotrexate is mainly excreted in bile, and reductions in its biliary clearance result in nonlinear PK profiles with disproportional increases in exposure (Bremnes, et al., 1989). The decrease in biliary excretion is caused by saturation of a transporter on the bile canalicular membrane. This transporter was confirmed to be multidrug resistance-associated protein 2 (MRP2) using Eisai hyperbilirubinemic rats, which are MRP2-deficient (Masuda et al., 1997). In addition, P-glycoprotein (P-gp) expressed on the brush border membrane of enterocytes can act as an efflux pump that limits intestinal absorption. Talinolol, a good substrate for P-gp, displays a nonlinear PK profile due to the saturation of P-gp transport activity in therapeutic dose ranges in healthy subjects (Bolger et al., 2009).
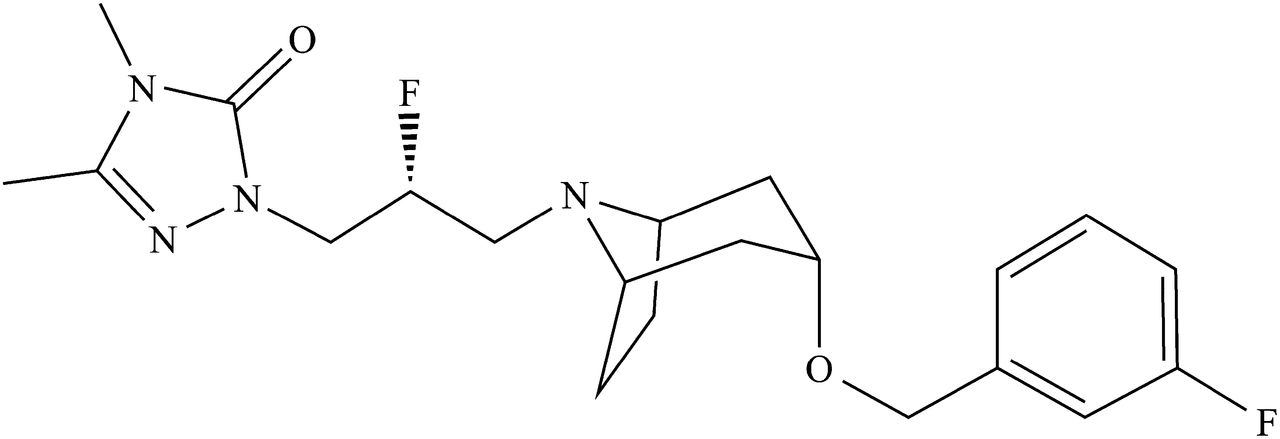
As mentioned previously, many reports have attempted to elucidate the mechanisms responsible for nonlinear PK profiles, but these studies describe the nonlinearity of specific drugs in a specific species. Reports about species differences in the mechanisms of nonlinearity are very limited (Sugimoto et al., 1999; Roller et al., 2009). The purpose of this study was to clarify the determinant factor of the mechanism of nonlinearity in different species. E2074, 2-[(2R)-2-Fluoro-3-{(3r)-[(3-fluorobenzyl)oxy]-8-azabicyclo[3.2.1]oct-8-yl}propyl]-4,5-dimethyl-2,4-dihydro-3H-1,2,4-triazol-3-one (Fig. 1), a novel sodium channel inhibitor, was used as a model compound in this study because nonlinear PK profiles with more than dose-proportional increases in exposure were observed in preclinical species. Because one of the presumed causes of nonlinearity is decreased metabolic clearance, kinetic parameters—the Michaelis–Menten constant (Km) and maximum velocity (Vmax)—were estimated using liver microsomes to assess the possibility of saturated hepatic metabolism. In addition to hepatic metabolism, the impact of intestinal metabolism on nonlinearity was also verified using intestinal microsomes in vitro and PK analysis with ketoconazole coadministration in vivo. E2074 is a P-gp substrate according to the in vitro data, and the effect of P-gp on its absorption was also assessed using mdr1a/1b knockout (KO) mice.
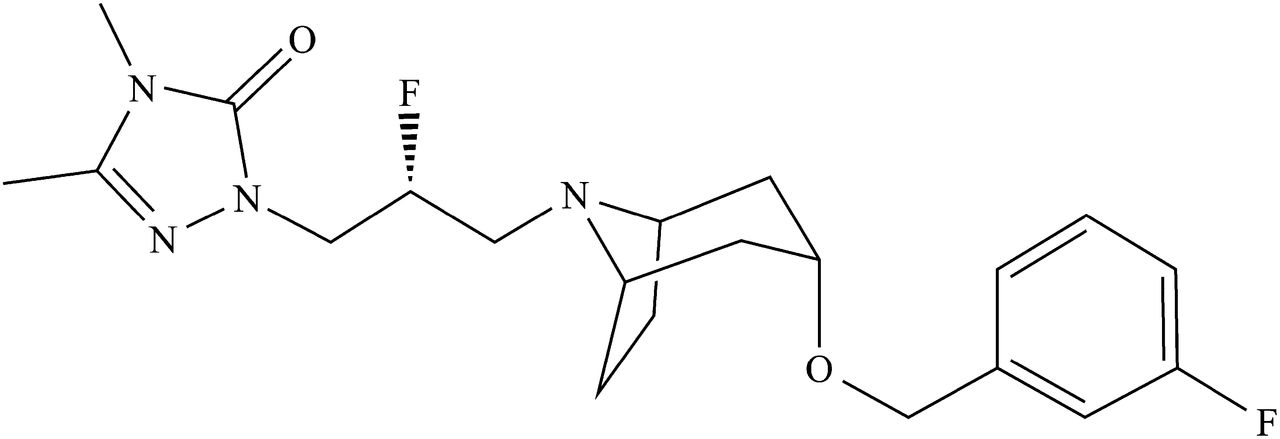
Chemical structure of E2074.
Materials and Methods
Chemicals.
E2074 was synthesized by Eisai Co. Ltd. (Ibaraki, Japan). Ketoconazole (KCZ), loperamide hydrochloride, and propranolol hydrochloride were purchased from Sigma Aldrich (St. Louis, MO). Pooled rat liver microsomes [Sprague-Dawley (SD) rats, male, 8 weeks old], dog liver microsomes (beagle, male, 0.5–3.0 years old), monkey liver microsomes (cynomolgus, male, 4–10 years old), rat intestinal microsomes (SD, male, 8 weeks old), dog intestinal microsomes (beagle, male, more than 6 months old), and monkey intestinal microsomes (cynomolgus, male, 4–8 years old) were purchased from XenoTech (Lenexa, KS). Recombinant human cytochrome P450 (rP450) was obtained from BD Gentest Products and Service (Woburn, MA). All other reagents and solvents were of analytical grade.
Animals.
Male SD rats were supplied by Charles River Laboratories Japan (Kanagawa, Japan). SD rats with jugular and portal vein double cannulation were provided by Charles River Laboratories Japan. Male cynomolgus monkeys were obtained from Guangxi Grandforest Scientific Primate Company, Ltd. (Guangxi, China). Male beagles were supplied by Narc Corporation (Chiba, Japan). Mdr1a/1b KO and wild-type (WT) mice were purchased from Taconic (Germantown, NY). All experimental protocols were approved by the Institutional Animal Care and Use Committee of Eisai.
In Vivo PK Studies.
For the PK of E2074 in rats (i.v.: 1 mg/1 ml/kg; oral: 3, 10, and 30 mg/5 ml/kg), dogs (i.v.: 1 mg/0.5 ml/kg; oral: 3 and 10 mg/3 ml/kg), monkeys (i.v.: 1 mg/0.5 ml/kg; oral: 3 and 10 mg/3 ml/kg), and mice (oral: 3 mg/10 ml/kg), the dosing solution was 5% glucose/1 M HCl (97:3, v/v). The oral dosing solution of KCZ was 5% glucose/1 M HCl (95:5, v/v). In the case of concomitant administration of E2074 and KCZ in monkeys, KCZ (5 mg/4 ml/kg) was administrated orally immediately before E2074 administration. The dog and monkey studies were conducted using a crossover design. Blood samples (0.25–0.5 ml) were collected using heparinized syringes via the jugular (rats) or cephalic vein (dogs and monkeys) at 5 (only for i.v.), 15, and 30 minutes, and 1, 2, 4, 6, and 8 hours after dosing. For double-cannulated rats, blood samples were collected using heparinized syringes through the jugular and portal vein cannulae at 5, 20, and 40 minutes, and 1, 1.5, 2, and 4 hours after dosing. For mice, blood samples (approximately 20 μl) were collected using heparinized capillaries through the tail vein at 15 and 30 minutes, and 1, 2, 4, 6, and 8 hours after dosing. Plasma was obtained by centrifugation at 12,000g for 5 minutes at 4°C (Kubota 1910; Kubota Co., Ltd., Tokyo, Japan). Aliquots of plasma (rat, dog, and monkey: 50 μl; mouse: 10 μl) were transferred to polypropylene tubes and stored at –15°C until analysis. To estimate brain penetration, mice were decapitated at 30 minutes after administration, and brain tissue was obtained. The brain tissue was stored at –15°C and homogenized with distilled water to prepare 20% (w/v) homogenates before analysis.
In Vitro Metabolic Studies.
To estimate the enzyme kinetic parameters in liver and intestinal microsomes, the reaction solution comprising 0.1 mM EDTA, 100 mM phosphate buffer (pH 7.4), 25 µl of NADPH-generating system, E2074 (0.025, 0.074, 0.25, 0.74, 2.5, 7.4, 24.6, 49.2, and 73.8 μM), and liver or intestinal microsomes for each animal (0.5, 0.2, and 1 mg/ml for rat/ monkey liver, dog liver, and monkey intestine, respectively) in a final volume of 250 µl was used. The NADPH-generating system was prepared as a mixture containing 3.3 mM β-NADP+, 80 mM glucose 6-phosphate, 60 mM MgCl2, and 1 unit/ml glucose 6-phosphate dehydrogenase. After the reaction solution lacking the NADPH-generating system was preincubated at 37°C for 5 minutes, the reaction was initiated by adding the NADPH-generating system or 60 mM MgCl2, and the mixture was incubated at 37°C for 7, 30, and 15 minutes for rat, dog, and monkey microsomes, respectively. Each sample was prepared in duplicate.
In the rP450s assays, the reaction solution consisted of 0.1 mM EDTA, 100 mM phosphate buffer (pH 7.4), 25 µl of NADPH-generating system, E2074 (0.25 μM), and each rP450 isoform (25 pmol P450/ml) in a final volume of 250 µl. After the reaction solution lacking the NADPH-generating system was preincubated at 37°C for 5 minutes, the reaction was initiated by adding the NADPH-generating system, and the mixture was used immediately or incubated at 37°C for 30 minutes. Each sample was prepared in duplicate.
Protein Binding and Blood-to-Plasma Partition Ratio.
The in vitro unbound fraction in plasma (fp) and liver microsomes (fmic) was estimated using equilibrium dialysis. E2074 was added to plasma and liver microsomes (0.5 and 0.2 mg/ml for rat/monkey and dog microsomes, respectively) at a concentration of 0.25 μM. We added 1 ml of the matrix to the donor side of a dialysis chamber, and 1 ml of phosphate-buffered saline (pH 7.4) was added to the receiver side chamber. The dialysis chamber was maintained at 37°C (plasma) or 4°C (liver microsomes) for 24 hours. The incubated samples were collected from both sides of the chamber and stored at −15°C until analysis.
We calculated fp and fmic as the receiver side to donor side concentration ratio. To estimate the blood to plasma partition ratio (Rb), E2074 was added to blood at a concentration of 0.25 μM in a final volume of 0.5 ml. After the blood was incubated at 37°C for 10 minutes, an aliquot of the blood sample was collected, and the remaining sample was centrifuged to obtain plasma. The blood and plasma samples were stored at −15°C until analysis. Rb was calculated from the blood and plasma concentrations.
Transport Assay Using Mouse mdr1a-Overexpressing Cell Line.
We purchased mdr1a-expressing (porcine kidney epithelial LLC-PK1 cells transformed with mouse mdr1a cDNA) and control cells (LLC-PK1 cells) from the Netherlands Cancer Institute (Amsterdam, Netherlands). Cells were seeded at a density of 4 × 105 cells/cm2 on a filter well in cell culture insert plates (Millicell-24 Cell Culture Plate PET, pore size 1 μm, membrane area 0.7 cm2; EMD Millipore, Billerica, MA) and incubated in a CO2 incubator (37°C, 5% CO2) for 4 days. Medium199 containing 10% fetal bovine serum, 50 units/ml penicillin, and 50 μg/ml streptomycin was used for cell culture. On the day of the experiment, cells were washed with 10 mM HEPES/Hanks’ balanced salt solution (HBSS) (pH 7.4) before the transport assay. In the plates seeded with cells, both the apical and basal sides were preincubated with 10 mM HEPES/HBSS for 2 hours at 37°C. Then, the solution on the apical or basal side was replaced with 10 mM HEPES/HBSS containing E2074 or loperamide as a positive control (1 μM). After the cells were incubated for 2 hours at 37°C, 100 μl of the medium were collected from the compartment opposite of the one spiked with E2074.
Extraction Procedure.
Plasma, blood, microsomal samples, and medium samples were deproteinized with acetonitrile containing 0.3 μM propranolol as an internal standard (IS) followed by centrifugation. The obtained supernatant was filtrated, and then the filtrate was analyzed by high-performance liquid chromatography with tandem mass spectrometry (LC/MS/MS).
Analytical Procedure.
The concentration of E2074 was measured using a Waters LC/MS/MS system (Milford, MA). The ionization mode was positive electrospray ionization. Chromatography was performed using an Acquity UPLC BEH C18 column (1.7 μm, 2.1 mm i.d. × 50 mm; Waters). The mobile phases were water containing 0.1% formic acid (A) and acetonitrile containing 0.1% formic acid (B). The initial condition was 100% (A)/0% (B), and (B) was increased linearly to 50% over 3 minutes. The flow rate was 0.3 ml/min. The injection volume of the extracted sample was 5 μl. The selected ion monitoring conditions were as follows:
PK Analysis.
The PK parameters of E2074, excluding the maximum concentration in plasma (Cmax) and time to reach Cmax (tmax), were determined by model-independent analysis in WinNonlin (ver 6.1; Pharsight Corporation, St. Louis, MO). Cmax and tmax were taken from the observed values. Bioavailability (BA) was calculated according to the following equation:
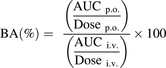 (1)
(1)Data Analysis.
The peaks of E2074 and IS in samples were integrated using MassLynx (Waters). The peak area ratio (PAR) of E2074 to IS was calculated, and the residual ratio was calculated using the following equation: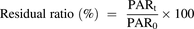 (2)where PARt represents the mean PAR after incubation and PAR0 represents the mean PAR at 0 minutes.
(2)where PARt represents the mean PAR after incubation and PAR0 represents the mean PAR at 0 minutes.
The enzyme kinetic parameters in rat and monkey microsomes were calculated from the Michaelis–Menten equation with a Hill coefficient for rats and monkeys or biphasic components for dogs as follows: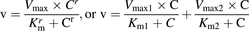 (3)where v is the metabolic velocity of E2074, C is the E2074 concentration, and Km, Vmax, and r are the Michaelis–Menten constant, maximum velocity, and Hill coefficient, respectively. The v value was directly calculated from the following equation: v = ke/microsomal concentration × C, where ke is the elimination rate constant calculated by the residual ratio and incubation time at the concentration C. The data set of v and C was used to estimate Km and Vmax values using the aforementioned Michaelis–Menten equation. Fitting was performed using SCIENTIST (Version 2.01; MicroMath Research, St. Louis, MO). The intrinsic clearance (CLint,u) was estimated by the following equation:
(3)where v is the metabolic velocity of E2074, C is the E2074 concentration, and Km, Vmax, and r are the Michaelis–Menten constant, maximum velocity, and Hill coefficient, respectively. The v value was directly calculated from the following equation: v = ke/microsomal concentration × C, where ke is the elimination rate constant calculated by the residual ratio and incubation time at the concentration C. The data set of v and C was used to estimate Km and Vmax values using the aforementioned Michaelis–Menten equation. Fitting was performed using SCIENTIST (Version 2.01; MicroMath Research, St. Louis, MO). The intrinsic clearance (CLint,u) was estimated by the following equation: (4)The predicted hepatic blood clearance (CLh,vitro) was calculated from CLint,u by the dispersion model with a dispersion number of 0.17 (Roberts and Rowland, 1986a,b). The hepatic blood flow rate (55.2, 30.9, and 43.6 ml/min/kg for rats, dogs, and monkeys, respectively), liver weight (40, 32, and 30 g/kg for rats, dogs, and monkeys, respectively), and microsomal protein (45 mg/g liver) were used as scaling factors (Davies and Morris, 1993; Marathe et al., 2010).
(4)The predicted hepatic blood clearance (CLh,vitro) was calculated from CLint,u by the dispersion model with a dispersion number of 0.17 (Roberts and Rowland, 1986a,b). The hepatic blood flow rate (55.2, 30.9, and 43.6 ml/min/kg for rats, dogs, and monkeys, respectively), liver weight (40, 32, and 30 g/kg for rats, dogs, and monkeys, respectively), and microsomal protein (45 mg/g liver) were used as scaling factors (Davies and Morris, 1993; Marathe et al., 2010).
For P-gp susceptibility, the apparent permeability (Papp) of E2074 across the cell monolayer was calculated using the following equation: (5)where the incubation time was 2 hours and the membrane area was 0.3 × 106 cm2.
(5)where the incubation time was 2 hours and the membrane area was 0.3 × 106 cm2.
Results
Pharmacokinetics of E2074 in Rats, Dogs, and Monkeys.
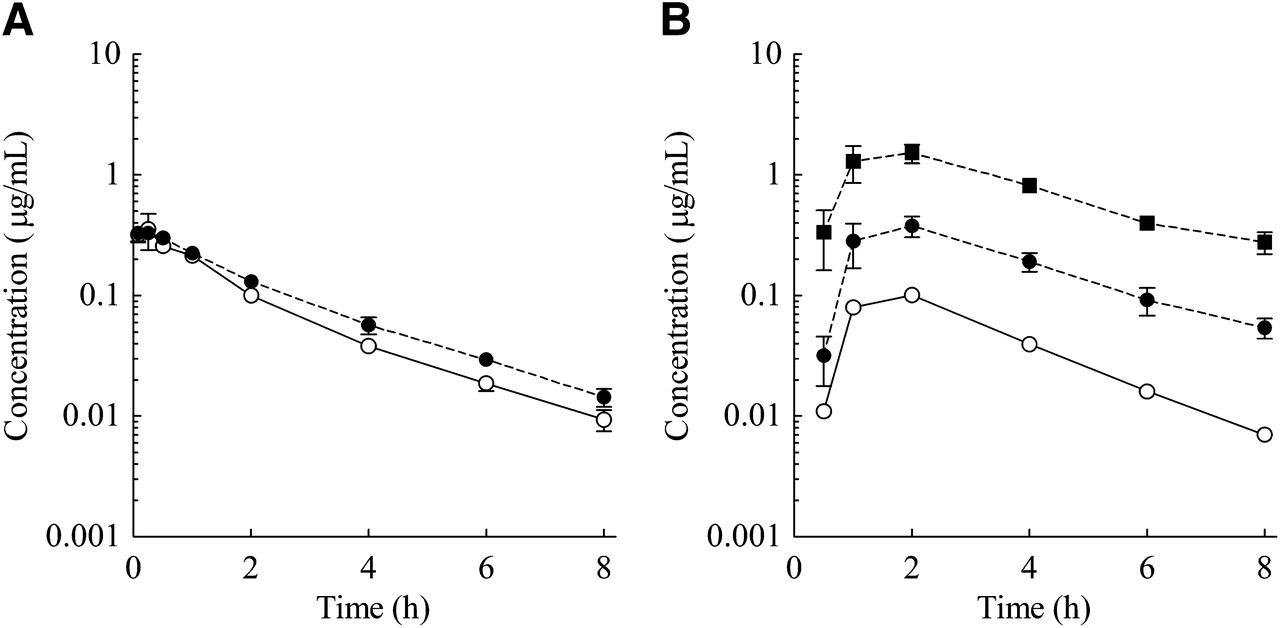
The plasma concentration–time profiles and PK parameters are shown in Fig. 2 and Table 1, respectively. After i.v. dosing at 1 mg/kg, E2074 was characterized in all species by moderate to high plasma clearance (3.21 l/h per kg in rats, 1.70 l/h per kg in dogs, and 1.35 l/h per kg in monkeys) and a moderate volume of distribution (2.54 l/kg in rats, 1.84 l/kg in dogs, and 3.98 l/kg in monkeys). The half-life (t1/2) of the drug was 0.85 hours in rats, 1.04 hours in dogs, and 3.18 hours in monkeys. After oral administration to rats at 3, 10, and 30 mg/kg, the area under the plasma concentration curve (AUC) and Cmax increased more than dose proportionally, and the resultant BA increased from 2.6 to 15.1%. In addition, in dogs and monkeys after oral administration at 3 and 10 mg/kg, the AUC and Cmax increased more than dose proportionally, and the resultant BA increased from 1.2 to 43.7% in dogs and from 7.8 to 20.9% in monkeys.
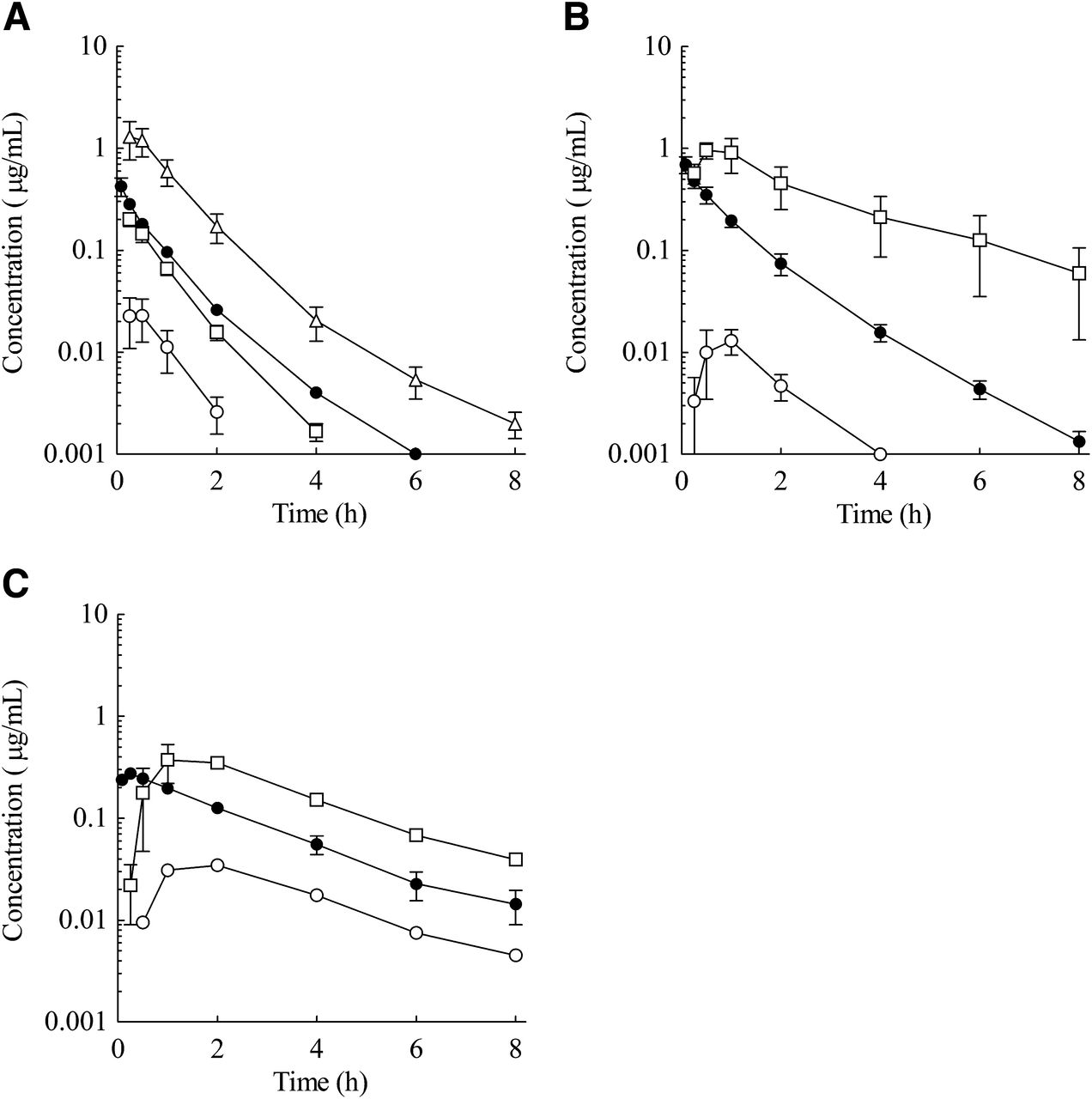
Plasma concentration–time profiles of E2074 in rats (A), dogs (B), and monkeys (C) (●, i.v. 1 mg/kg; ○, oral 3 mg/kg; □, oral 10 mg/kg; △, oral 30 mg/kg). Each point represents the mean ± S.E. of five rats for the rat oral 3 mg/kg dose; the monkey oral 3 mg/kg dose represents the mean of two animals, and the other represents the mean of three animals.
Pharmacokinetic parameters of E2074 in rats, dogs, and monkeys
The pharmacokinetic parameters represent the mean ± S.E. of three animals or five rats, or the mean of two monkeys.
In Vitro Metabolism.
E2074 was metabolized by liver microsomes in all species tested. Monkey intestinal microsomes, but not rat or dog intestinal microsomes, displayed E2074 metabolic activity. The enzyme kinetic parameters of E2074 for hepatic and intestinal metabolism were estimated in rat, dog, and monkey liver microsomes and monkey intestinal microsomes (Fig. 3; Table 2). The hepatic metabolism of E2074 in rats and monkeys was typified by a sigmoidal Michaelis–Menten reaction with a Hill coefficient. The Km, Vmax, and r values were 7.23 μM, 1.42 nmol/min per mg protein, and 1.10, respectively, for rats and 42.44 μM, 3.58 nmol/min/mg protein, and 1.04, respectively, for monkeys. The metabolism in dogs was represented by a Michaelis–Menten reaction with biphasic components. The Km and Vmax values were 0.41 μM and 0.07 nmol/min per mg protein, respectively, for high-affinity component and 21.5 μM and 0.72 nmol/min per mg protein, respectively, for the low-affinity component. In microsomes, fmic was in unity for all species tested. The fp values were 0.675, 0.601, and 0.676 in rats, dogs and monkeys, respectively, and the corresponding Rb values were 0.89, 0.86, and 1.01, respectively (Table 3). The CLint,u values calculated from Km and Vmax were 0.196, 0.208, and 0.084 ml/min per mg protein in rats, dogs, and monkeys, respectively, and the corresponding CLh,vitro values were 3.19, 1.83, and 2.00 l/h/kg, respectively (Table 3). The intestinal metabolism in monkeys displayed a classic Michaelis–Menten pattern (r = 1) with a Km of 34.60 μM and a Vmax of 0.66 nmol/min per mg protein. The P450 enzyme involved in E2074 metabolism was evaluated by rP450s, and CYP3A4 was revealed to play a major role (Fig. 4).

Michaelis–Menten (A, B, and C) and Eadie–Hofstee plots (D, E, and F) of E2074 metabolism in rat (A and D), dog (B and E), and monkey (C and F) liver and intestinal microsomes (●, observed value of liver microsomes; ○, observed value of intestinal microsomes; solid lines, fitting curve of liver microsomes; dotted line, fitting curve of intestinal microsomes).
Kinetic parameters of E2074 in liver and intestinal microsomes
The figures in parentheses are the standard deviation of estimates.
Estimation of hepatic clearance of E2074 by using the dispersion model
Pharmacokinetic parameters of E2074 in monkeys with or without concomitant oral dose of KCZ
The monkeys were different individuals than those found in Table 1. The KCZ dose was 5 mg/kg orally. The pharmacokinetic parameters represent the mean ± S.E. (three monkeys) or mean (two monkeys). The figures in parentheses are the ratio without KCZ in the same dosing route.

Comparison of E2074 metabolic activity in recombinant human P450s. The protein concentration of each P450 was 25 pmol/ml, and the E2074 concentration was 0.25 μM. The data are presented as the mean of two experiments.
Portal Vein Concentration in Double-Cannulated Rats.
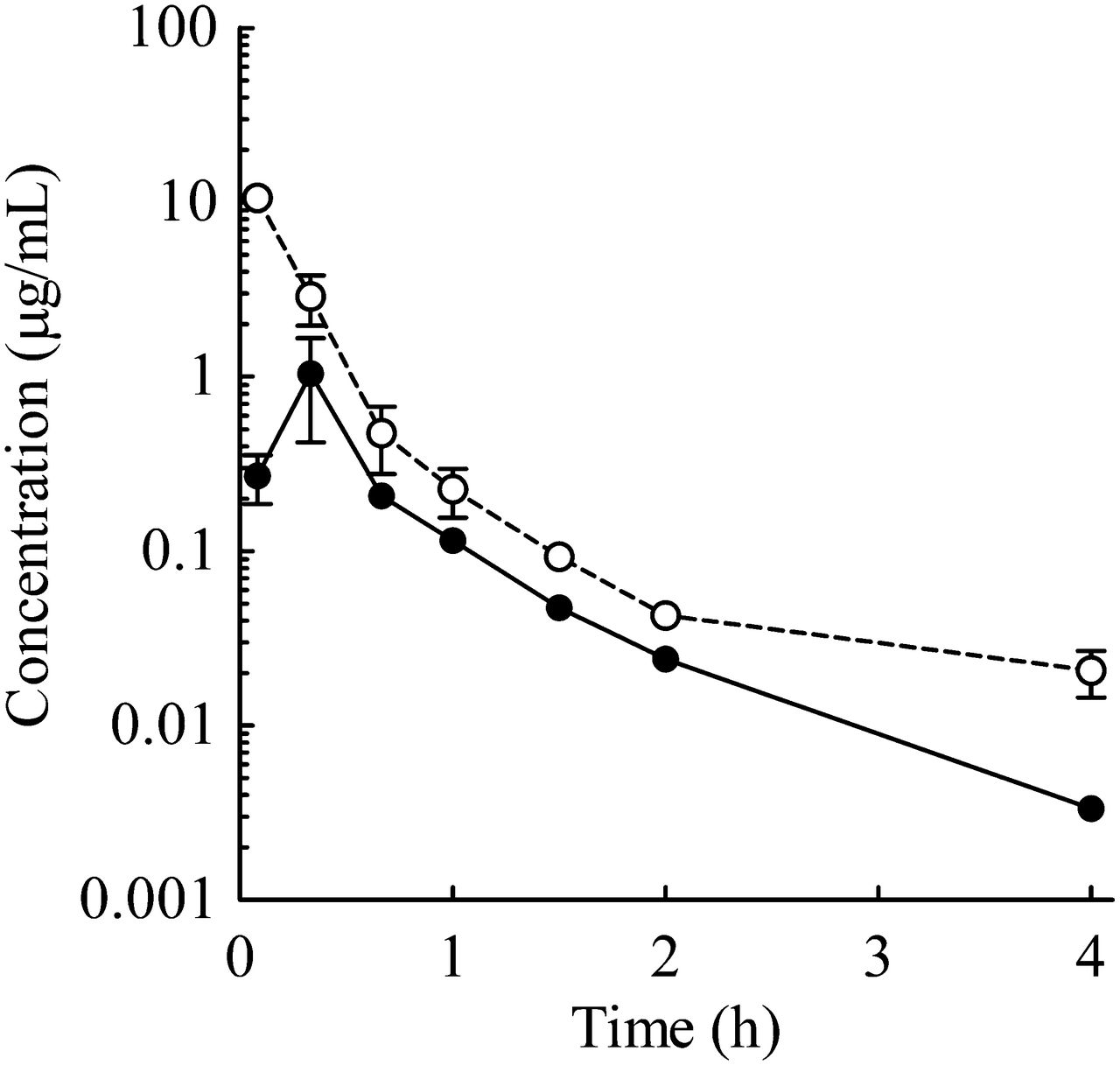
The Cmax values of the portal and jugular veins were 10.6 ± 1.4 and 1.04 ± 0.54 μg/ml, respectively, at an oral dose of 10 mg/kg in double-cannulated rats (Fig. 5).

Plasma concentration–time profiles of E2074 (oral 10 mg/kg) in cannulated rats (○, portal vein concentration; ●, systemic concentration). Each point represents the mean ± S.E. of three rats. The Cmax values of the portal vein and jugular vein were 10.6 ± 1.4 and 1.04 ± 0.54 μg/ml, respectively.
Inhibition of Intestinal Metabolism in Monkeys.
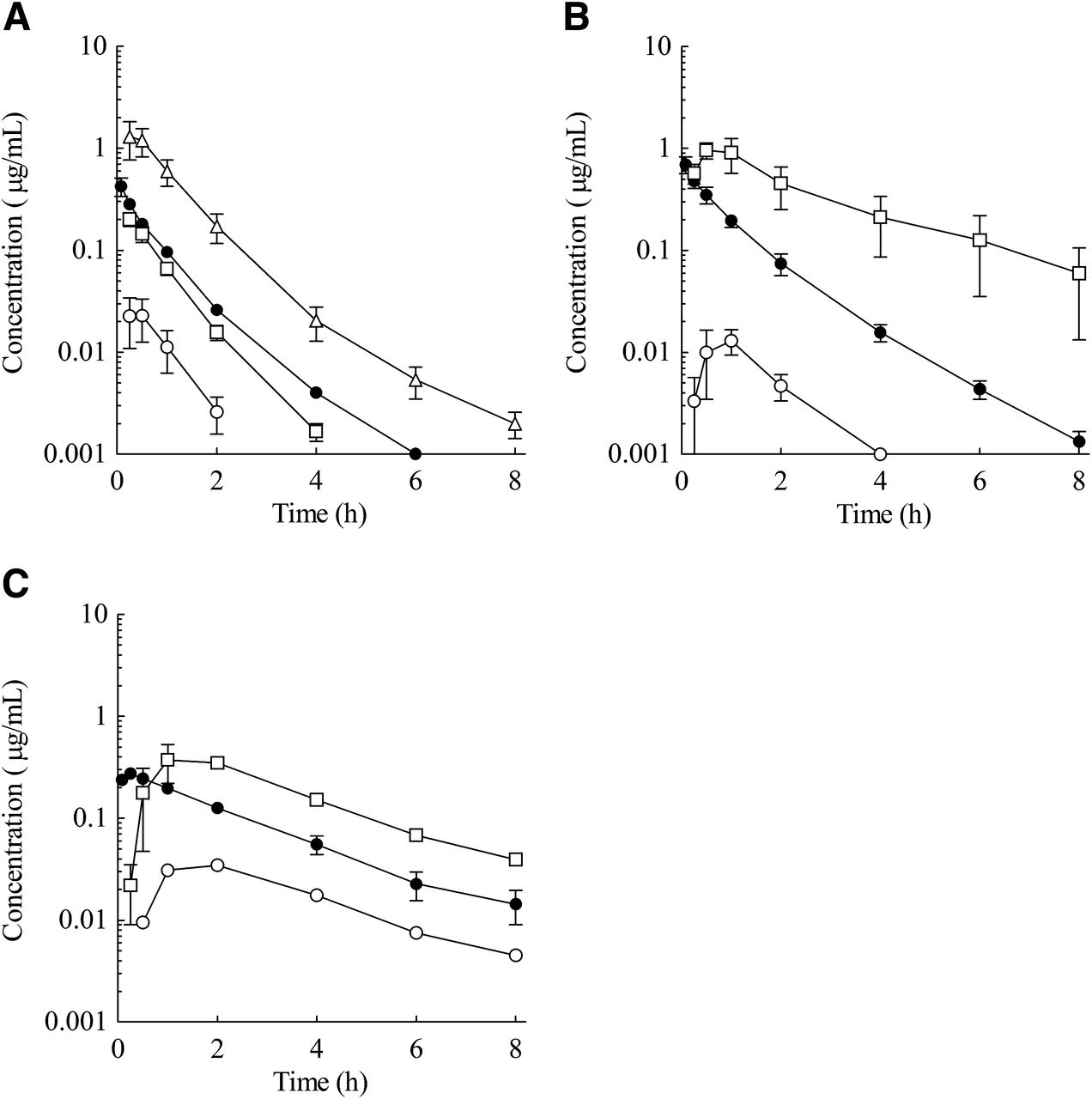
As CYP3A4 was shown to be the major metabolic enzyme among human rP450s tested, the effect of CYP3A4 inhibition on the PK profile of E2074 was evaluated by concomitant oral administration of KCZ (5 mg/kg) to investigate the contribution of intestinal metabolism of E2074 in monkeys. For i.v. administration, the PK profile of E2074 was not changed by KCZ (Fig. 6A). The t1/2, clearance (CL), and Vss values were 1.93 hours, 1.47 l/h/kg, and 3.07 l/kg, respectively, without KCZ and 1.96 hours, 1.22 l/h/kg, and 3.29 l/kg, respectively, with KCZ (Table 4). Conversely, KCZ coadministration significantly affected the oral PK profile of E2074 (Fig. 6B). At a dose of 3 mg/kg, Cmax and AUC were increased by 3.9-fold and 4.6-fold, respectively, by concomitant KCZ administration, but no remarkable change was observed for t1/2 (1.59 and 2.20 hours without or with KCZ, respectively; Table 4). The clear nonlinear PK profile of E2074 was demonstrated in monkeys at oral doses of 3–10 mg/kg (Table 1); however, in the case of KCZ coadministration, dose-proportional increases of Cmax and AUC with similar dose normalized values were found in the same dose range (Table 4).

Plasma concentration–time profiles of E2074 in monkeys with or without concomitant oral KCZ administration. (A) E2074 concentrations after i.v. administration at 1 mg/kg (○, without KCZ; ●, with KCZ), (B) E2074 concentrations after oral administration (○, oral 3 mg/kg without KCZ; ●, oral 3 mg/kg with KCZ; ▪, oral 10 mg/kg with KCZ). The concomitant oral dose of KCZ was 5 mg/kg. Each point represents the mean ± S.E. of three animals; for oral 3 mg/kg without KCZ, each point represents the mean of two animals.
Comparing pharmacokinetic parameters of E2074 between WT and mdr1a/1b KO mice
E2074 dose was 3 mg/kg orally, and the pharmacokinetic parameters represent the mean ± S.E. of three mice.
Susceptibility to Mouse P-gp.
To evaluate the P-gp susceptibility of E2074, transcellular transport was assessed using a mouse P-gp–overexpressing cell line. The apical to basal and basal to apical permeabilities were 9.67 × 10−6 and 30.40 × 10−6 cm/s, respectively, resulting in a (basal to apical)/(apical to basal) ratio (flux ratio) of 3.14, compared with a flux ratio of 9.00 for the positive control loperamide. These findings indicated that E2074 was a P-gp substrate.
Pharmacokinetics in Mdr1a/1b KO Mice.
The PK profile of E2074 was evaluated after oral administration at 3 mg/kg to mdr1a/1b KO and WT mice. The PK profile was similar between KO and WT mice (Fig. 7). The Cmax values for WT and KO mice were 0.11 ± 0.01 and 0.10 ± 0.02 μg/ml, respectively, and the corresponding AUCs were 0.23 ± 0.00 and 0.24 ± 0.04 µg·h/ml, respectively (Table 5). The brain to plasma concentration ratios (Kp,brain) at 30 minutes after administration were 2.4 and 8.3 in WT and KO mice, respectively, indicating that mdr1a/1b KO increased Kp,brain by 3.5-fold.

Plasma concentration–time profiles of E2074 in mdr1a/1b KO mice and WT mice (○, KO mice; ●, WT mice). The oral E2074 dose was 3 mg/kg, and each point represents the mean ± S.E. of three mice.
Discussion
In preclinical animals, nonlinear PK profiles with greater than dose-proportional increases in AUC and Cmax were observed after oral administration of E2074, and BA increased as the dose level increased (Fig. 2 and Table 1). An evident prolongation of t1/2 at higher oral dose was observed only in dogs, suggesting that the major cause of the nonlinear PK profile of E2074 was saturation of the first-pass effect in rats and monkeys, and saturation of the elimination of E2074 after entering the systemic circulation also contributed to the nonlinear PK profile in dogs in the dose range studied. E2074 was metabolized by liver microsomes in rats, dogs, and monkeys, and the estimated CLh,vitro from CLint,u was comparable with in vivo blood clearance (CLb) after i.v. administration (Table 3).
The accomplishment of in vitro–in vivo extrapolation using liver microsomes indicated that the main elimination route of E2074 after i.v. dosing in all species tested was hepatic metabolism; thus, the major reason for the nonlinear PK profile of E2074 after oral administration was initially believed to be saturation of hepatic metabolism. The Km estimated from enzyme kinetic studies using liver microsomes is one of the key parameters for evaluating the saturation of hepatic metabolism, and the estimated Km values were 7.23 μM for rats, 0.41 μM for dogs, and 42.44 μM for monkeys (Table 2).
The other parameter for assessing the saturation of hepatic metabolism is the concentration at the site of metabolism in the liver after oral administration of E2074. Estimation of the concentration at the metabolic site is difficult, but unbound Cmax in the portal vein (Cmax,pv,u) could be used as a surrogate concentration. In double-cannulated rats, Cmax,pv,u values were 10-fold higher than unbound systemic Cmax values (Fig. 5); thus, the Cmax,pv,u values at E2074 doses of 3, 10, and 30 mg/kg in intact rats were assumed to be 0.4, 3.3, and 22.6 μM, respectively (Tables 1 and 3). The ratios of Cmax,pv,u to Km (7.23 μM) at the three aforementioned E2074 doses in rat liver microsomes were 0.06, 0.46, and 3.1, respectively, suggesting that saturation of first-pass hepatic metabolism at 10 and 30 mg/kg was a key cause of the nonlinear PK profile in rats. In dogs, there were no data for Cmax,pv,u, and the unbound systemic plasma Cmax values (0.02 and 1.57 μM at 3 and 10 mg/kg oral doses, respectively) were used for comparison with the Km values obtained in the enzyme kinetic study (Tables 1 and 3). The ratios of unbound systemic plasma Cmax to Km1 were 0.05 at 3 mg/kg and 3.84 at 10 mg/kg, and the nonlinearity at 10 mg/kg in dogs could be attributed to the saturation of hepatic metabolism.
Regarding the differences between monkeys and rats/dogs, the Km value (42.44 μM) in monkey liver microsomes was much higher than the unbound systemic Cmax (0.78 μM) at 10 mg/kg; even if Cmax,pv,u was 10-fold higher than the unbound plasma Cmax, as also observed in rats, the Km was still higher than the Cmax,pv,u. Thus, the main cause of the nonlinear PK profile in monkeys was possibly not the saturation of hepatic metabolism.
In addition, the BA value in monkeys after oral dosing at 3 mg/kg (7.8%) was much lower than estimated hepatic availability (approximately 50%) calculated from the total clearance and hepatic blood flow rate, indicating incomplete absorption and/or extensive metabolism in the intestine. The ClogP of E2074 was 2.52, and the solubility of the drug in phosphate-buffered saline (pH 7.0–7.2) was more than 2 mg/ml. From these physicochemical properties, it can be speculated that membrane permeability and solubility would not limit the intestinal absorption of E2074; thus, the low gastrointestinal availability mediated by intestinal metabolism would be the main cause of the low BA and nonlinear PK profile in monkeys.
CYP3A substrates such as midazolam and nifedipine are metabolized in the intestine (Holtbecker et al., 1996; Akabane et al., 2010), and intestinal metabolic activity tends to be higher in monkey than in humans (Sakuda et al., 2006). As E2074 was principally metabolized by CYP3A4 (Fig. 4), similar to midazolam, intestinal metabolism was suspected to be the cause of low BA of E2074 in monkeys.
Among the intestinal microsomes from rats, dogs, and monkeys, only monkey intestinal microsomes exhibited E2074 metabolic activity. The Km value of monkey intestinal microsomes was estimated to be 34.60 μM, which was comparable to that measured in liver microsomes (Table 2). Although the concentration of E2074 in the intestine was not correctly estimated, it was higher than the Km, and the intestinal metabolic activity at higher doses (10 mg/kg) was less than that at lower doses (3 mg/kg), thus causing the nonlinear PK profile after oral administration in monkeys.
Ogasawara et al. studied the effect of the CYP3A4 inhibitor KCZ on midazolam and simvastatin PK profiles in cynomolgus monkeys (Ogasawara et al., 2007, 2009). In their report, concomitant oral administration of KCZ made it possible to clarify the contribution of intestinal metabolism in vivo, so our study applied the same methodology. In Table 4, concomitant oral administration of KCZ at 5 mg/kg did not affect the PK profile of E2074 after i.v. dosing, indicating that KCZ did not influence the hepatic metabolism of E2074. The effect of KCZ on an E2074 oral dose of 3 mg/kg was clearly demonstrated by the increased dose-normalized Cmax and AUC. The different effects of KCZ on the PK profiles of E2074 after i.v. and oral administration strongly indicated a significant contribution of intestinal metabolism after oral administration in monkeys. In addition, the dose-normalized Cmax and AUC values were similar at 3 and 10 mg/kg with concomitant KCZ administration, explaining the linear PK profile between 3 and 10 mg/kg if intestinal metabolism was abolished in monkeys. These results demonstrated that the nonlinear PK profile of E2074 in monkeys was caused by the saturation of first-pass intestinal metabolism.
E2074 is a P-gp substrate (flux ratio: 3.14) based on the result in mouse P-gp–overexpressing cells. P-gp is present in the intestine and brain blood capillaries, and it acts as a barrier to drug absorption and brain penetration. The brain penetration and intestinal absorption of fexofenadine are restricted by P-gp in mice (Tahara et al., 2005). As KCZ is an inhibitor of P-gp and CYP3A4, the contribution of P-gp to the absorption of E2074 in the intestine was elucidated using mdr1a/1b KO mice.
There were no differences in the plasma concentration–time profile, Cmax, or AUC between KO and WT mice; however, the Kp,brain in KO mice was 3.5-fold higher than that in the WT mice (Fig. 7; Table 5). These results indicated that P-gp did not affect the intestinal absorption or systemic exposure of E2074, but it restricted the brain penetration of the drug in mice.
Intestinal absorption can be estimated by simultaneously measuring plasma concentrations in portal and systemic veins (Kuze et al., 2009). The intestinal absorption of E2074 was estimated in double-cannulated rats (Fig. 5), and the absorption was estimated to be almost complete (116%) in these rats (unpublished data), which further supports the very minor at most contribution of P-gp to E2074 absorption. These results suggest that the effect of P-gp on E2074 absorption in intestine is limited, and the saturation of P-gp transport activity would not be the cause of nonlinear PK profiles in monkeys as observed in rats and dogs.
In conclusion, we clarified the cause of nonlinear PK profiles of E2074 in animals, identifying the saturation of hepatic metabolism in rats and dogs, and the saturation of intestinal metabolism in monkeys as the primary mechanisms.
Acknowledgments
The authors thank Drs. Tatsuhiro Onogi (director of Department of Pharmacology), Fumihiro Ozaki, and Motohiro Soejima (Department of Medicinal Chemistry) for the synthesis of E2074, and Drs. Nancy Wong and Anne Cooper for useful discussions.
Authorship Contributions
Participated in research design: Nagaya, Takenaka.
Conducted experiments: Nagaya.
Performed data analysis: Nagaya.
Wrote or contributed to the writing of the manuscript: Nagaya, Takenaka, Kusano, Yoshimura.
Footnotes
- Received November 15, 2012.
- Accepted February 11, 2013.
This work was previously partly presented as a poster presentation at the following conference: Nagaya Y, Takenaka O, Kusano K, and Yoshimura T (2009) Clarification of species difference in the cause of nonlinear PK profile. 16th North American Meeting of the International Society for the Study of Xenobiotics; 2009 Oct 18–22; Baltimore, MD.
Abbreviations
- AUC
- area under the plasma concentration curve
- BA
- bioavailability
- CL
- clearance
- E2074
- 2-[(2R)-2-fluoro-3-{(3r)-[(3-fluorobenzyl)oxy]-8-azabicyclo[3.2.1]oct-8-yl}propyl]-4,5-dimethyl-2,4-dihydro-3H-1,2,4-triazol-3-one
- fmic
- unbound fraction in liver microsomes
- fp
- unbound fraction in plasma
- HBSS
- Hank’s balanced salts solution
- IS
- internal standard
- i.v.
- intravenous administration
- KCZ
- ketoconazole
- Km
- Michaelis–Menten constant
- KO
- knockout
- LC/MS/MS
- high-performance liquid chromatography with tandem mass spectrometry
- MK-826
- carbapenem antibiotic
- MRP2
- multidrug resistance-associated protein 2
- P450
- cytochrome P450
- PAR
- peak area ratio
- P-gp
- P-glycoprotein
- PK
- pharmacokinetics
- r
- Hill coefficient
- Rb
- blood to plasma partition ratio
- rP450
- recombinant human cytochrome P450
- t1/2
- half-life
- tmax
- time to reach Cmax
- Vmax
- maximum velocity
- Vss
- steady-state volume of distribution
- WT
- wild-type
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}